High-Resolution CT Change over Time in Patients with Idiopathic Pulmonary Fibrosis on Antifibrotic Treatment

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Methods
2.1. Study Population and Study Design
2.2. Radiological and Functional Analysis
2.3. Statistical Analysis
3. Results
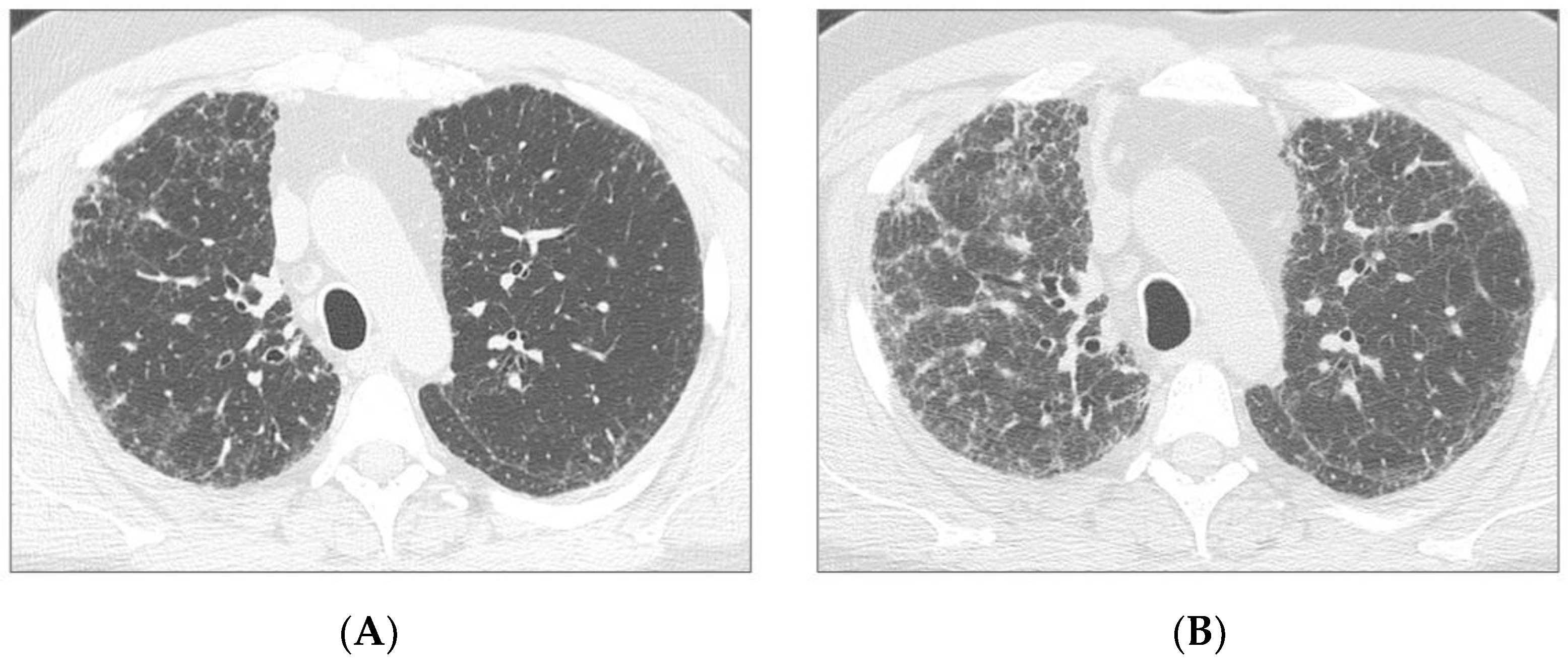
3.1. Clinical, Functional and Radiological Evaluation at Baseline
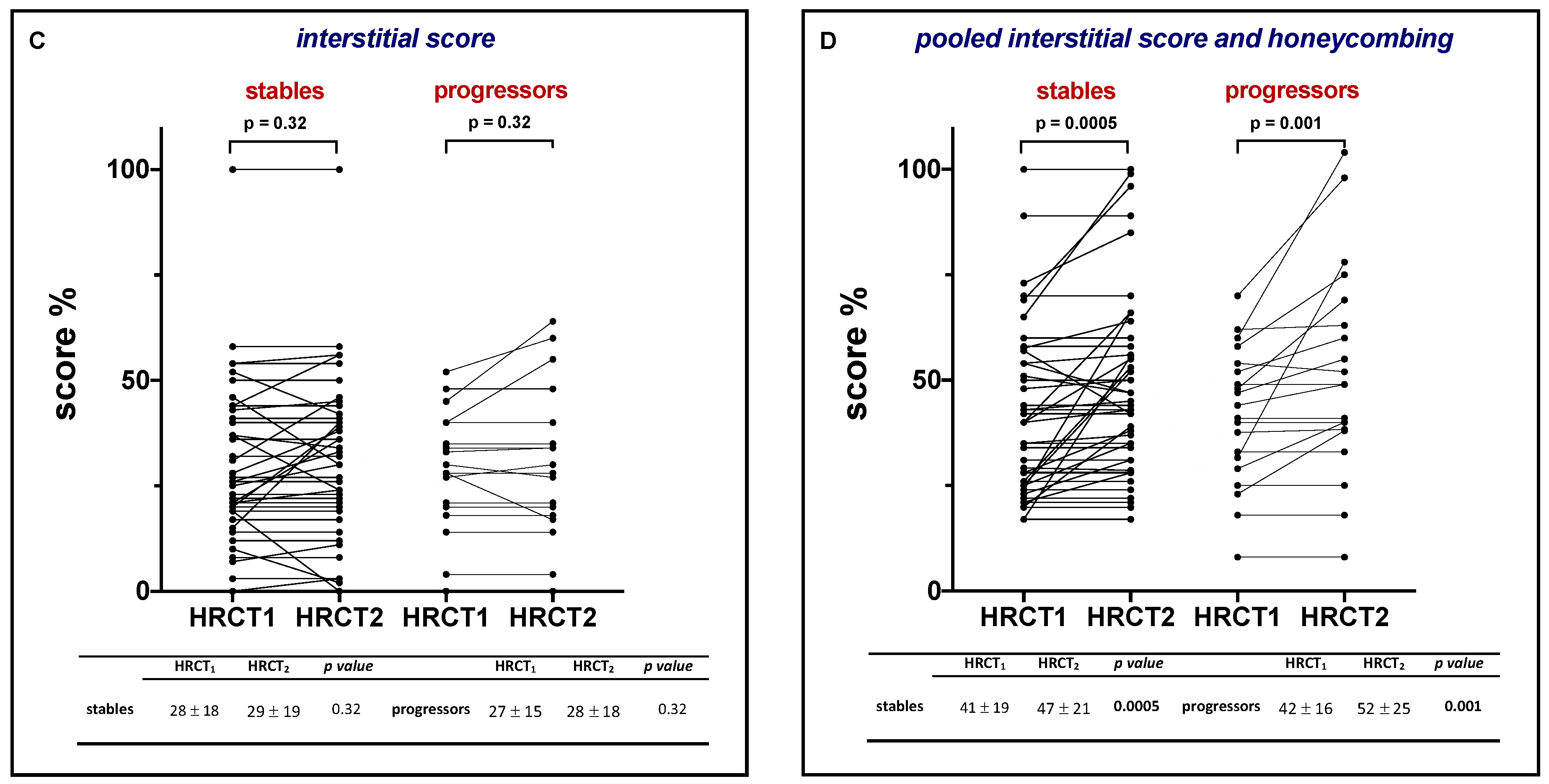
3.2. Functional and Radiological evaluation
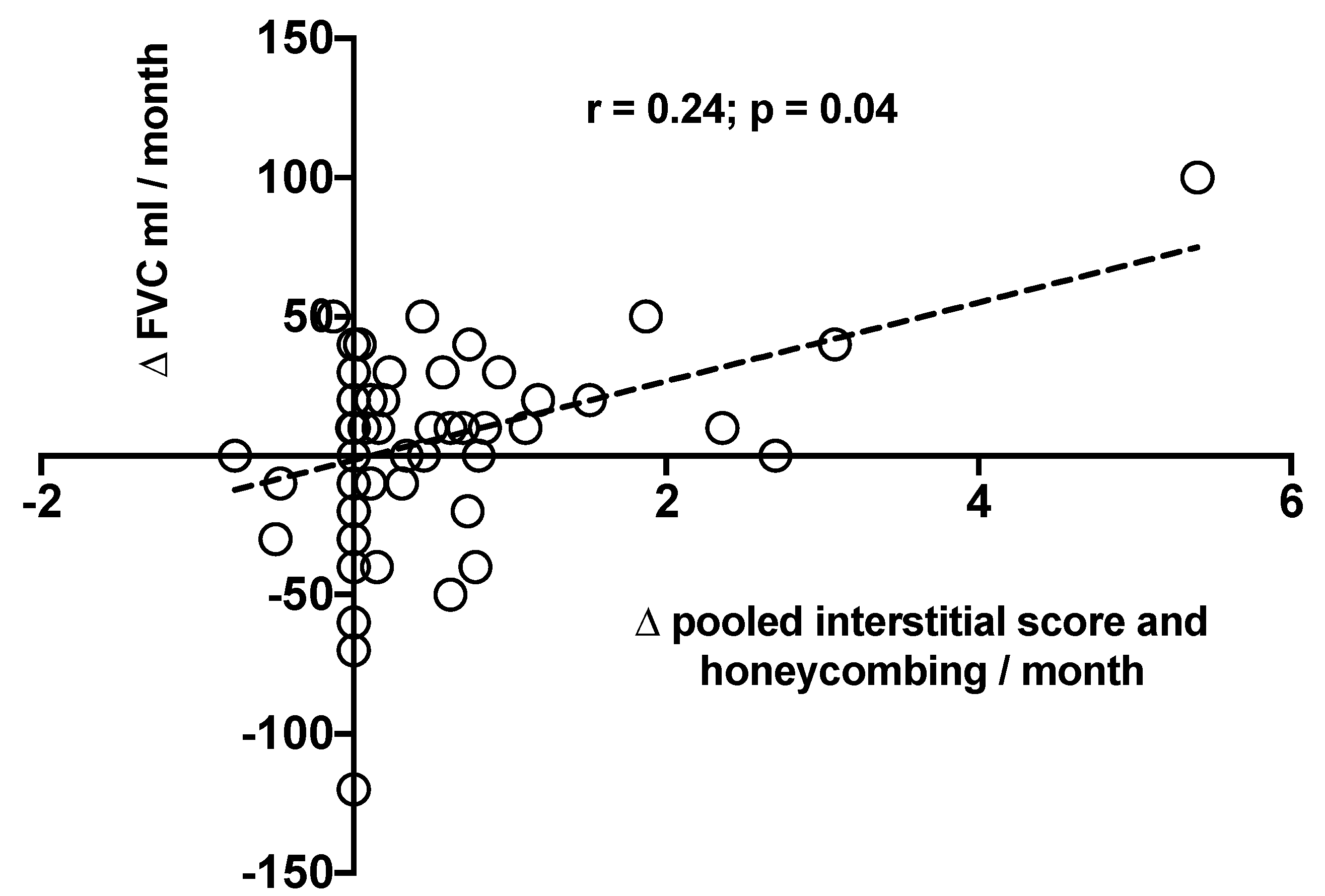
3.3. Functional and Radiological Correlations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IPF | Idiopathic Pulmonary Fibrosis |
| UIP | Usual Interstitial Pneumonia |
| HRCT | High-Resolution Computed Tomography |
| GGO | Ground Glass Opacities |
| FVC | Forced Vital Capacity |
| DLCO | diffusing capacity of the lung for carbon monoxide |
| 6MWT | six-minute walking test |
| L | liters |
| mL | milliliters |
| FEV1 | forced expiratory volume in one second |
| IS | Interstitial Score |
| AS | Alveolar Score |
| HC | honeycombing |
References
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morelli, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Garcia, C.A.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir Crit Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef] [PubMed]
- Du Bois, R.M.; Weycker, D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Kartashov, A.; King, T.E., Jr.; Lancaster, L.; Noble, P.W.; Sahn, S.A.; et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: Test properties and minimal clinically important difference. Am. J. Respir. Crit. Care Med. 2011, 184, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Du Bois, R.M.; Weycker, D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Kartashov, A.; Lancaster, L.; Noble, P.W.; Raghu, G.; Sahn, S.A.; et al. Ascertainment of individual risk of mortality for patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.U.; Desai, S.R.; Rubens, M.B.; Goh, N.S.; Cramer, D.; Nicholson, A.G.; Colby, T.V.; du Bois, R.M.; Hansell, D.M. Idiopathic pulmonary fibrosis: A composite physiologic index derived from disease extent observed by computed tomography. Am. J. Respir. Crit. Care Med. 2003, 167, 962–969. [Google Scholar] [CrossRef]
- Ley, B.; Elicker, B.M.; Hartman, T.E.; Ryerson, C.J.; Vittinghoff, E.; Ryu, J.H.; Lee, J.S.; Jones, K.D.; Richeldi, L.; King, T.E., Jr.; et al. Idiopathic Pulmonary Fibrosis: CT and Risk of Death. Radiology 2014, 273, 570–579. [Google Scholar] [CrossRef]
- Salisbury, M.L.; Lynch, D.A.; van Beek, E.J.; Kazerooni, E.A.; Guo, J.; Xia, M.; Murray, S.; Anstrom, K.J.; Yow, E.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis: The Association between the Adaptive Multiple Features Method and Fibrosis Outcomes. Am. J. Respir. Crit. Care Med. 2017, 195, 921–929. [Google Scholar] [CrossRef]
- Lynch, D.A.; Godwin, J.D.; Safrin, S.; Starko, K.M.; Hormel, P.; Brown, K.K.; Raghu, G.; King, T.E., Jr.; Bradford, W.Z.; Schwartz, D.A.; et al. High-resolution computed tomography in idiopathic pulmonary fibrosis: Diagnosis and prognosis. Am. J. Respir. Crit. Care Med. 2005, 172, 488–493. [Google Scholar] [CrossRef]
- Sumikawa, H.; Johkoh, T.; Colby, T.V.; Ichikado, K.; Suga, M.; Taniguchi, H.; Kondoh, Y.; Ogura, T.; Arakawa, H.; Fujimoto, K.; et al. Computed tomography findings in pathological usual interstitial pneumonia: Relationship to survival. Am. J. Respir. Crit. Care Med. 2008, 177, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Sverzellati, N.; Devaraj, A.; Desai, S.R.; Quigley, M.; Wells, A.U.; Hansell, D.M. Method for minimizing observer variation for the quantitation of high-resolution computed tomographic signs of lung disease. J. Comput. Assist. Tomogr. 2011, 35, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Hansell, D.M.; Goldin, J.G.; King, T.E., Jr.; Lynch, D.A.; Richeldi, L.; Wells, A.U. CT staging and monitoring of fibrotic interstitial lung diseases in clinical practice and treatment trials: A Position Paper from the Fleischner society. Lancet Respir. Med. 2015, 3, 483–496. [Google Scholar] [CrossRef]
