Epigenome-Wide Association Analysis of Differentially Methylated Signals in Blood Samples of Patients with Non-Small-Cell Lung Cancer

 , and
, and
Abstract
1. Introduction
2. Methods
2.1. Study Subjects and Preparation of Tissue Samples
2.2. Preparation of Genomic DNA and DNA Methylation Profiling
2.3. Epigenome-Wide Association Study
2.4. Statistical Analysis
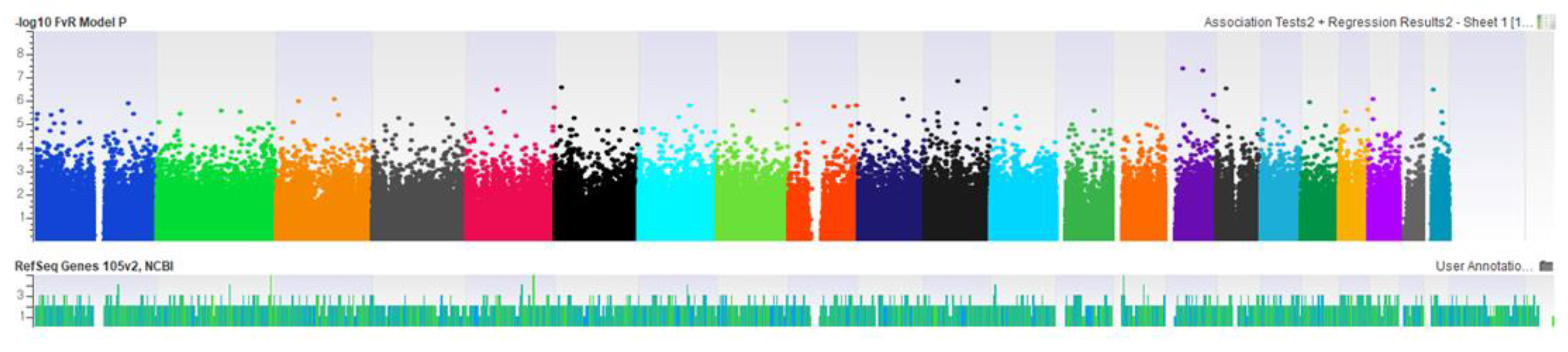
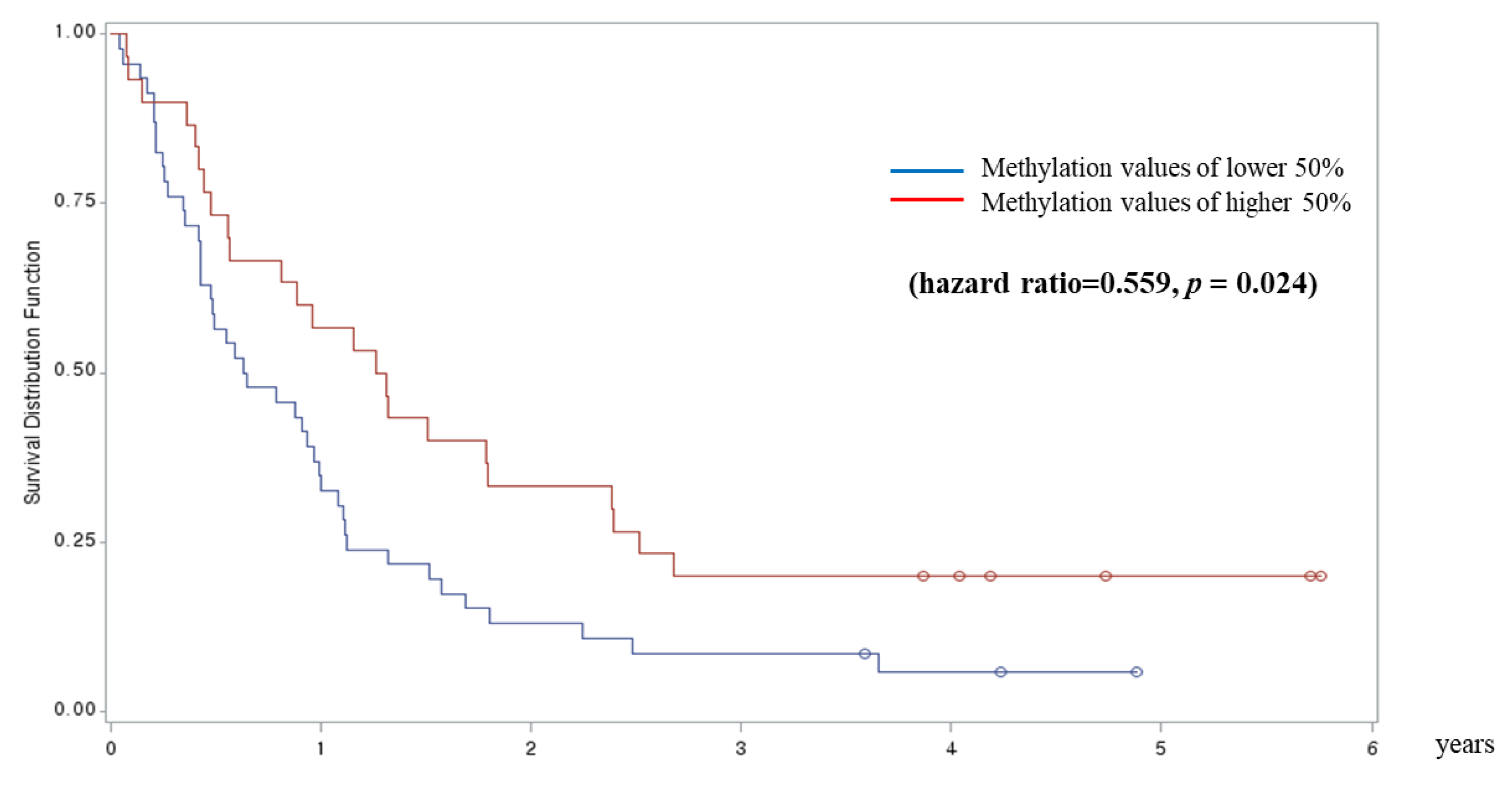
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Horn, L.; Jackman, D.; Spigel, D.; Antonia, S.; Hellmann, M.; Powderly, J.; Heist, R.; Sequist, L.V.; Smith, D.C.; et al. Five-year follow-up of nivolumab in previously treated advanced non-small-cell lung cancer: Results from the CA209-003 study. J. Clin. Oncol. 2018, 36, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Henschke, C.I.; Yankelevitz, D.F.; Libby, D.M.; Pasmantier, M.W.; Smith, J.P.; Miettinen, O.S.; International Early Lung Cancer Action Program Investigators. Survival of patients with stage I lung cancer detected on CT screening. N. Engl. J. Med. 2006, 355, 1763–1771. [Google Scholar] [PubMed]
- Burotto, M.; Thomas, A.; Subramaniam, D.; Giaccone, G.; Rajan, A. Biomarkers in early-stage non-small-cell lung cancer: Current concepts and future directions. J. Thorac. Oncol. 2014, 9, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Balgkouranidou, I.; Liloglou, T.; Lianidou, E.S. Lung cancer epigenetics: Emerging biomarkers. Biomarkers Med. 2013, 7, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Brock, M.V.; Hooker, C.M.; Ota-Machida, E.; Han, Y.; Guo, M.; Ames, S.; Glöckner, S.; Piantadosi, S.; Gabrielson, E.; Pridham, G.; et al. DNA methylation markers and early recurrence in stage I lung cancer. N. Engl. J. Med. 2008, 358, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Mendez-Gonzalez, J.; Nadal, E.; Chen, G.; Carmona, F.J.; Sayols, S.; Moran, S.; Heyn, H.; Vizoso, M.; Gomez, A.; et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 4140–4147. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J.; Shackelford, R.E.; El-Osta, H. Epigenetics in non-small cell lung cancer: From basics to therapeutics. Transl. Lung Cancer Res. 2016, 5, 155–171. [Google Scholar] [CrossRef]
- Zhang, Y.; Schöttker, B.; Ordóñez-Mena, J.; Holleczek, B.; Yang, R.; Burwinkel, B.; Butterbach, K.; Brenner, H. F2RL3 methylation, lung cancer incidence and mortality. Int. J. Cancer 2015, 137, 1739–1748. [Google Scholar] [CrossRef]
