Investigation of the Ergopeptide Epimerization Process


Abstract
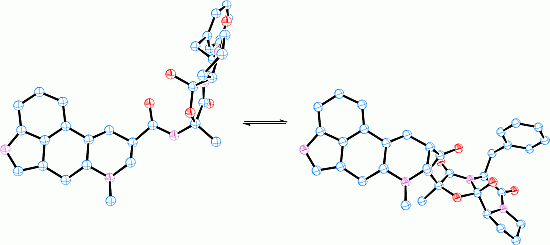
:
1. Introduction
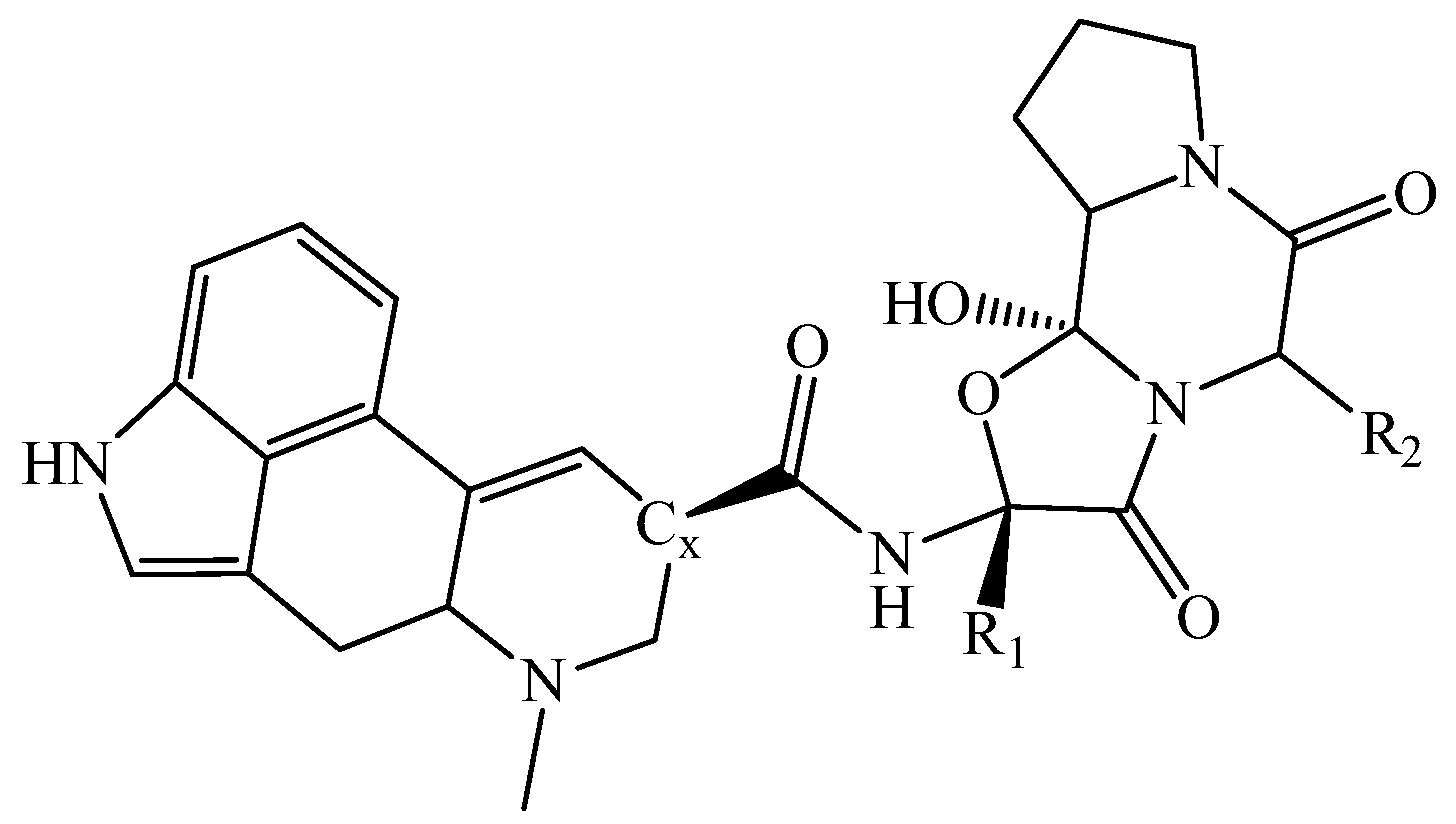
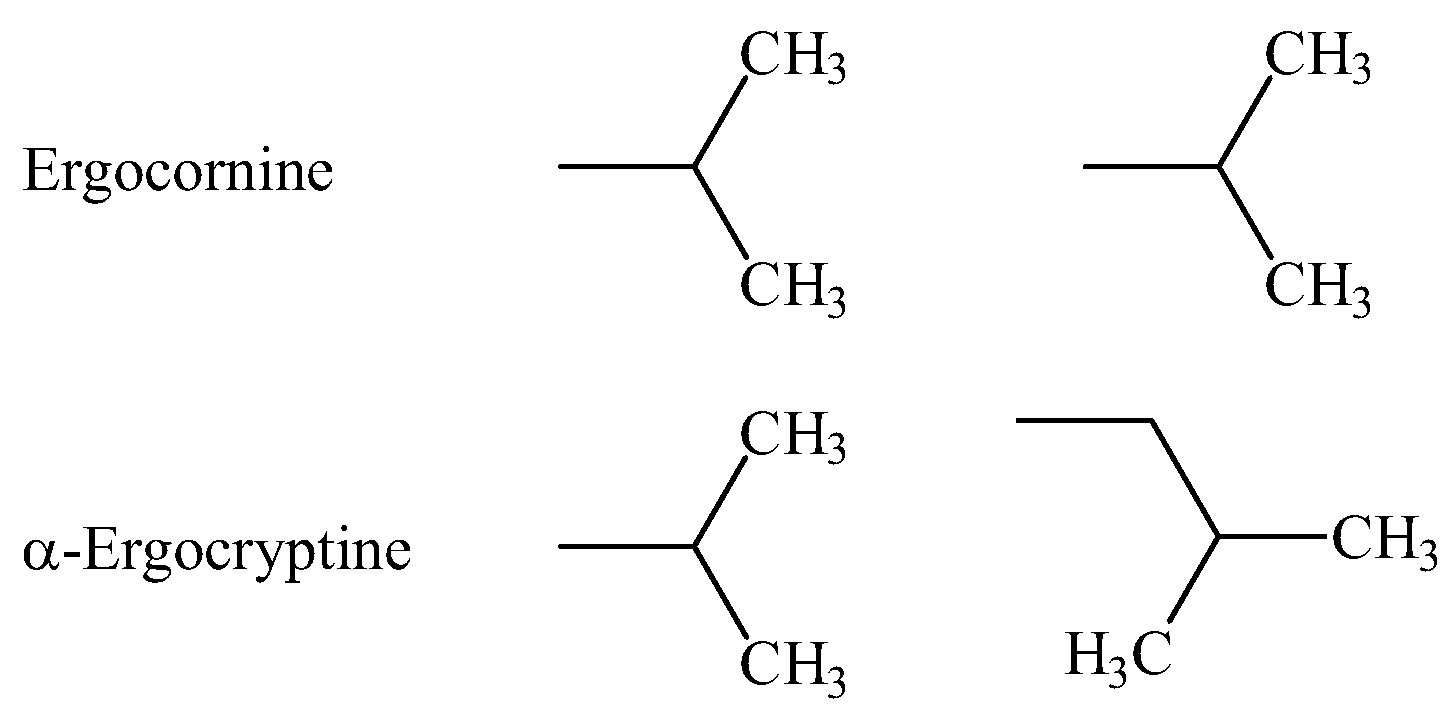
2. Quantum Chemical and Classical Simulation of Ergopeptides
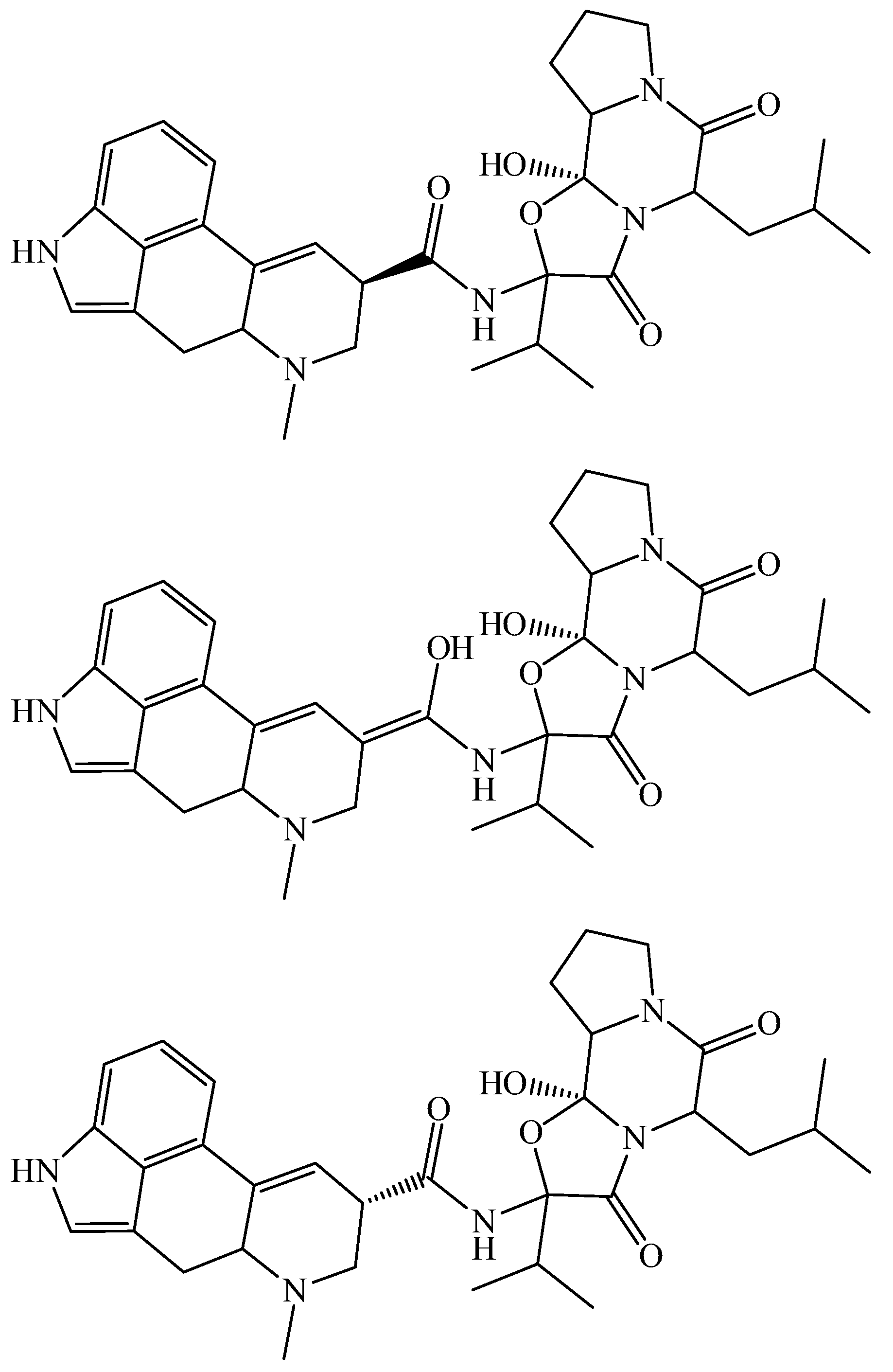

{kind=link}
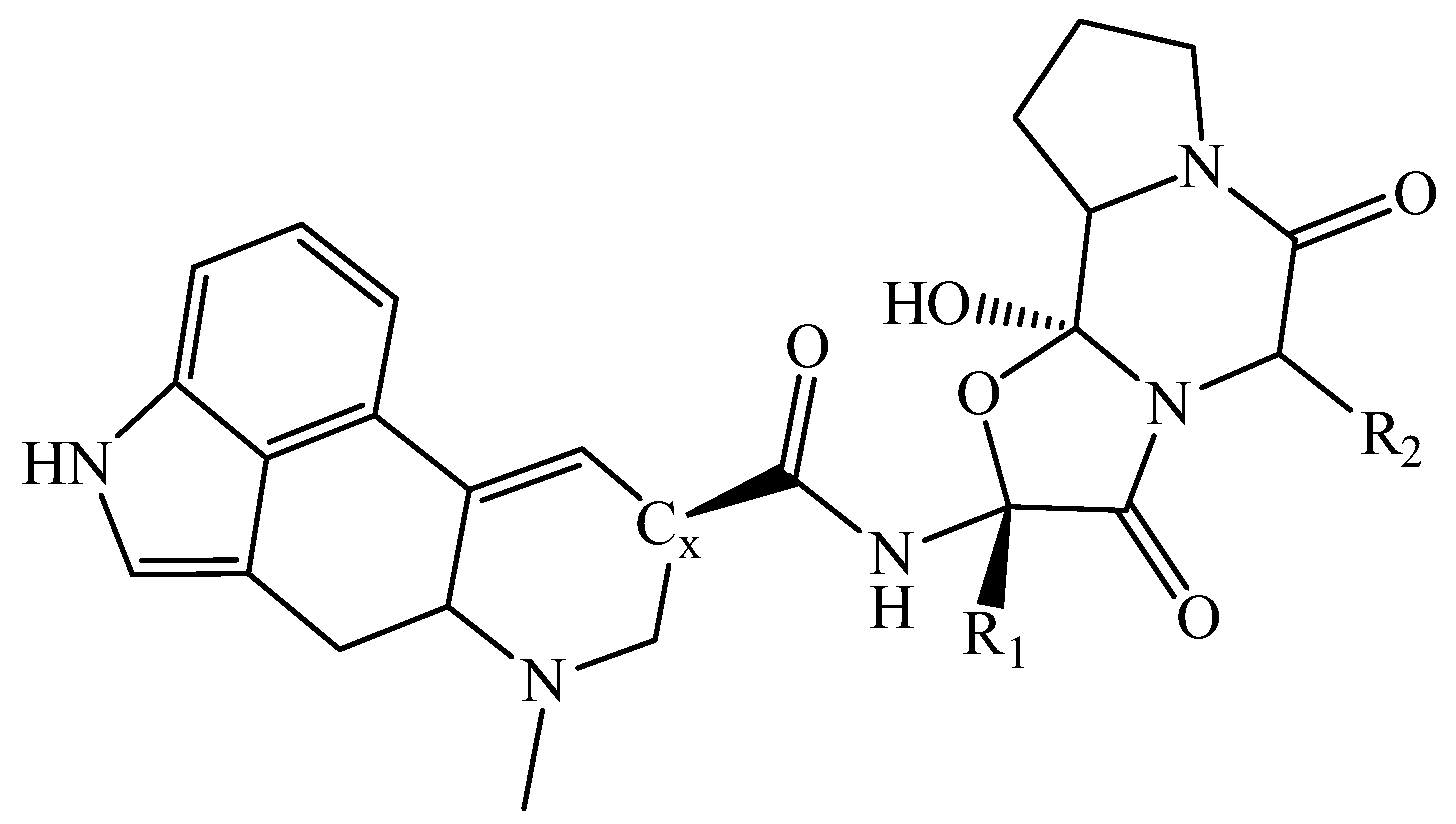{kind=link}
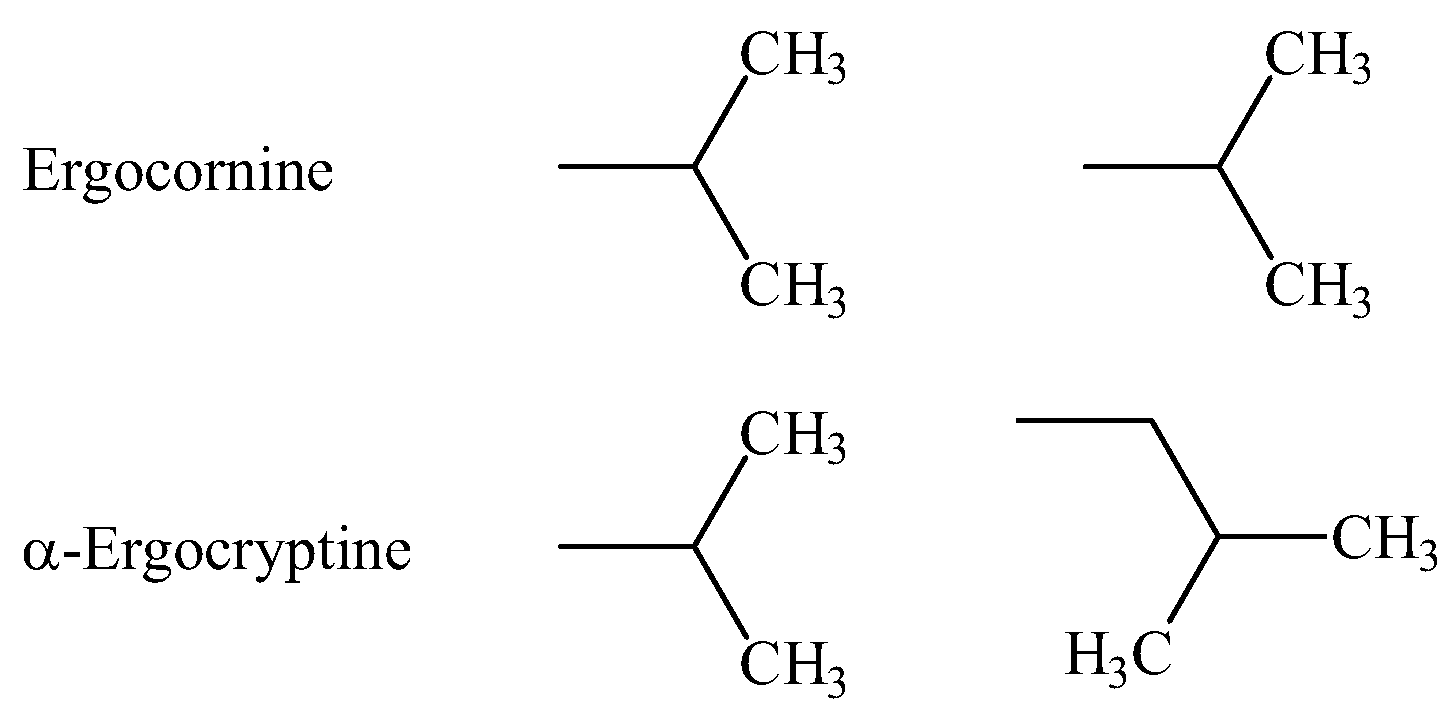{kind=link}
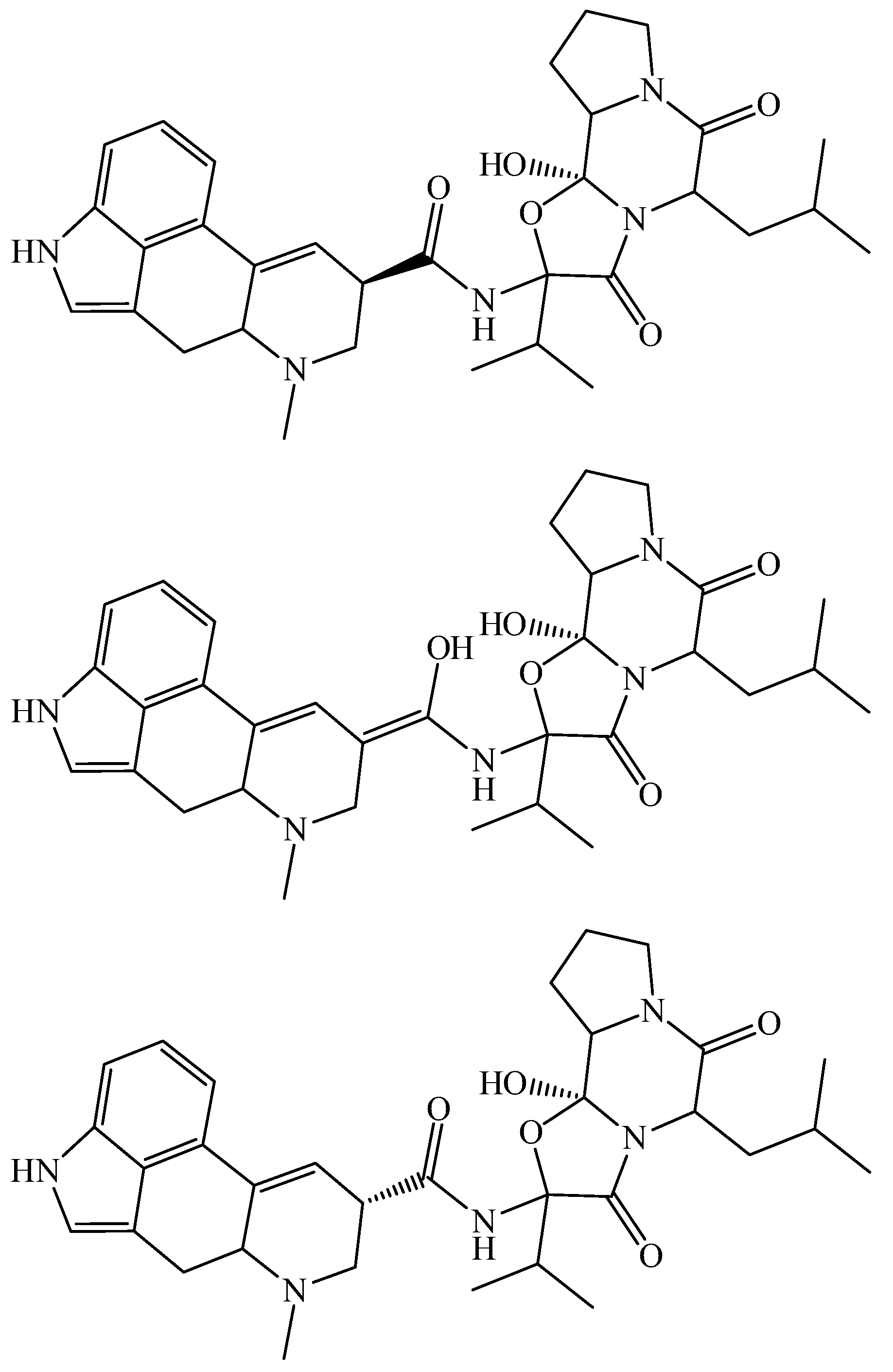{kind=link}
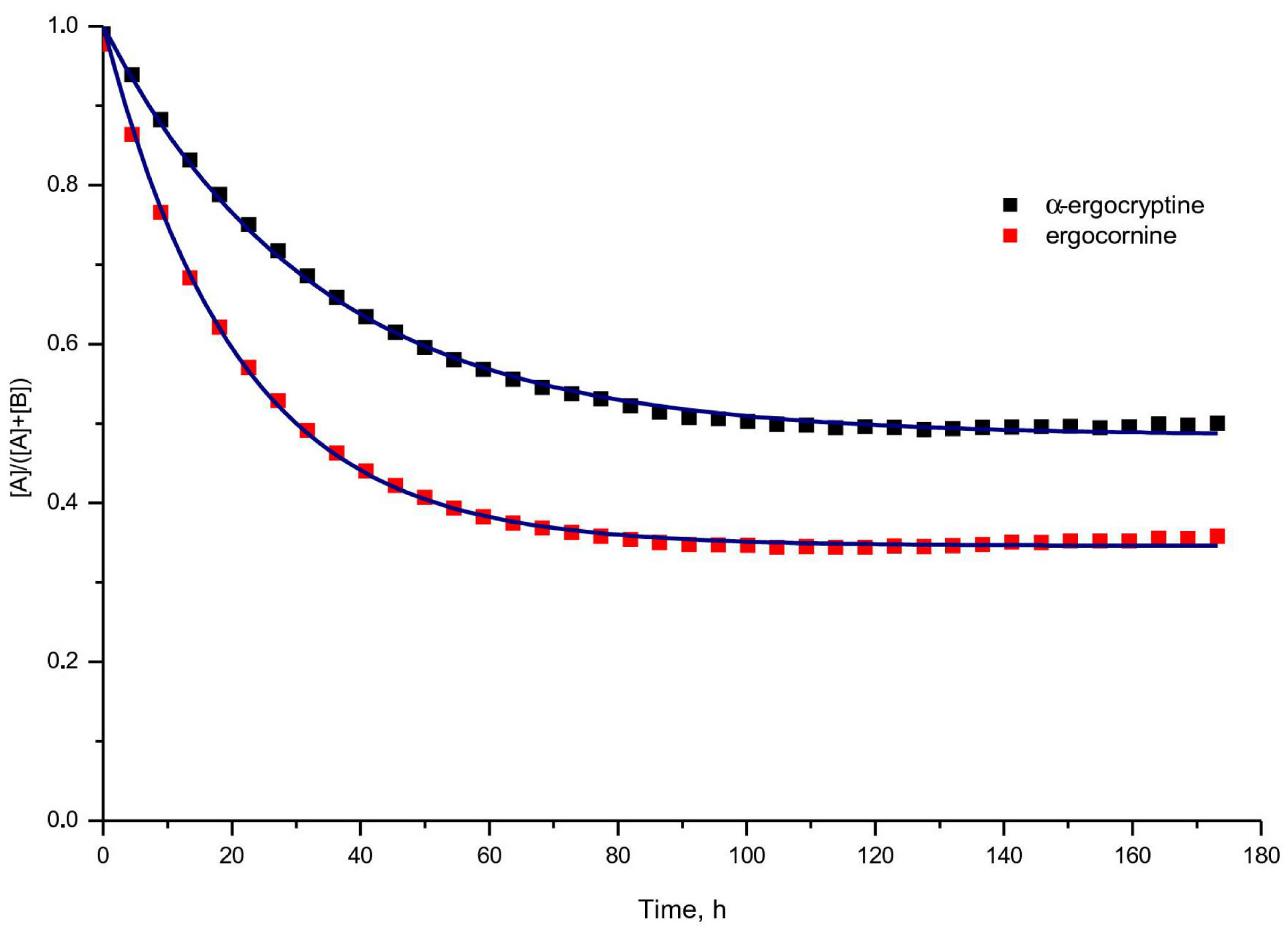{kind=link}
| Epimers | Energy in a.u. |
|---|---|
| R-α-ergocryptine | −1893.9465 |
| R-ergocornine | −1854.6201 |
| S-α-ergocryptine | −1893.9364 |
| S-ergocornine | −1854.6401 |
| I-α-ergocryptine | −1893.9113 |
| I-ergocornine | −1854.5965 |