- Fell, C.D.; Martinez, F.J.; Liu, L.X.; Murray, S.; Han, M.K.; Kazerooni, E.A.; Gross, B.H.; Myers, J.; Travis, W.D.; Colby, T.V. Clinical predictors of a diagnosis of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 181, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Cocconcelli, E.; Balestro, E.; Biondini, D.; Barbiero, G.; Polverosi, R.; Calabrese, F.; Pezzuto, F.; Lacedonia, D.; Rea, F.; Schiavon, M.; et al. High-Resolution Computed Tomography (HRCT) Reflects Disease Progression in Patients with Idiopathic Pulmonary Fibrosis (IPF): Relationship with Lung Pathology. J. Clin. Med. 2019, 8, 399. [Google Scholar] [CrossRef]
- Altman, D.G. Practical Statistics for Medical Research; Chapman and Hall: London, UK, 1991; Volume 10, pp. 1635–1636. [Google Scholar]
- King, T.E.J.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef]
- Okuda, R.; Hagiwara, E.; Baba, T.; Kitamura, H.; Kato, T.; Ogura, T. Safety and efficacy of pirfenidone in idiopathic pulmonary fibrosis in clinical practice. Respir. Med. 2013, 107, 1431–1437. [Google Scholar] [CrossRef]
- Bando, M. Pirfenidone: Clinical trials and clinical practice in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2016, 54, 298–304. [Google Scholar] [CrossRef]
- Brunnemer, E.; Wälscher, J.; Tenenbaum, S.; Hausmanns, J.; Schulze, K.; Seiter, M.; Heussel, C.P.; Warth, A.; Herth, F.J.F.; Kreuter, M. Real-World Experience with Nintedanib in Patients with Idiopathic Pulmonary Fibrosis. Respiration 2018, 95, 301–309. [Google Scholar] [CrossRef]