- Baglietto, L.; Ponzi, E.; Haycock, P.; Hodge, A.; Bianca Assumma, M.; Jung, C.H.; Chung, J. DNA methylation changes measured in pre-diagnostic peripheral blood samples are associated with smoking and lung cancer risk. Int. J. Cancer 2017, 140, 50–61. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Niu, L.; Li, L.; Taylor, J.A. ENmix: A novel background correction method for Illumina HumanMethylation450 BeadChip. Nucleic Acids Res. 2016, 44, e20. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, J.; Bäcklin, C.L.; Wahlberg, P.; Busche, S.; Berglund, E.C.; Eloranta, M.L.; Flaegstad, T.; Forestier, E.; Frost, B.M.; Harila-Saari, A.; et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013, 14, r105. [Google Scholar] [CrossRef] [PubMed]
- McCartney, D.L.; Walker, R.M.; Morris, S.W.; McIntosh, A.M.; Porteous, D.J.; Evans, K.L. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom. Data 2016, 9, 22–24. [Google Scholar] [CrossRef]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef]
- Kim, H.C.; Jung, C.Y.; Cho, D.G.; Jeon, J.H.; Lee, J.E.; Ahn, J.S.; Kim, S.J.; Kim, Y.; Kim, Y.C.; Kim, J.E.; et al. Clinical characteristics and prognostic factors of lung cancer in Korea: A pilot study of data from the Korean Nationwide Lung Cancer Registry. Tuberc. Respir. Dis. 2019, 82, 118–125. [Google Scholar] [CrossRef]
- Croswell, J.M.; Baker, S.G.; Marcus, P.M.; Clapp, J.D.; Kramer, B.S. Cumulative incidence of false-positive test results in lung cancer screening: A randomized trial. Ann. Intern. Med. 2010, 152, 505–512. [Google Scholar] [CrossRef]
- Field, J.K.; Smith, R.A.; Aberle, D.R.; Oudkerk, M.; Baldwin, D.R.; Yankelevitz, D.; Pedersen, J.H.; Swanson, S.J.; Travis, W.D.; Wisbuba, I.I.; et al. International association for the study of lung cancer computed tomography screening workshop 2011 report. J. Thorac. Oncol. 2012, 7, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Hubers, A.J.; Brinkman, P.; Boksem, R.J.; Rhodius, R.J.; Witte, B.I.; Zwinderman, A.H.; Heideman, D.A.; Duin, S.; Koning, R.; Steenbergen, R.D.; et al. Combined sputum hypermethylation and eNose analysis for lung cancer diagnosis. J. Clin. Pathol. 2014, 67, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Kneip, C.; Schmidt, B.; Seegebarth, A.; Weickmann, S.; Fleischhacker, M.; Liebenberg, V.; Field, J.K.; Dietrich, D. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J. Thorac. Oncol. 2011, 6, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Maddala, T.; Hubbell, E.; Aravanis, A.; Zhang, N.; Venn, O.; Valouev, A.; Shen, L.; Patel, S.; Jamshidi, A.; et al. Genome-wide sequencing for early stage lung cancer detection from plasma cell-free DNA (cfDNA): The Circulating Cancer Genome Atlas (CCGA) study. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Fasanelli, F.; Baglietto, L.; Ponzi, E.; Guida, F.; Campanella, G.; Johansson, M.; Grankvist, K.; Johansson, M.; Assumma, M.B.; Naccarati, A.; et al. Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat. Commun. 2015, 6, 10192. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.; Tao, M.H.; Chen, J.; Scelo, G.; Bencko, V.; Fabianova, E.; Foretova, L.; Janout, V.; Lissowska, J.; Mates, D.; et al. No association between global DNA methylation in peripheral blood and lung cancer risk in nonsmoking women: Results from a multicenter study in Eastern and Central Europe. Eur. J. Cancer Prev. 2018, 27, 1–5. [Google Scholar] [CrossRef]
- Park, S.L.; Patel, Y.M.; Loo, L.W.M.; Mullen, D.J.; Offringa, I.A.; Maunakea, A.; Stram, D.O.; Siegmund, K.; Murphy, S.E.; Tiirikainen, M.; et al. Association of internal smoking dose with blood DNA methylation in three racial/ethnic populations. Clin. Epigenet. 2018, 10, 110. [Google Scholar] [CrossRef]
- Yan, J.; Wei, Q.; Jian, W.; Qiu, B.; Wen, J.; Liu, J.; Fu, B.; Zhou, X.; Zhao, T. IMP3 predicts invasion and prognosis in human lung adenocarcinoma. Lung 2016, 94, 137–146. [Google Scholar] [CrossRef]
- Yanagita, K.; Nagashio, R.; Jiang, S.X.; Kuchitsu, Y.; Hachimura, K.; Ichinoe, M.; Igawa, S.; Fukuda, E. Cytoskeleton-associated protein 4 is a novel serodiagnostic marker for lung cancer. Am. J. Pathol. 2018, 188, 1328–1333. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Patients | Controls | |
|---|---|---|
| Total number | 150 | 150 |
| Male (%) | 86 (57.3) | 86 (57.3) |
| Age, years (mean ± standard error) | 56.1 ± 7.95 | 55.6 ± 7.94 |
| Smoking state (current/past/nonsmokers) | 48/30/72 | 48/30/72 |
| Histological type, (%) | ||
| Adenocarcinoma (% of EGFR mutant) | 108 (59.3) | |
| Squamous cell carcinoma | 42 | |
| Stage (I/II/III/IV) | 46/14/31/59 | — |
| Target ID | Chr a | Position | Gene ID | Location | Beta b | Beta (SE) | -Value c |