| Ergotalkaloid | Water | ACN | |||
|---|---|---|---|---|---|
| ϕ | Var | ϕ | Var | ||
| R-α-ergocryptine | 139.7 | 9.7 | 142.7 | 8.3 | |
| R-ergocornine | 134.3 | 16.7 | 129.8 | 21.1 | |
| S-α-ergocryptine | 105.9 | 6.2 | 76.1 | 6.0 | |
| S-ergocornine | 105.7 | 6.5 | 106.5 | 6.3 | |
| I-α-ergocryptine | 169.6 | 7.9 | 103.5 | 4.3 | |
| I-ergocornine | 168.0 | 8.8 | 170.0 | 7.5 | |
3. Laboratory Experiments
3.1. Experimental Procedure
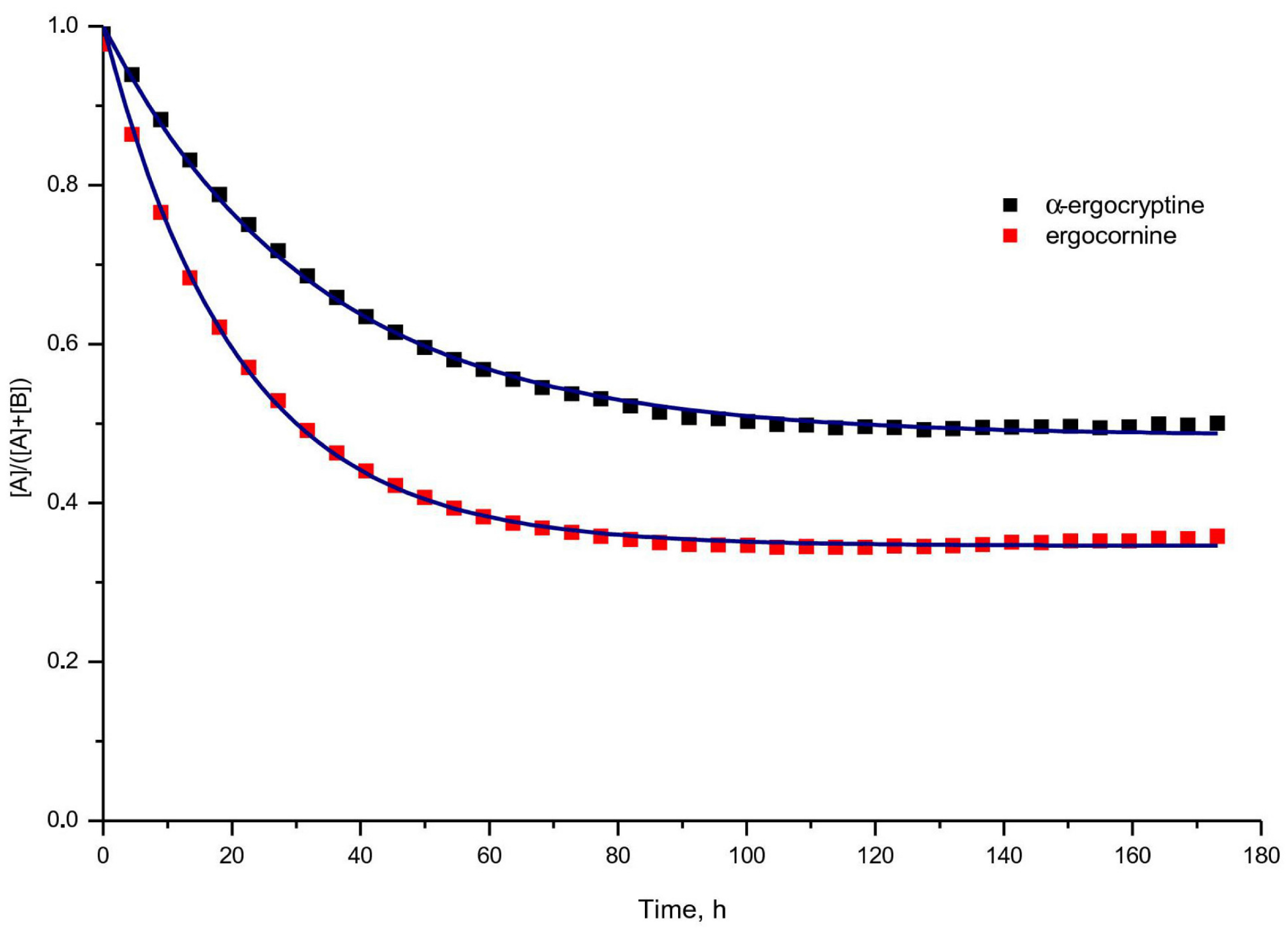
3.2. Standard Kinetic Analysis
| Solvent Mixture | Ergot Alkaloid | k,10 h | k’,10 | K | Fit-Function |
|---|---|---|---|---|---|
| i | α-ergo-cryptine | 15.67 ± 0.16 | 14.75 ± 0.22 | 1.06 ± 0.02 | 0.9982 |
| ergo-cornine | 31.54 ± 0.31 | 16.70 ± 0.23 | 1.89 ± 0.03 | 0.9983 | |