- Best, A.C.; Lynch, A.M.; Bozic, C.M.; Miller, D.; Grunwald, G.K.; Lynch, D.A. Quantitative CT indexes in idiopathic pulmonary fibrosis: Relationship with physiologic impairment. Radiology 2003, 228, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Best, A.C.; Meng, J.; Lynch, A.M.; Bozic, C.M.; Miller, D.; Grunwald, G.K.; Lynch, D.A. Idiopathic pulmonary fibrosis: Physiologic tests, quantitative CT indexes, and CT visual scores as predictors of mortality. Radiology 2008, 246, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Boon, K.; Bailey, N.W.; Yang, J.; Steel, M.P.; Groshong, S.; Kervitsky, D.; Brown, K.K.; Schwarz, M.I.; Schwartz, D.A. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF). PLoS ONE 2009, 4, e5134. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Carrillo, G.; Estrada, A.; Mejia, M.; Becerril, C.; Cisneros, J.; Gaxiola, M.; Pérez-Padilla, R.; Navarro, C.; Richards, T.; et al. Accelerated variant of idiopathic pulmonary fibrosis: Clinical behavior and gene expression pattern. PLoS ONE 2007, 2, e482. [Google Scholar] [CrossRef] [PubMed]
- Balestro, E.; Calabrese, F.; Turato, G.; Lunardi, F.; Bazzan, E.; Marulli, G.; Biondini, D.; Rossi, E.; Sanduzzi, A.; Rea, F.; et al. Immune inflammation and disease progression in idiopathic pulmonary fibrosis. PLoS ONE 2016, 11, e0154516. [Google Scholar] [CrossRef]
- Biondini, D.; Balestro, E.; Lacedonia, D.; Cerri, S.; Milaneschi, R.; Luppi, F.; Cocconcelli, E.; Bazzan, E.; Clini, E.; Foschino Barbaro, M.P.; et al. Pretreatment rate of decay in forced vital capacity predicts long-term response to pirfenidone in patients with idiopathic pulmonary fibrosis. Sci. Rep. 2018, 8, 5961. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M.; Costabel, U.; Richeldi, L.; Cottin, V.; Wijsenbeek, M.; Bonella, F.; Bendstrup, E.; Maher, T.M.; Wachtlin, D.; Stowasser, S.; et al. Statin Therapy and Outcomes in Trials of Nintedanib in Idiopathic Pulmonary Fibrosis. Respiration 2018, 95, 317–326. [Google Scholar] [CrossRef]