|---|---|---|---|---|---|---|---|
| cg12169243 | 15 | 35825579 | DPH6 | Body | 127.250 | 30.577 | 4.3E–08 |
| cg25429010 | 15 | 75932582 | IMP3 | 5′UTR | −529.523 | 128.213 | 5.2E–08 |
| Target ID | Chr a | Position | Gene | Location | Beta b | Beta (SE) | -Value c |
|---|---|---|---|---|---|---|---|
| cg09245319 | 2 | 227049703 | — | — | −227.7 | 66.0 | 3.5E–09 |
| cg17183999 | 16 | 8998406 | USP7 | Body | −433.8 | 124.7 | 1.3E–08 |
| cg06366994 | 4 | 166298963 | CPE | TSS1500 | −51.9 | 13.3 | 2.0E–08 |
| cg24992236 | 14 | 101536808 | MEG9 | Body | 133.9 | 34.9 | 2.2E–08 |
| cg22144719 | 9 | 138106031 | — | — | −55.5 | 14.0 | 2.2E–08 |
| cg22448179 | 7 | 55166882 | EGFR | Body | −93.7 | 26.3 | 7.2E–08 |
| Target ID | Chr a | Position | Gene | Location | Beta b | Beta (SE) | -Value c |
|---|---|---|---|---|---|---|---|
| cg25021476 | 15 | 55488413 | RSL24D1 | Body | −771.0 | 182.1 | 3.9E–08 |
| cg04989085 | 12 | 47629182 | FAM113B | Body | 190.3 | 47.0 | 8.7E–08 |
| cg20905681 | 12 | 106641994 | CKAP4 | TSS1500 | 905.8 | 228.2 | 1.3E–07 |
| cg26379694 | 5 | 152732350 | — | — | 35.4 | 9.0 | 1.3E–07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Y.; Choi, H.-M.; Cheong, H.S.; Shin, H.D.; Choi, C.M.; Kim, W.J. Epigenome-Wide Association Analysis of Differentially Methylated Signals in Blood Samples of Patients with Non-Small-Cell Lung Cancer. J. Clin. Med. 2019, 8, 1307. https://doi.org/10.3390/jcm8091307
Hong Y, Choi H-M, Cheong HS, Shin HD, Choi CM, Kim WJ. Epigenome-Wide Association Analysis of Differentially Methylated Signals in Blood Samples of Patients with Non-Small-Cell Lung Cancer. Journal of Clinical Medicine. 2019; 8(9):1307. https://doi.org/10.3390/jcm8091307
Chicago/Turabian StyleHong, Yoonki, Hye-Mi Choi, Hyun Sub Cheong, Hyoung Doo Shin, Chang Min Choi, and Woo Jin Kim. 2019. "Epigenome-Wide Association Analysis of Differentially Methylated Signals in Blood Samples of Patients with Non-Small-Cell Lung Cancer" Journal of Clinical Medicine 8, no. 9: 1307. https://doi.org/10.3390/jcm8091307
APA StyleHong, Y., Choi, H.-M., Cheong, H. S., Shin, H. D., Choi, C. M., & Kim, W. J. (2019). Epigenome-Wide Association Analysis of Differentially Methylated Signals in Blood Samples of Patients with Non-Small-Cell Lung Cancer. Journal of Clinical Medicine, 8(9), 1307. https://doi.org/10.3390/jcm8091307