
Population Genomics, Transcriptional Response to Heat Shock, and Gut Microbiota of the Hong Kong Oyster Magallana hongkongensis

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
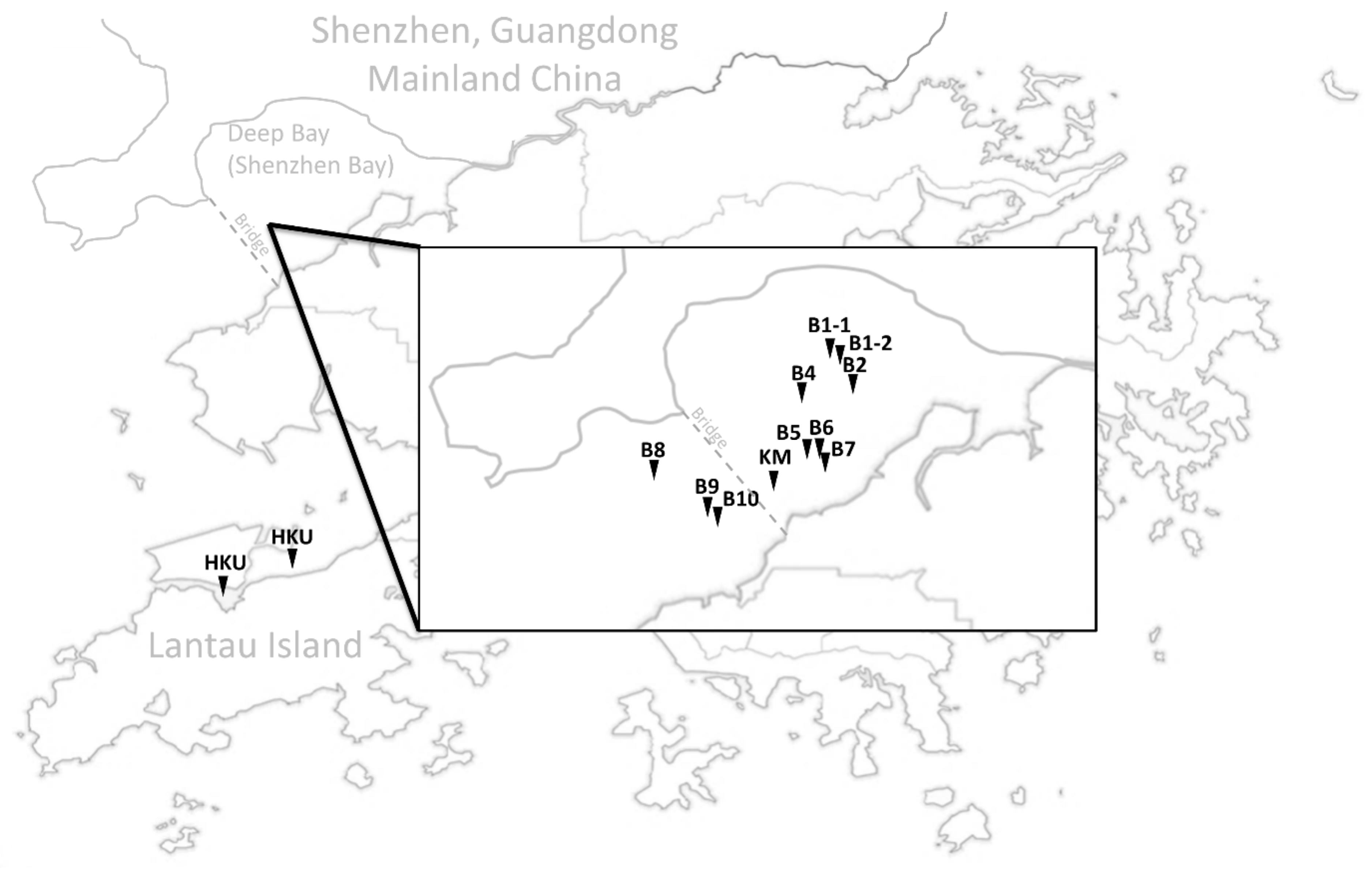
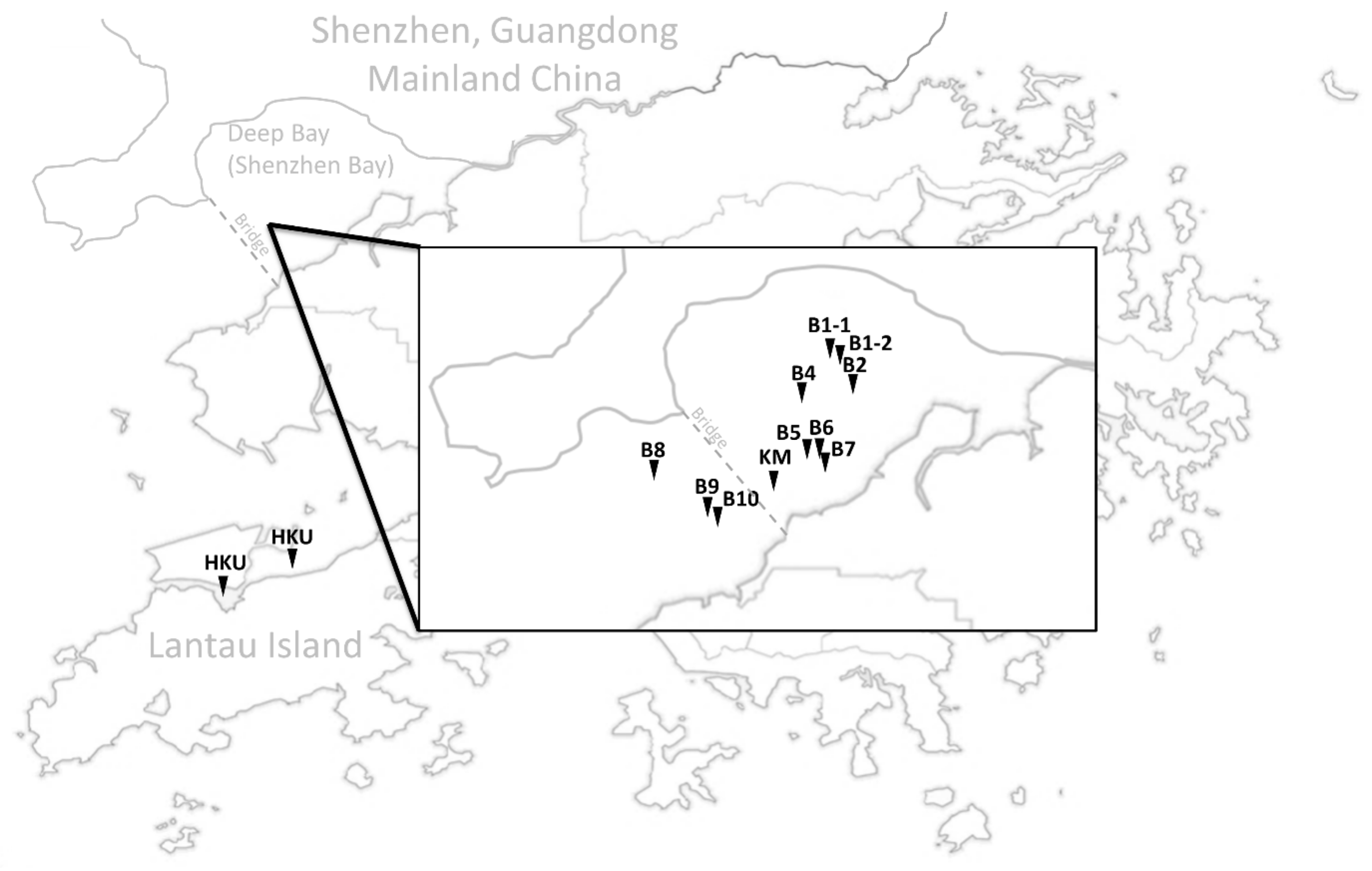