- Lee, H.Y.; Lee, K.S.; Jeong, Y.J.; Hwang, J.H.; Kim, H.J.; Chung, M.P.; Han, J. High-resolution CT findings in fibrotic idiopathic interstitial pneumonias with little honeycombing: Serial changes and prognostic implications. AJR Am. J. Roentgenol. 2012, 199, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Goldin, J.G. Interpretation of HRCT Scans in the Diagnosis of IPF: Improving Communication between Pulmonologists and Radiologists. Lung 2018, 196, 561–567. [Google Scholar] [CrossRef]
- Launay, D.; Remy-Jardin, M.; Michon-Pasturel, U.; Mastora, I.; Hachulla, E.; Lambert, M.; Delannoy, V.; Queyrel, V.; Duhamel, A.; Matran, R.; et al. High resolution computed tomography in fibrosing alveolitis associated with systemic sclerosis. J. Rheumatol. 2006, 33, 1789–1801. [Google Scholar]
- Remy-Jardin, M.; Giraud, F.; Remy, J.; Copin, M.C.; Gosselin, B.; Duhamel, A. Importance of ground-glass attenuation in chronic diffuse infiltrative lung disease: Pathologic-CT correlation. Radiology 1993, 189, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bouros, D.; Wells, A.U.; Nicholson, A.G.; Colby, T.V.; Polychronopoulos, V.; Pantelidis, P.; Haslam, P.L.; Vassilakis, D.A.; Black, C.M.; du Bois, R.M. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am. J. Respir. Crit. Care Med. 2002, 165, 1581–1586. [Google Scholar] [CrossRef]
- Iwasawa, T.; Ogura, T.; Sakai, F.; Kanauchi, T.; Komagata, T.; Baba, T.; Gotoh, T.; Morita, S.; Yazawa, T.; Inoue, T. CT analysis of the effect of pirfenidone in patients with idiopathic pulmonary fibrosis. Eur. J. Radiol. 2014, 83, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Bartholmai, B.J.; Rajagopalan, S.; van Moorsel, C.H.M.; van Es, H.W.; van Beek, F.T.; Struik, M.H.L.; Kokosi, M.; Egashira, R.; Brun, A.L.; et al. Predicting Outcomes in Idiopathic Pulmonary Fibrosis Using Automated Computed Tomographic Analysis. Am. J. Respir. Crit. Care Med. 2018, 198, 767–776. [Google Scholar] [CrossRef]