| ii | α-ergo-cryptine | 1.64 ± 0.01 | 0.58 ± 0.07 | 2.83 ± 0.34 | 0.9997 |
| ergo-cornine | 3.70 ± 0.07 | 2.20 ± 0.25 | 1.68 ± 0.19 | 0.9959 |
3.3. Experimental Setting
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Richards, I.S. Principles and Practice of Toxicology in Public Health; Jones & Bartlett: Sudbury, MA, USA, 2008; p. 64. [Google Scholar]
- Lüllmann, H.; Mohr, K.; Hein, L. Pharmakologie und Toxikologie; Thieme: Stuttgart, Germany, 2006; p. 283. [Google Scholar]
- Schardl, C.L.; Panaccione, D.G.; Tudzynski, P. Ergot alkaloids–biology and molecular biology. Alkaloids: Chem. Biol. 2006, 63, 45–86. [Google Scholar]
- Christ, C.D.; Mark, A.E.; van Gunsteren, W.F. Basic ingredients of free energy calculations: A review. J. Comput. Chem. 2010, 31, 1569–1582. [Google Scholar] [CrossRef] [PubMed]
- Pohorille, A.; Jarzynski, C.; Chipot, C. Good Practices in Free-Energy Calculations. J. Phys. Chem. B 2010, 114, 10235–10253. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Mark, A.E. Calculation of the free energy of solvation for neutral analogs of amino acid side chains. J. Comput. Chem. 2002, 23, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Becker, R.; Köppen, R.; Durmaz, V. Classical hybrid Monte-Carlo simulations of the interconversion of hexabromocyclododecane. J. Mol. Simul. 2008, 34, 727–736. [Google Scholar] [CrossRef]
- Kriegl, J.M.; Nienhaus, K.; Deng, P.; Fuchs, J.; Nienhaus, G.U. Ligand Dynamics in a Protein Internal Cavity. Proc. Natl. Acad. Sci. USA 2003, 100, 7069–7074. [Google Scholar] [CrossRef] [PubMed]