2.1. Samples Collection, DNA Extraction, Species Identification, Sequencing
2.2. Bioinformatic Analyses
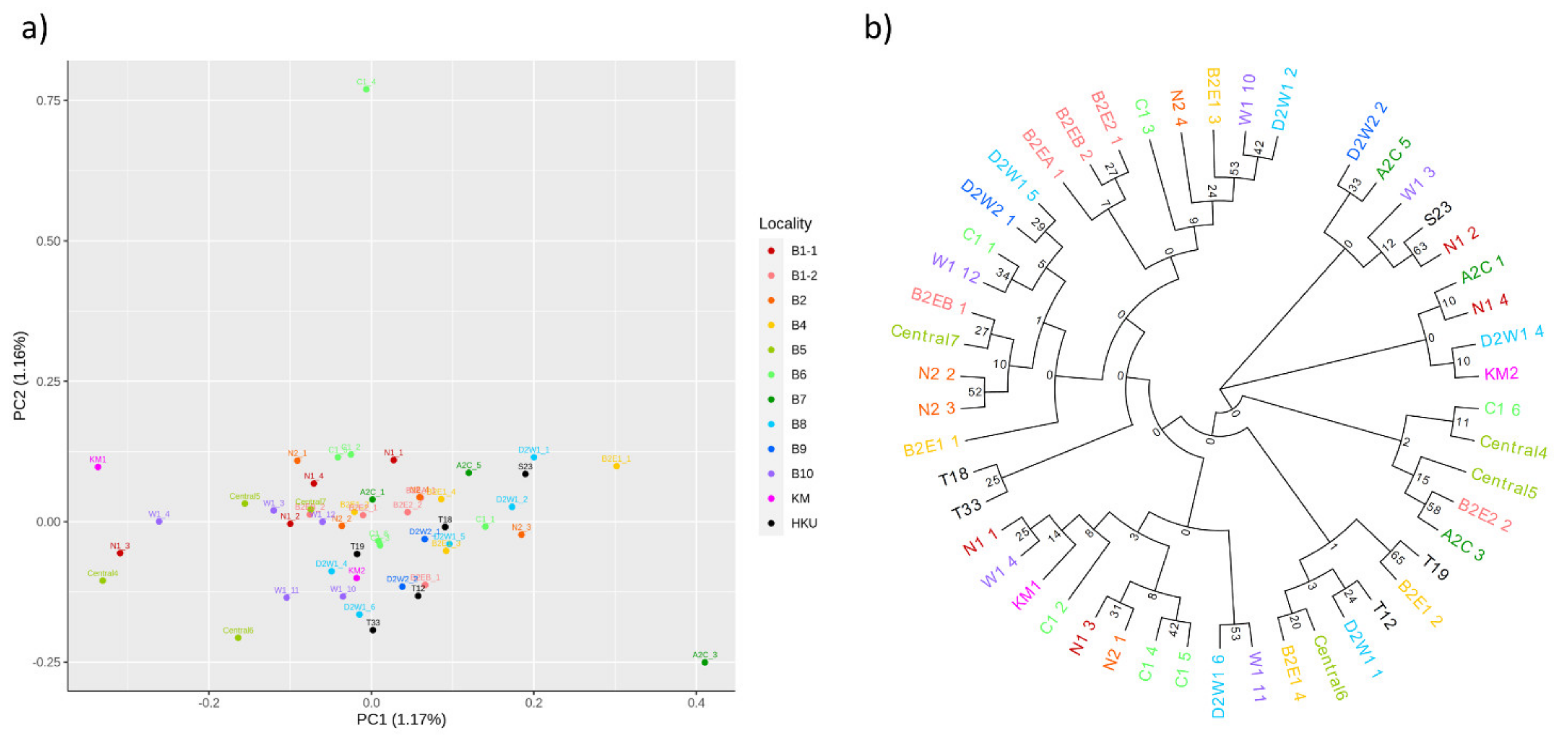
2.3. Population Genomic Analyses
2.4. Annotation of Homeobox Gene and Heat Shock Protein Family
2.5. Heat Shock Treatment and RNA-seq
2.6. Gene Model Prediction and Transcriptome Profiling