- Robbie, H.; Wells, A.U.; Jacob, J.; Walsh, S.L.F.; Nair, A.; Srikanthan, A.; Tazoniero, P.; Devaraj, A. Visual and Automated CT Measurements of Lung Volume Loss in Idiopathic Pulmonary Fibrosis. AJR Am. J. Roentgenol. 2019, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kim, G.H.; Salisbury, M.L.; Barber, D.; Bartholmai, B.J.; Brown, K.K.; Conoscenti, C.S.; De Backer, J.; Flaherty, K.R.; Gruden, J.F.; et al. Computed Tomographic Biomarkers in Idiopathic Pulmonary Fibrosis: The Future of Quantitative Analysis. Am. J. Respir. Crit. Care Med. 2018, 199, 12–21. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entire | Stables | Progressors | p-Value | |
|---|---|---|---|---|
| Population | ||||
| (n = 68) | (n = 48) | (n = 20) | ||
| Male—n (%) | 55 (81) | 37 (77) | 18 (90) | 0.31 |
| Female—n (%) | 13 (19) | 11 (23) | 2 (10) | 0.31 |
| Age at diagnosis—years | 66 (44–78) | 68 (46–78) | 61 (44–78) | 0.07 |
| Smoking history—pack years | 15 (0–80) | 15 (0–80) | 15 (0–55) | 0.31 |
| Current—n (%) | 9 (13) | 7 (15) | 2 (10) | 1.00 |
| Former—n (%) | 40 (59) | 29 (60) | 11 (55) | 1.00 |
| Nonsmokers—n (%) | 19 (28) | 12 (25) | 7 (35) | 0.55 |
| Clinical-radiological diagnosis—n (%) | 35 (51) | 27 (56) | 8 (40) | 0.29 |
| Histological diagnosis—n (%) | 33 (49) | 21 (44) | 12 (60) | 0.29 |
| FVC at diagnosis—L | 2.76 (1.19–5.68) | 2.6 (1.19–5.29) | 2.97 (1.68–5.68) | 0.04 |
| FVC at diagnosis—% pred. | 78 (44–120) | 78 (44–120) | 78 (50–107) | 0.40 |
| FEV1 at diagnosis—L | 2.21 (1.02-4.45) | 2.19 (1.02–4.45) | 2.50 (1.40–3.70) | 0.06 |
| FEV1 at diagnosis—% pred. | 83 (40–127) | 83 (40–127) | 86 (49–122) | 0.27 |
| DLCO at diagnosis—% pred. | 57 (34–114) | 53 (34–114) | 65 (37–97) | 0.02 |
| 6MWT at diagnosis—mt | 400 (125–600) | 400 (125–600) | 408 (250–540) | 0.50 |
| FVC decline per year—mL | 86 (−1381–1155) | 37 (−1381–371) | 413 (135–1155) | <0.0001 |