- Duane, S.; Kennedy, A.D.; Pendleton, B.J.; Roweth, D. Hybrid Monte Carlo. Phys. Lett. B 1987, 195, 216–222. [Google Scholar] [CrossRef]
- Brass, A.; Pendleton, B.J.; Chen, Y.; Robson, B. Hybrid Monte Carlo Simulations Theory and Initial Comparison with Molecular Dynamics. Biopolymers 1993, 33, 1307–1315. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field: I–V. J. Comp. Chem. 1996, 17, 490–641. [Google Scholar] [CrossRef]
- Gelman, A.; Rubin, D. Inference from Iterative Simulation using Multiple Sequences. Statist. Sci. 1992, 7, 457–511. [Google Scholar] [CrossRef]
- Hestenes, M.R.; Stiefel, E. Methods of Conjugate Gradients for Solving Linear Systems. J. Res. Nat. Bur. Stand. 1952, 49, 409–436. [Google Scholar] [CrossRef]
- Fletcher, R.; Reeves, C.M. Function minimization by conjugate gradients. Comput. J. 1962, 7, 149–154. [Google Scholar] [CrossRef]
- Durmaz, V.; Becker, R.; Weber, M. How to Simulate Affinities for Host-Guest Systems Lacking Binding Mode Information: Application in the Liquid Chromatographic Separation of Hexabromocyclododecane Stereoisomers. J. Mol. Model. 2012, 18, 2399–2408. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollman, P.A. Application of RESP Charges To Calculate Conformational Energies, Hydrogen Bond Energies, and Free Energies of Solvation. J. Am. Chem. Soc. 1993, 115, 9620–9631. [Google Scholar] [CrossRef]
- Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665–9678. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, A.M.; Lyubartsev, A.P. New Six-site Acetonitrile Model for Simulations of Liquid Acetonitrile and its Aqueous Mixtures. J. Comput. Chem. 2007, 28, 2020–2026. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. Comput. Phys. Commun. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8592. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Storm, I.D.; Rasmussen, P.H.; Strobel, B.W.; Hansen, H.C.B. Ergot alkaloids in rye flour determined by solid-phase cation-exchange and high-pressure liquid chromatography with fluorescence detection. Food Addit. Contam. 2008, 25, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Klaffke, H.S.; Krauthause, W.; Wittkowski, R. Determination of ergot alkaloids in rye and rye flour. Mycotoxin Res. 2006, 22, 197–200. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Andrae, K.; Merkel, S.; Durmaz, V.; Fackeldey, K.; Köppen, R.; Weber, M.; Koch, M. Investigation of the Ergopeptide Epimerization Process. Computation 2014, 2, 102-111. https://doi.org/10.3390/computation2030102
Andrae K, Merkel S, Durmaz V, Fackeldey K, Köppen R, Weber M, Koch M. Investigation of the Ergopeptide Epimerization Process. Computation. 2014; 2(3):102-111. https://doi.org/10.3390/computation2030102
Chicago/Turabian StyleAndrae, Karsten, Stefan Merkel, Vedat Durmaz, Konstantin Fackeldey, Robert Köppen, Marcus Weber, and Matthias Koch. 2014. "Investigation of the Ergopeptide Epimerization Process" Computation 2, no. 3: 102-111. https://doi.org/10.3390/computation2030102