2.7. Gut Microbiota Analysis
2.8. Data Availability
3. Results
3.1. Population Relationships of Local Oysters
3.2. SNPs and INDELs in Homeobox Genes and Heat Shock Protein Genes
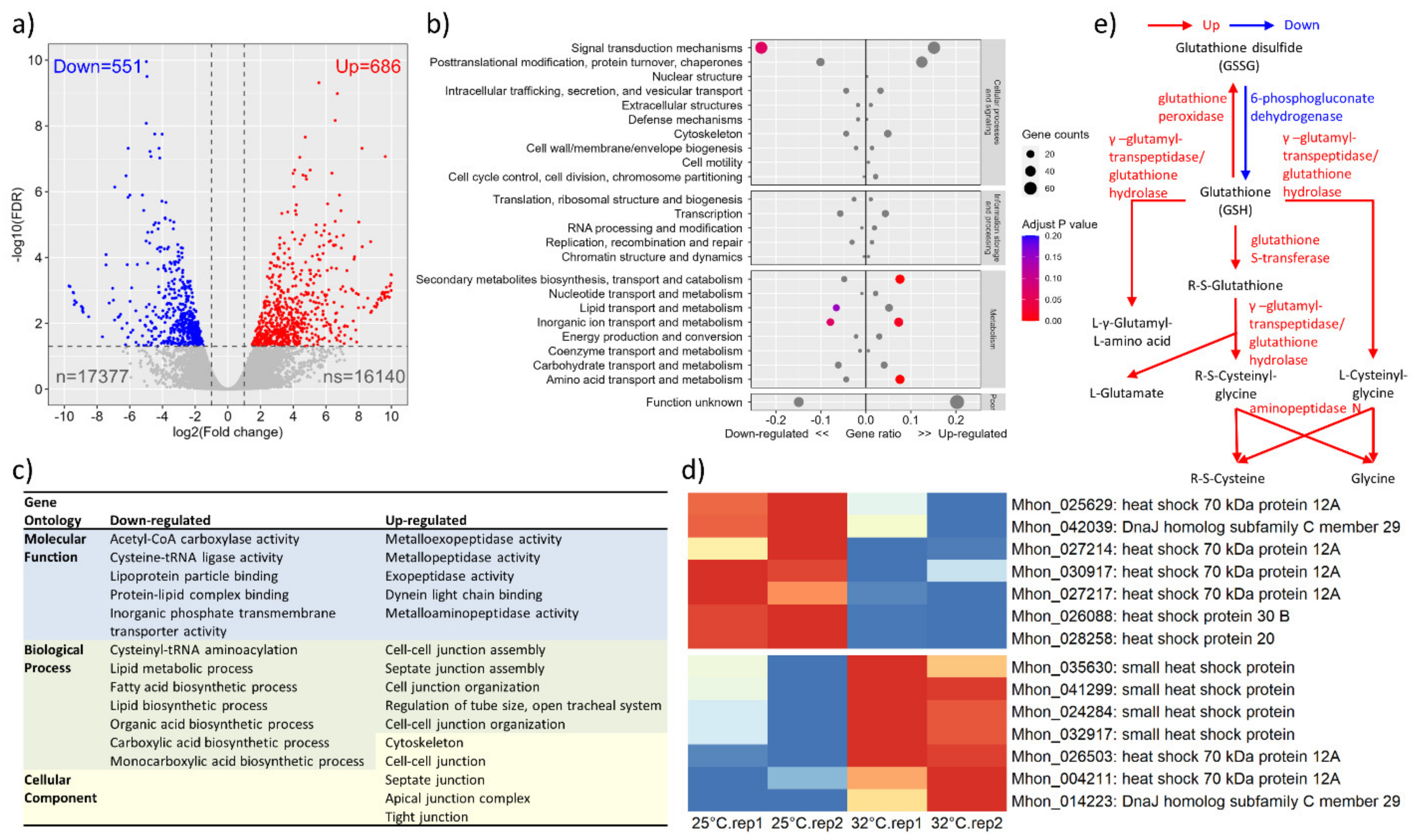
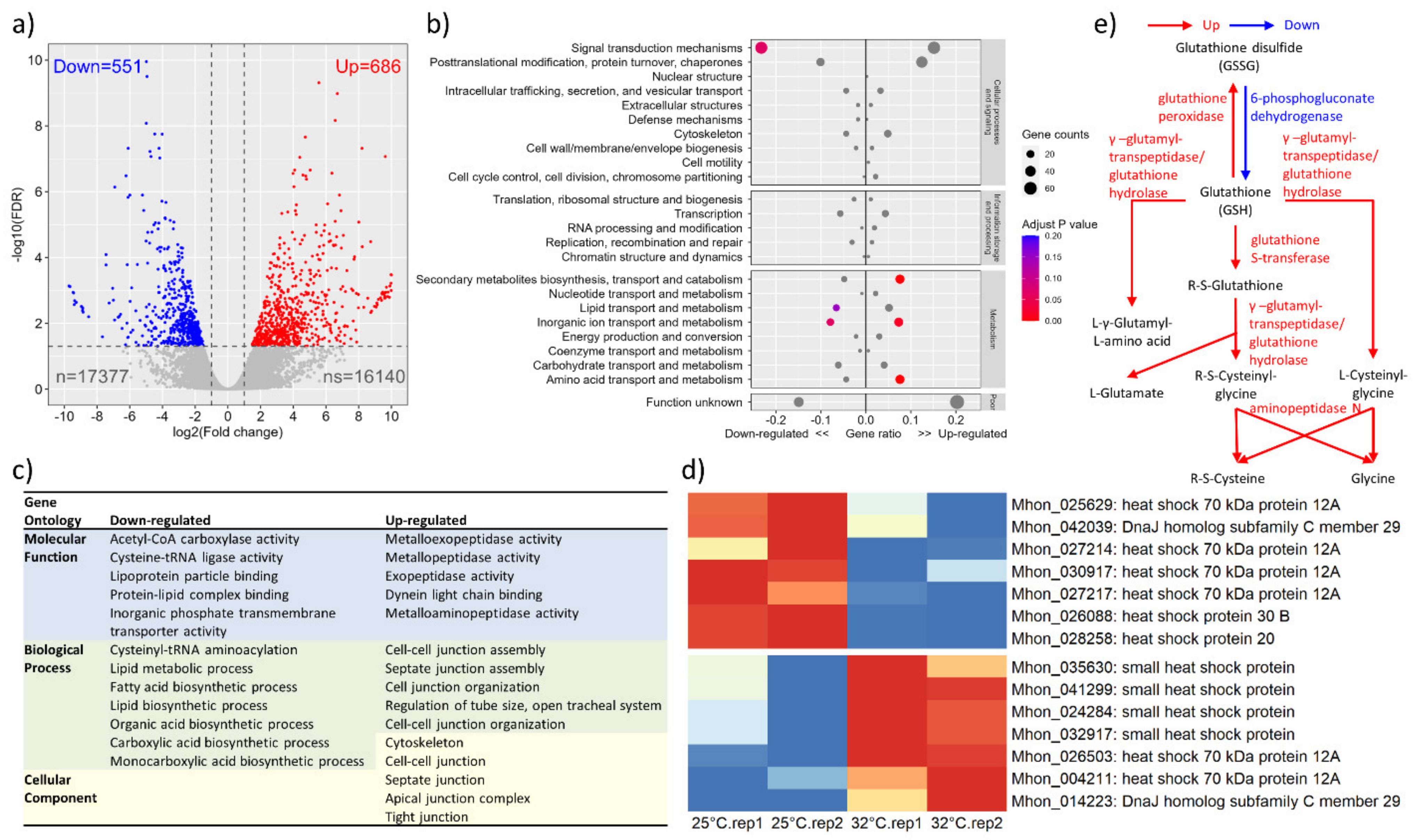
3.3. Transcriptome Profile in Response to Heat Shock