| FVC decline per year—% pred. | 2 (−25–29) | 0 (−25–4.7) | 9 (5–29) | <0.0001 |
| Deaths—n (%) | 16 (23) | 8 (17) | 8 (40) | 0.05 |
| Alveolar score in HRCT1—% | 21 (0–90) | 21 (0–90) | 22 (0–44) | 0.68 |
| Honeycombing in HRCT1—% | 7 (0–70) | 6 (0–70) | 9 (0–50) | 0.32 |
| Interstitial score in HRCT1—% | 26 (0–100) | 26 (0–100) | 28 (0–52) | 0.92 |
| Pooled interstitial score and honeycombing—% | 40 (8–100) | 38 (17–100) | 43 (8–70) | 0.52 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balestro, E.; Cocconcelli, E.; Giraudo, C.; Polverosi, R.; Biondini, D.; Lacedonia, D.; Bazzan, E.; Mazzai, L.; Rizzon, G.; Lococo, S.; et al. High-Resolution CT Change over Time in Patients with Idiopathic Pulmonary Fibrosis on Antifibrotic Treatment. J. Clin. Med. 2019, 8, 1469. https://doi.org/10.3390/jcm8091469
Balestro E, Cocconcelli E, Giraudo C, Polverosi R, Biondini D, Lacedonia D, Bazzan E, Mazzai L, Rizzon G, Lococo S, et al. High-Resolution CT Change over Time in Patients with Idiopathic Pulmonary Fibrosis on Antifibrotic Treatment. Journal of Clinical Medicine. 2019; 8(9):1469. https://doi.org/10.3390/jcm8091469
Chicago/Turabian StyleBalestro, Elisabetta, Elisabetta Cocconcelli, Chiara Giraudo, Roberta Polverosi, Davide Biondini, Donato Lacedonia, Erica Bazzan, Linda Mazzai, Giulia Rizzon, Sara Lococo, and et al. 2019. "High-Resolution CT Change over Time in Patients with Idiopathic Pulmonary Fibrosis on Antifibrotic Treatment" Journal of Clinical Medicine 8, no. 9: 1469. https://doi.org/10.3390/jcm8091469
APA StyleBalestro, E., Cocconcelli, E., Giraudo, C., Polverosi, R., Biondini, D., Lacedonia, D., Bazzan, E., Mazzai, L., Rizzon, G., Lococo, S., Turato, G., Tinè, M., Cosio, M. G., Saetta, M., & Spagnolo, P. (2019). High-Resolution CT Change over Time in Patients with Idiopathic Pulmonary Fibrosis on Antifibrotic Treatment. Journal of Clinical Medicine, 8(9), 1469. https://doi.org/10.3390/jcm8091469