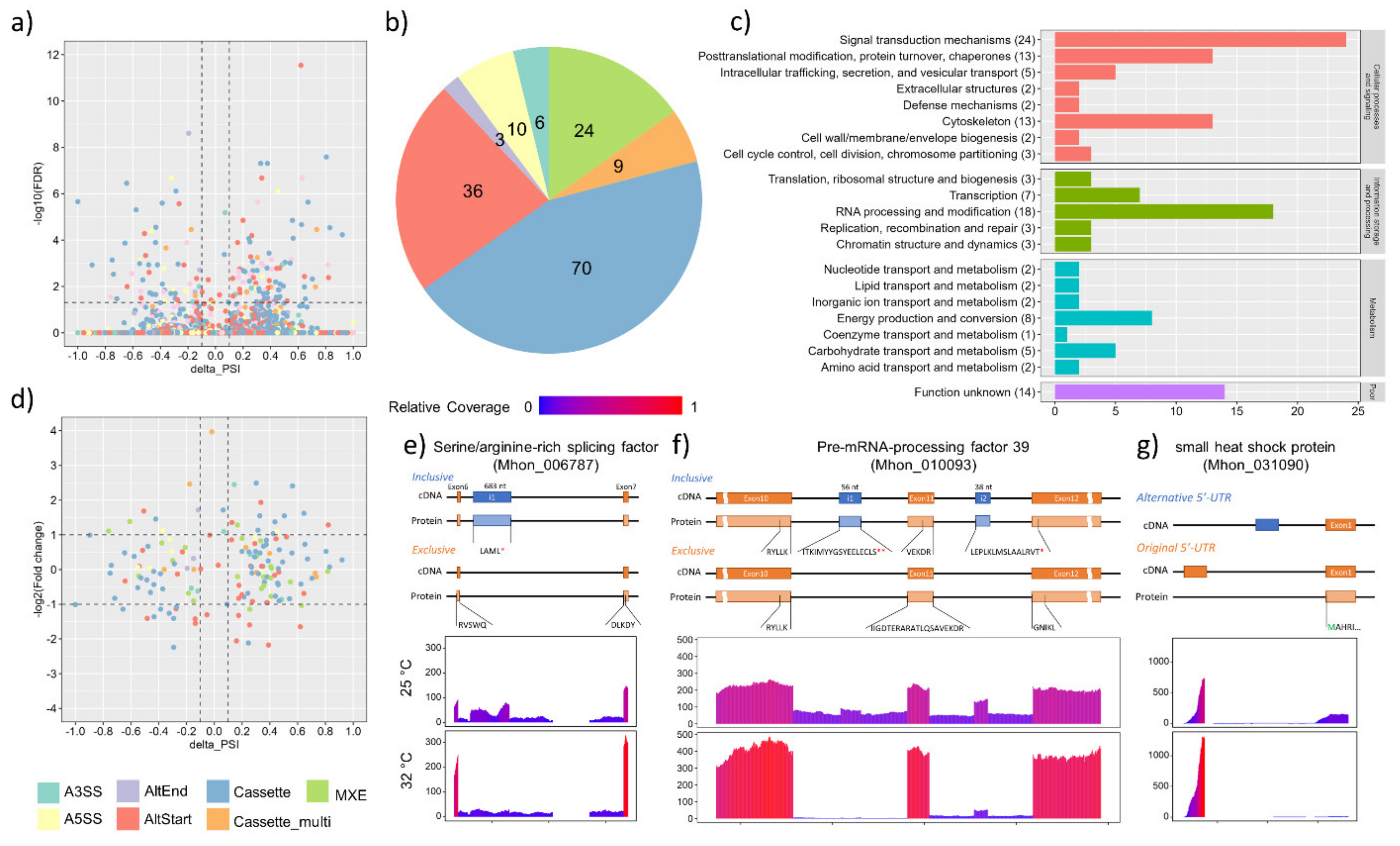
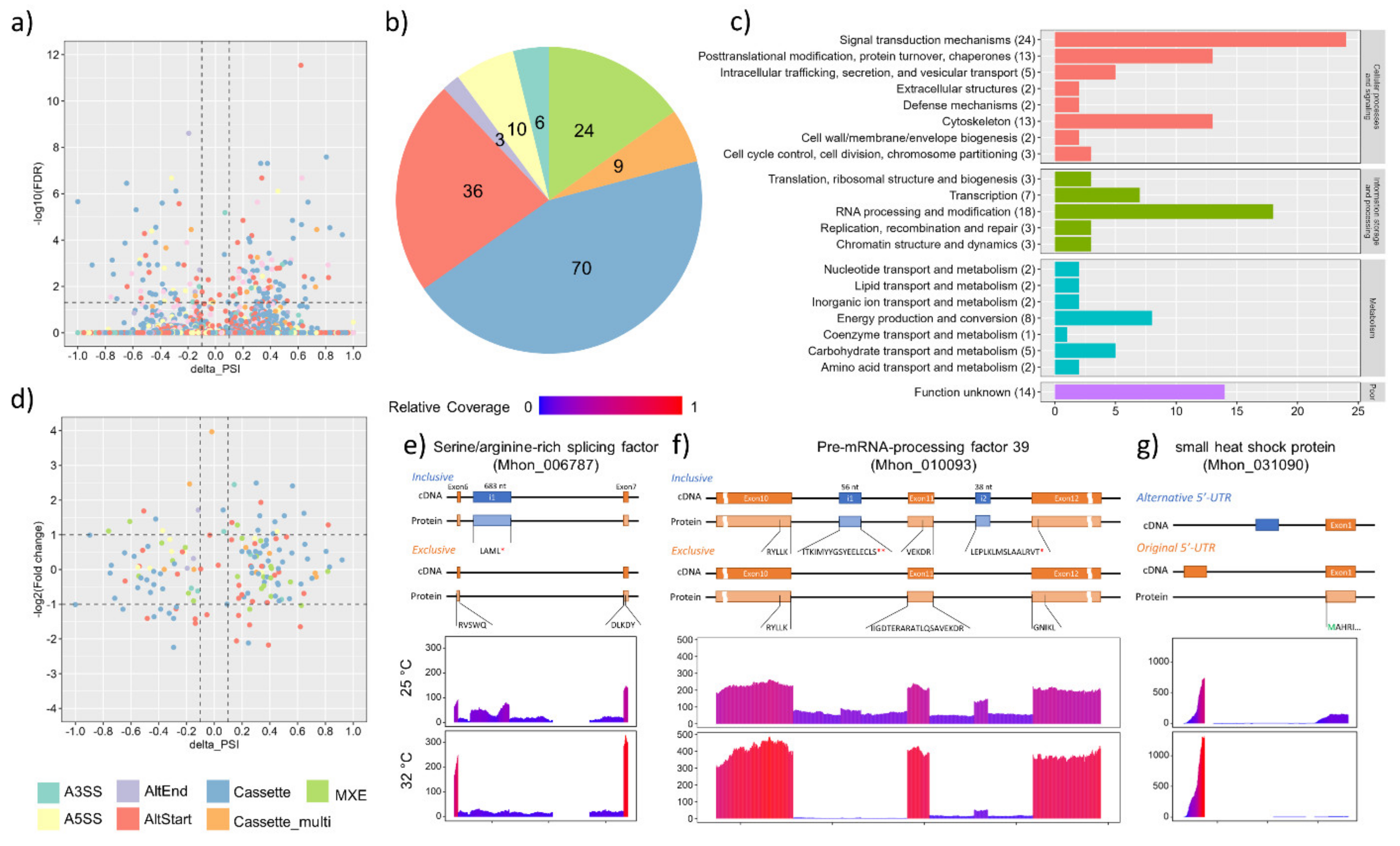
3.4. Alternative Splicing Events under Heat Shock Treatment
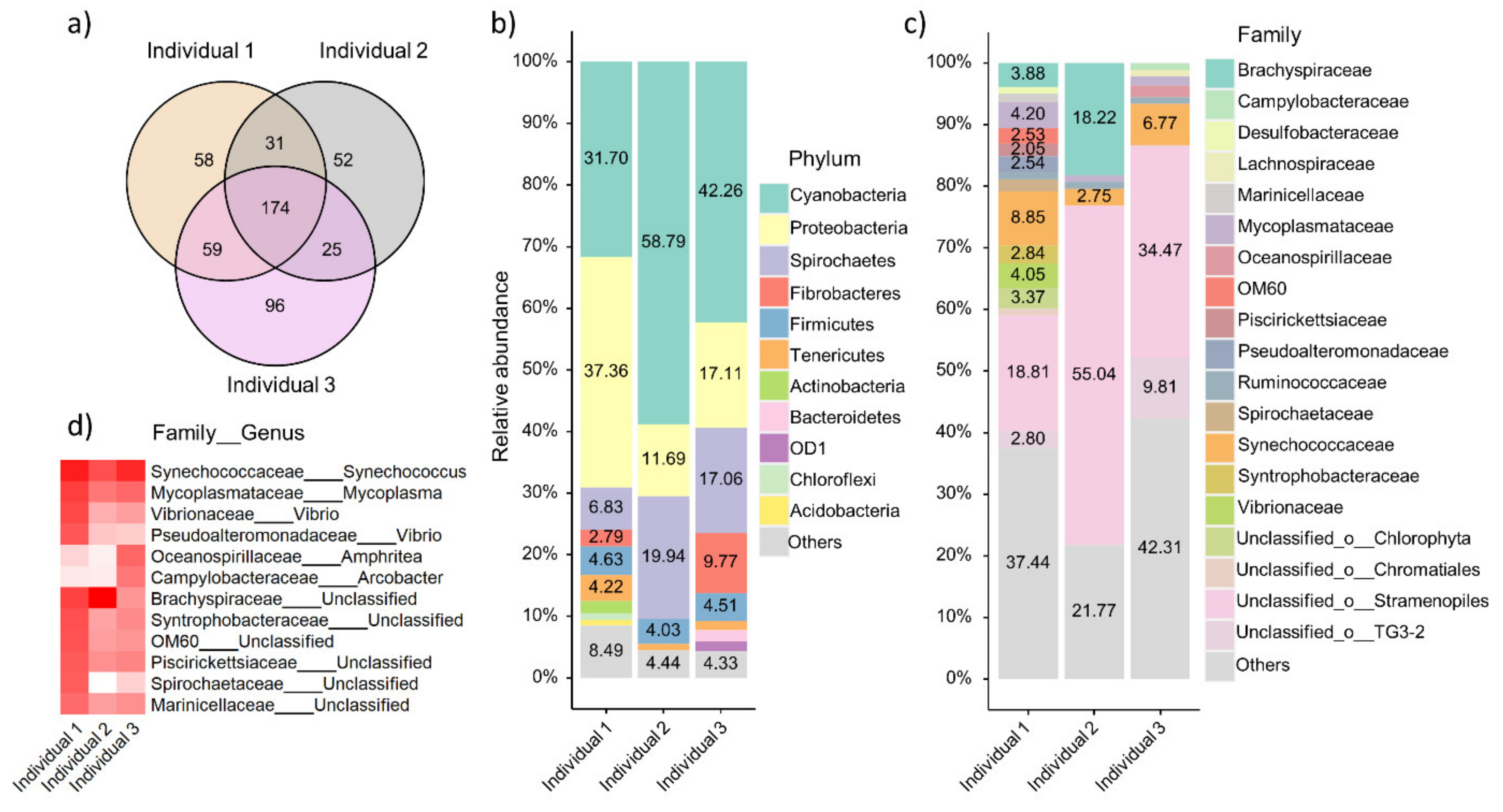
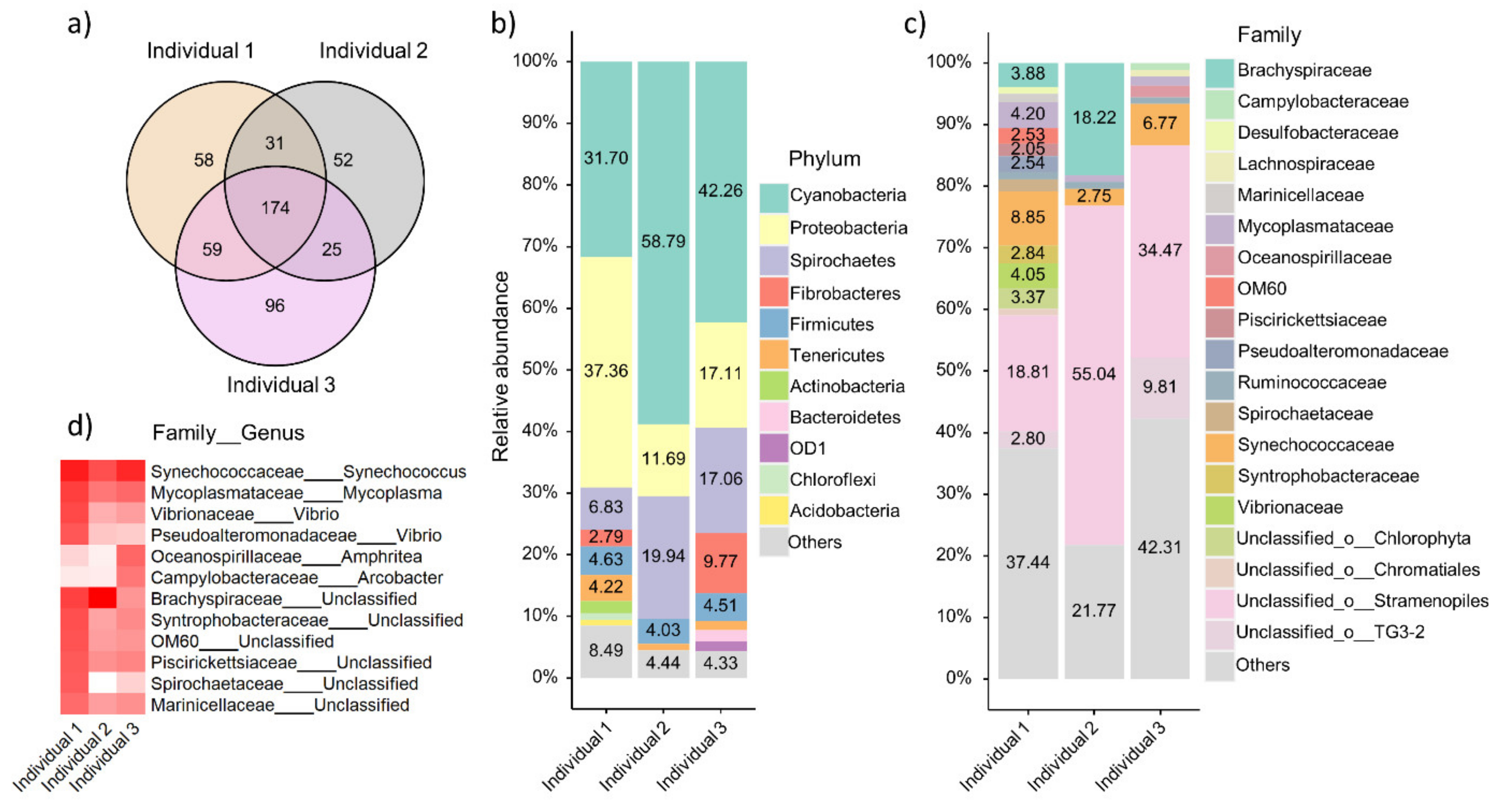
3.5. Taxonomic Composition of Local Oyster Gut Microbiome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Report on the Public Engagement on Promotion of Sustainable Consumption of Biological Resources. 2017. Available online: https://www.enb.gov.hk/sites/default/files/susdev/html/en/council/pscbr_sdc_e.pdf (accessed on 7 February 2022).
- Lam, K.; Morton, B. Mitochondrial DNA and morphological identification of a new species of Crassostrea (Bivalvia: Ostreidae) cultured for centuries in the Pearl River Delta, Hong Kong, China. Aquaculture 2003, 228, 1–13. [Google Scholar] [CrossRef]
- Salvi, D.; Macali, A.; Mariottini, P. Molecular phylogenetics and systematics of the bivalve family Ostreidae based on rRNA sequence-structure models and multilocus species tree. PLoS ONE 2014, 9, e108696. [Google Scholar] [CrossRef]
- Peng, D.; Zhang, S.; Zhang, H.; Pang, D.; Yang, Q.; Jiang, R.; Lin, Y.; Mu, Y.; Zhu, Y. The oyster fishery in China: Trend, concerns and solutions. Mar. Policy 2021, 129, 104524. [Google Scholar] [CrossRef]
- Lam, K.; Morton, B. The oysters of Hong Kong (Bivalvia: Ostreidae and Gryphaeidae). Raffles Bull. Zool. 2004, 52, 11–28. [Google Scholar]
- Talmage, S.C.; Gobler, C.J. Effects of past, present, and future ocean carbon dioxide concentrations on the growth and survival of larval shellfish. Proc. Natl. Acad. Sci. USA 2010, 107, 17246–17251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenz, M.; da Gama, B.A.P.; Gerner, N.V.; Gobin, J.; Gröner, F.; Harry, A.; Jenkins, S.R.; Kraufvelin, P.; Mummelthei, C.; Sareyka, J.; et al. Non-native marine invertebrates are more tolerant towards environmental stress than taxonomically related native species: Results from a globally replicated study. Environ. Res. 2011, 111, 943–952. [Google Scholar] [CrossRef]
- King, W.L.; Jenkins, C.; Seymour, J.R.; Labbate, M. Oyster disease in a changing environment: Decrypting the link between pathogen, microbiome and environment. Mar. Environ. Res. 2019, 143, 124–140. [Google Scholar] [CrossRef]
- Thomas, Y.; Pouvreau, S.; Alunno-Bruscia, M.; Barillé, L.; Gohin, F.; Bryère, P.; Gernez, P. Global change and climate-driven invasion of the Pacific oyster (Crassostrea gigas) along European coasts: A bioenergetics modelling approach. J. Biogeogr. 2016, 43, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Qin, J.G.; Abbott, C.A.; Li, X.; Benkendorff, K. Synergistic impacts of heat shock and spawning on the physiology and immune health of Crassostrea gigas: An explanation for summer mortality in Pacific oysters. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2007, 293, 2353–2362. [Google Scholar] [CrossRef] [Green Version]
- Howie, A.H.; Bishop, M.J. Contemporary oyster reef restoration: Responding to a changing world. Front. Ecol. Evol. 2021, 9, 518. [Google Scholar] [CrossRef]
- Wang, R.X.; Wang, A.L.; Wang, J.Y. Antibiotic resistance monitoring in heterotrophic bacteria from anthropogenic-polluted seawater and the intestines of oyster Crassostrea hongkongensis. Ecotoxicol. Environ. Saf. 2014, 109, 27–31. [Google Scholar] [CrossRef]
- Li, Y.; Nong, W.; Baril, T.; Yip, H.Y.; Swale, T.; Hayward, A.; Ferrier, D.E.K.; Hui, J.H.L. Reconstruction of ancient homeobox gene linkages inferred from a new high-quality assembly of the Hong Kong oyster (Magallana hongkongensis) genome. BMC Genomics 2020, 21, 1–17. [Google Scholar] [CrossRef]
- Peng, J.; Li, Q.; Xu, L.; Wei, P.; He, P.; Zhang, X.; Zhang, L.; Guan, J.; Zhang, X.; Lin, Y.; et al. Chromosome-level analysis of the Crassostrea hongkongensis genome reveals extensive duplication of immune-related genes in bivalves. Mol. Ecol. Resour. 2020, 20, 980–994. [Google Scholar] [CrossRef]
- Sgrò, C.M.; Lowe, A.J.; Hoffmann, A.A. Building evolutionary resilience for conserving biodiversity under climate change. Evol. Appl. 2011, 4, 326–337. [Google Scholar] [CrossRef]
- Gaitán-Espitia, J.D.; Hobday, A.J. Evolutionary principles and genetic considerations for guiding conservation interventions under climate change. Glob. Chang. Biol. 2021, 27, 475–488. [Google Scholar] [CrossRef]
- Cowen, R.K.; Sponaugle, S. Larval dispersal and marine population connectivity. Ann. Rev. Mar. Sci. 2009, 1, 443–466. [Google Scholar] [CrossRef] [Green Version]
- Shlesinger, T.; Loya, Y. Depth-dependent parental effects create invisible barriers to coral dispersal. Commun. Biol. 2021, 4, 1–10. [Google Scholar] [CrossRef]
- Lau, S.C.Y.; Thomas, M.; Hancock, B.; Russell, B.D. Restoration potential of Asian oysters on heavily developed coastlines. Restor. Ecol. 2020, 28, 1643–1653. [Google Scholar] [CrossRef]
- Silliman, K. Population structure, genetic connectivity, and adaptation in the Olympia oyster (Ostrea lurida) along the west coast of North America. Evol. Appl. 2019, 12, 923–939. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wu, X.; Yu, Z. Genetic diversity and substantial population differentiation in Crassostrea hongkongensis revealed by mitochondrial DNA. Mar. Genom. 2013, 11, 31–37. [Google Scholar] [CrossRef]
- Helm, M.M.; Bourne, N.; Lovatelli, A. Hatchery Culture of Bivalves. A Practical Manual; Food and Agriculture Organization of the United Nations: Rome, Italy, 2004; Volume 471, ISBN 92-5-105224-7. [Google Scholar]
- Kawecki, T.J.; Ebert, D. Conceptual issues in local adaptation. Ecol. Lett. 2004, 7, 1225–1241. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Hou, Z.; Wang, H.; Sun, M.; Liu, X.; Liu, B.; Guo, X. Intraspecific variation in mitogenomes of five Crassostrea species provides insight into oyster diversification and speciation. Mar. Biotechnol. 2016, 18, 242–254. [Google Scholar] [CrossRef]
- Nash, S.; Rahman, M.S. Short-term heat stress impairs testicular functions in the American oyster, Crassostrea virginica: Molecular mechanisms and induction of oxidative stress and apoptosis in spermatogenic cells. Mol. Reprod. Dev. 2019, 86, 1444–1458. [Google Scholar] [CrossRef]
- Tan, Y.; Cong, R.; Qi, H.; Wang, L.; Zhang, G.; Pan, Y.; Li, L. Transcriptomics analysis and re-sequencing reveal the mechanism underlying the thermotolerance of an artificial selection population of the Pacific oyster. Front. Physiol. 2021, 12, 509. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, L.; Zhou, Z.; Liu, Y.; Dong, M.; Wang, W.; Song, X.; Wang, M.; Gao, Q.; Song, L. Transcriptomic analysis of oyster Crassostrea gigas larvae illustrates the response patterns regulated by catecholaminergic system upon acute heat and bacterial stress. Dev. Comp. Immunol. 2017, 73, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Fang, X.; Guo, X.; Li, L.; Luo, R.; Xu, F.; Yang, P.; Zhang, L.; Wang, X.; Qi, H.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Dai, H.; Guo, X.; Zhang, Z.; Zhang, K.; Wang, C.; Wang, X.; Wang, W.; Chen, H.; Li, X.; et al. Genome of the estuarine oyster provides insights into climate impact and adaptive plasticity. Commun. Biol. 2021, 4, 1–12. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Mao, F.; Tong, Y.; Liu, Y.; Zhang, Y.; Yu, Z. Characterization and identification of differentially expressed genes involved in thermal adaptation of the Hong Kong oyster Crassostrea hongkongensis by digital gene expression profiling. Front. Mar. Sci. 2017, 4, 112. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, D. The pacific oyster Crassostrea gigas. Part I. Feeding behaviour of larvae and adults. Aquaculture 1983, 31, 195–219. [Google Scholar] [CrossRef]
- Apprill, A. Marine animal microbiomes: Toward understanding host-microbiome interactions in a changing ocean. Front. Mar. Sci. 2017, 4, 222. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Lokmer, A.; Corre, E.; Auguet, J.-C.; Petton, B.; Toulza, E.; Montagnani, C.; Tanguy, G.; Pecqueur, D.; Salmeron, C.; et al. Oyster hemolymph is a complex and dynamic ecosystem hosting bacteria, protists and viruses. Anim. Microbiome 2020, 2, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Piquer, J.; Bowman, J.P.; Ross, T.; Tamplin, M.L. Molecular analysis of the bacterial communities in the live Pacific oyster (Crassostrea gigas) and the influence of postharvest temperature on its structure. J. Appl. Microbiol. 2012, 112, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Nicolae, V.; Akileh, R.; Liu, T. A brief review of oyster-associated microbiota. Microbiol. Res. J. Int. 2017, 20, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Stevick, R.J.; Post, A.F.; Gómez-Chiarri, M. Functional plasticity in oyster gut microbiomes along a eutrophication gradient in an urbanized estuary. Anim. Microbiome 2021, 3, 1–17. [Google Scholar] [CrossRef]
- Lokmer, A.; Goedknegt, M.A.; Thieltges, D.W.; Fiorentino, D.; Kuenzel, S.; Baines, J.F.; Mathias Wegner, K. Spatial and temporal dynamics of pacific oyster hemolymph microbiota across multiple scales. Front. Microbiol. 2016, 7, 1367. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Liao, Y.; Ye, J.; Yang, C.; Adzigbli, L.; Wang, Q.; Du, X.; Deng, Y. Microbiota Diversity in Pearl Oyster Pinctada fucata martensii Intestine and Its Aquaculture Environment. Front. Mar. Sci. 2021, 8, 271. [Google Scholar] [CrossRef]
- Scanes, E.; Parker, L.M.; Seymour, J.R.; Siboni, N.; Dove, M.C.; O’Connor, W.A.; Ross, P.M. Microbiomes of an oyster are shaped by metabolism and environment. Sci. Rep. 2021, 11, 1–7. [Google Scholar] [CrossRef]
- Pierce, M.L.; Ward, J.E. Gut microbiomes of the eastern oyster (Crassostrea virginica) and the blue mussel (Mytilus edulis): Temporal variation and the influence of marine aggregate-associated microbial communities. mSphere 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Pierce, M.L.; Ward, J.E. Microbial ecology of the bivalvia, with an emphasis on the family Ostreidae. J. Shellfish. Res. 2018, 37, 793–806. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–501. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2014, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Dray, S.; Dufour, A.B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Knaus, B.J.; Grünwald, N.J. vcfR: A package to manipulate and visualize variant call format data in R. In Proceedings of the Molecular Ecology Resources; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; Volume 17, pp. 44–53. [Google Scholar]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, B.; Gao, S.; Lercher, M.J.; Hu, S.; Chen, W.H. Evolview v3: A webserver for visualization, annotation, and management of phylogenetic trees. Nucleic Acids Res. 2019, 47, W270–W275. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018, 47, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; McCarthy, D.; Ritchie, M.; Robinson, M.; Smyth, G. EdgeR: Differential Analysis of Sequence Read Count Data. 2008, pp. 1–119. Available online: https://www.bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf (accessed on 20 October 2021).
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovations 2021, 2, 100141. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant graphics for data analysis; Springer-Verlag: New York, NY, USA, 2016. [Google Scholar]
- Wu, W.; Zong, J.; Wei, N.; Cheng, J.; Zhou, X.; Cheng, Y.; Chen, D.; Guo, Q.; Zhang, B.; Feng, Y. CASH: A constructing comprehensive splice site method for detecting alternative splicing events. Brief. Bioinform. 2018, 19, 905–917. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Pea-Llopis, S.; Ferrando, M.D.; Pea, J.B. Impaired glutathione redox status is associated with decreased survival in two organophosphate-poisoned marine bivalves. Chemosphere 2002, 47, 485–497. [Google Scholar] [CrossRef]
- Mello, D.F.; Arl, M.; Trevisan, R.; Dafre, A.L. How important are glutathione and thiol reductases to oyster hemocyte function? Fish Shellfish. Immunol. 2015, 46, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Shi, H.; Yu, H.; Li, Q. Characterization of polymorphic microsatellite markers and genetic diversity in the Hong Kong oyster Crassostrea hongkongensis using paired-end Illumina shotgun sequencing. Genes Genom. 2015, 37, 615–620. [Google Scholar] [CrossRef]
- Vendrami, D.L.J.; Houston, R.D.; Gharbi, K.; Telesca, L.; Gutierrez, A.P.; Gurney-Smith, H.; Hasegawa, N.; Boudry, P.; Hoffman, J.I. Detailed insights into pan-European population structure and inbreeding in wild and hatchery Pacific oysters (Crassostrea gigas) revealed by genome-wide SNP data. Evol. Appl. 2019, 12, 519–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohfritsch, A.; Bierne, N.; Boudry, P.; Heurtebise, S.; Cornette, F.; Lapègue, S. Population genomics shed light on the demographic and adaptive histories of European invasion in the Pacific oyster, Crassostrea gigas. Evol. Appl. 2013, 6, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, B.J.G.; Rycroft, C.; Ferchaud, A.L.; Saunders, R.; Li, L.; Liu, S.; Chan, A.M.; Otto, S.P.; Suttle, C.A.; Miller, K.M. Relative genomic impacts of translocation history, hatchery practices, and farm selection in Pacific oyster Crassostrea gigas throughout the Northern Hemisphere. Evol. Appl. 2020, 13, 1380–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Song, K.; Li, C.; Liu, S.; Shi, R.; Li, B.; Wang, T.; Li, A.; Que, H.; Li, L.; et al. Genome-wide association analysis of nutrient traits in the oyster Crassostrea gigas: Genetic effect and interaction network. BMC Genom. 2019, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wu, F.; Qi, H.; Meng, J.; Wang, W.; Liu, M.; Li, L.; Zhang, G. Whole-genome resequencing reveals the single nucleotide polymorphisms associated with shell shape in Crassostrea gigas. Aquaculture 2022, 547, 737502. [Google Scholar] [CrossRef]
- Qiu, D.; Huang, L.; Zhang, J.; Lin, S. Phytoplankton dynamics in and near the highly eutrophic Pearl River Estuary, South China Sea. Cont. Shelf Res. 2010, 30, 177–186. [Google Scholar] [CrossRef]
- Liu, S.M.; Qi, X.H.; Li, X.; Ye, H.R.; Wu, Y.; Ren, J.L.; Zhang, J.; Xu, W.Y. Nutrient dynamics from the Changjiang (Yangtze River) estuary to the East China Sea. J. Mar. Syst. 2016, 154, 15–27. [Google Scholar] [CrossRef]
- Feely, R.A.; Alin, S.R.; Newton, J.; Sabine, C.L.; Warner, M.; Devol, A.; Krembs, C.; Maloy, C. The combined effects of ocean acidification, mixing, and respiration on pH and carbonate saturation in an urbanized estuary. Estuar. Coast. Shelf Sci. 2010, 88, 442–449. [Google Scholar] [CrossRef]
- Casas, S.M.; La Peyre, J.F. Heat shock protein 70 levels and post-harvest survival of eastern oysters following sublethal heat shock in the laboratory or conditioning in the field. Cell Stress Chaperones 2020, 25, 369–378. [Google Scholar] [CrossRef]
- Yang, C.Y.; Sierp, M.T.; Abbott, C.A.; Li, Y.; Qin, J.G. Responses to thermal and salinity stress in wild and farmed Pacific oysters Crassostrea gigas. Comp. Biochem. Physiol. -Part A Mol. Integr. Physiol. 2016, 201, 22–29. [Google Scholar] [CrossRef]
- Xu, F.; Marlétaz, F.; Gavriouchkina, D.; Liu, X.; Sauka-Spengler, T.; Zhang, G.; Holland, P.W.H. Evidence from oyster suggests an ancient role for Pdx in regulating insulin gene expression in animals. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Xu, Q.; Zhu, C.; Fan, Y.; Song, Z.; Xing, S.; Liu, W.; Yan, J.; Sang, T. Population transcriptomics uncovers the regulation of gene expression variation in adaptation to changing environment. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Gao, Q.; Liu, C.; Wang, L.; Zhou, Z.; Gong, C.; Zhang, A.; Zhang, H.; Qiu, L.; Song, L. The transcriptional response of the Pacific oyster Crassostrea gigas against acute heat stress. Fish Shellfish Immunol. 2017, 68, 132–143. [Google Scholar] [CrossRef]
- Nie, H.; Jiang, K.; Li, N.; Li, D.; Yan, X. Transcriptomic analysis of Ruditapes philippinarum under aerial exposure and reimmersion reveals genes involved in stress response and recovery capacity of the Manila clam. Aquaculture 2020, 524, 735271. [Google Scholar] [CrossRef]
- Huang, B.; Zhang, L.; Tang, X.; Zhang, G.; Li, L. Genome-wide analysis of alternative splicing provides insights into stress adaptation of the Pacific oyster. Mar. Biotechnol. 2016, 18, 598–609. [Google Scholar] [CrossRef]
- Liu, M.; Guo, X. A novel and stress adaptive alternative oxidase derived from alternative splicing of duplicated exon in oyster Crassostrea virginica. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lareau, L.F.; Brenner, S.E. Regulation of splicing factors by alternative splicing and NMD is conserved between kingdoms yet evolutionarily flexible. Mol. Biol. Evol. 2015, 32, 1072–1079. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, T.; Douglass, S.; Gabunilas, J.; Pellegrini, M.; Chanfreau, G.F. Widespread use of non-productive alternative splice sites in Saccharomyces cerevisiae. PLoS Genet. 2014, 10, e1004249. [Google Scholar] [CrossRef] [Green Version]
- Ganie, S.A.; Reddy, A.S.N.N. Stress-induced changes in alternative splicing landscape in rice: Functional significance of splice isoforms in stress tolerance. Biology 2021, 10, 309. [Google Scholar] [CrossRef]
- Tabrez, S.S.; Sharma, R.D.; Jain, V.; Siddiqui, A.A.; Mukhopadhyay, A. Differential alternative splicing coupled to nonsense-mediated decay of mRNA ensures dietary restriction-induced longevity. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Sommer, F.; Bäckhed, F. The gut microbiota-masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Wang, X.; Yan, A. Microbial profiles of retail pacific oysters (Crassostrea gigas) from Guangdong Province, China. Front. Microbiol. 2021, 12, 1685. [Google Scholar] [CrossRef]
- Li, Y.-F.; Xu, J.-K.; Chen, Y.-W.; Ding, W.-Y.; Shao, A.-Q.; Liang, X.; Zhu, Y.-T.; Yang, J.-L. Characterization of gut microbiome in the mussel Mytilus galloprovincialis in response to thermal stress. Front. Physiol. 2019, 10, 1086. [Google Scholar] [CrossRef] [Green Version]
- Lokmer, A.; Mathias Wegner, K. Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 2015, 9, 670–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Man, A.W.C.; Zhou, Y.; Xia, N.; Li, H. Involvement of gut microbiota, microbial metabolites and interaction with polyphenol in host immunometabolism. Nutrients 2020, 12, 3054. [Google Scholar] [CrossRef] [PubMed]
- Raza, M.F.; Wang, Y.; Cai, Z.; Bai, S.; Yao, Z.; Awan, U.A.; Zhang, Z.; Zheng, W.; Zhang, H. Gut microbiota promotes host resistance to low-temperature stress by stimulating its arginine and proline metabolism pathway in adult Bactrocera dorsalis. PLoS Pathog. 2020, 16, e1008441. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.M.; Neish, A.S. Redox signaling mediated by the gut microbiota. Free Radic. Biol. Med. 2017, 105, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Houwenhuyse, S.; Stoks, R.; Mukherjee, S.; Decaestecker, E. Locally adapted gut microbiomes mediate host stress tolerance. ISME J. 2021, 15, 2401–2414. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Shoaie, S.; Bergentall, M.; Ghaffari, P.; Zhang, C.; Larsson, E.; Bäckhed, F.; Nielsen, J. The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 2015, 11, 834. [Google Scholar] [CrossRef]
- Deng, F.; Zhao, B.C.; Yang, X.; Lin, Z.B.; Sun, Q.S.; Wang, Y.F.; Yan, Z.Z.; Liu, W.F.; Li, C.; Hu, J.J.; et al. The gut microbiota metabolite capsiate promotes Gpx4 expression by activating TRPV1 to inhibit intestinal ischemia reperfusion-induced ferroptosis. Gut Microbes 2021, 13, 1–21. [Google Scholar] [CrossRef]
- Lin, H.; Decuypere, E.; Buyse, J. Acute heat stress induces oxidative stress in broiler chickens. Comp. Biochem. Physiol.-A Mol. Integr. Physiol. 2006, 144, 11–17. [Google Scholar] [CrossRef]
- Driedonks, N.; Xu, J.; Peters, J.L.; Park, S.; Rieu, I. Multi-level interactions between heat shock factors, heat shock proteins, and the redox system regulate acclimation to heat. Front. Plant Sci. 2015, 6, 999. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Lightfoot, R.; Stevens, J.L. Activation of heat shock factor by alkylating agents is triggered by glutathione depletion and oxidation of protein thiols. J. Biol. Chem. 1996, 271, 4805–4812. [Google Scholar] [CrossRef] [Green Version]
- Rokutan, K.; Hirakawa, T.; Teshima, S.; Honda, S.; Kishi, K. Glutathione depletion impairs transcriptional activation of heat shock genes in primary cultures of guinea pig gastric mucosal cells. J. Clin. Invest. 1996, 97, 2242–2250. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hsp Families | Number of Blasted Genes (n) |
|---|---|
| HSPE | 2 |
| HSPB | 20 |
| HSP40 | 50 |
| HSP70 | 123 |
| HSPC(HSP90) | 5 |
| HSP110 | 2 |
| Element | Value |
|---|---|
| Assembled genome size (bp) | 757,082,711 |
| Scaffold N50 (bp) | 72,332,161 |
| Number of Proteins | 40,086 |
| Number of longest Proteins only | 36,296 |
| Sum of Amino Acids (aa) | 15,406,827 |
| Mean of Proteins (aa) | 424 |
| Sum of Exons (bp) | 115,218,162 |
| Mean of Exons (bp) | 239 |
| Sum of Introns (bp) | 492,870,204 |
| Mean of Introns (bp) | 1201 |
| Numer of gene loci | 36,239 |
| Sum of gene region (bp) | 306,530,342 |
| % of gene loci in genome | 40.49% |
| Average gene region (bp) | 8459 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.; Huang, E.Y.Y.; Nong, W.; Law, S.T.S.; Yu, Y.; Cheung, K.; Li, Y.; Wong, C.F.; Yip, H.Y.; Joyce, P.W.S.; et al. Population Genomics, Transcriptional Response to Heat Shock, and Gut Microbiota of the Hong Kong Oyster Magallana hongkongensis. J. Mar. Sci. Eng. 2022, 10, 237. https://doi.org/10.3390/jmse10020237
Xie Y, Huang EYY, Nong W, Law STS, Yu Y, Cheung K, Li Y, Wong CF, Yip HY, Joyce PWS, et al. Population Genomics, Transcriptional Response to Heat Shock, and Gut Microbiota of the Hong Kong Oyster Magallana hongkongensis. Journal of Marine Science and Engineering. 2022; 10(2):237. https://doi.org/10.3390/jmse10020237
Chicago/Turabian StyleXie, Yichun, Elaine Y. Y. Huang, Wenyan Nong, Sean T. S. Law, Yifei Yu, Khan Cheung, Yiqian Li, Cheuk Fung Wong, Ho Yin Yip, Patrick W. S. Joyce, and et al. 2022. "Population Genomics, Transcriptional Response to Heat Shock, and Gut Microbiota of the Hong Kong Oyster Magallana hongkongensis" Journal of Marine Science and Engineering 10, no. 2: 237. https://doi.org/10.3390/jmse10020237