Endophenotypes for Age-Related Macular Degeneration: Extending Our Reach into the Preclinical Stages of Disease

Abstract
:1. Introduction
2. Experimental Section
2.1. Subjects

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Marker | Genotyping Success Rate | Chromosome | Location | Gene |
|---|---|---|---|---|---|
| 1 | rs10737680 | 0.979 | 1 | 196,710,325 | CFH |
| 2 | rs6795735 | 0.985 | 3 | 64,719,689 | ADAMTS9-AS2 |
| 3 | rs13081855 | 0.9625 | 3 | 99,762,695 | COL8A1 |
| 4 | rs4698775 | 0.9985 | 4 | 109,669,323 | CFI |
| 5 | rs3130783 | 0.958 | 6 | 30,806,580 | C2-CFB-SKIV2L |
| 6 | rs429608 | 0.997 | 6 | 31,962,685 | SKIV2L |
| 7 | rs943080 | 0.997 | 6 | 43,858,890 | VEGFA |
| 8 | rs3812111 | 1 | 6 | 116,122,572 | COL10A1 |
| 9 | rs13278062 | 1 | 8 | 23,225,458 | TNFRSF10B-LOC389641 |
| 10 | rs334353 | 0.9925 | 9 | 99,146,083 | TGFBR1 |
| 11 | rs10490924 | 0.9895 | 10 | 122,454,932 | ARMS2 |
| 12 | rs9542236 | 1 | 13 | 31,245,188 | B3GALTL |
| 13 | rs8017304 | 1 | 14 | 68,318,360 | RAD51B |
| 14 | rs920915 | 0.997 | 15 | 58,396,268 | LIPC |
| 15 | rs1864163 | 0.994 | 16 | 56,963,321 | CETP |
| 16 | rs4420638 | 0.964 | 19 | 44,919,689 | APOC1/APOE |
| 17 | rs8135665 | 0.994 | 22 | 38,080,269 | SLC16A8 |
2.2. Genetic Analysis
2.3. Microperimetry
2.4. Optical Coherence Tomography
3. Results
3.1. Characteristics of the Study Population
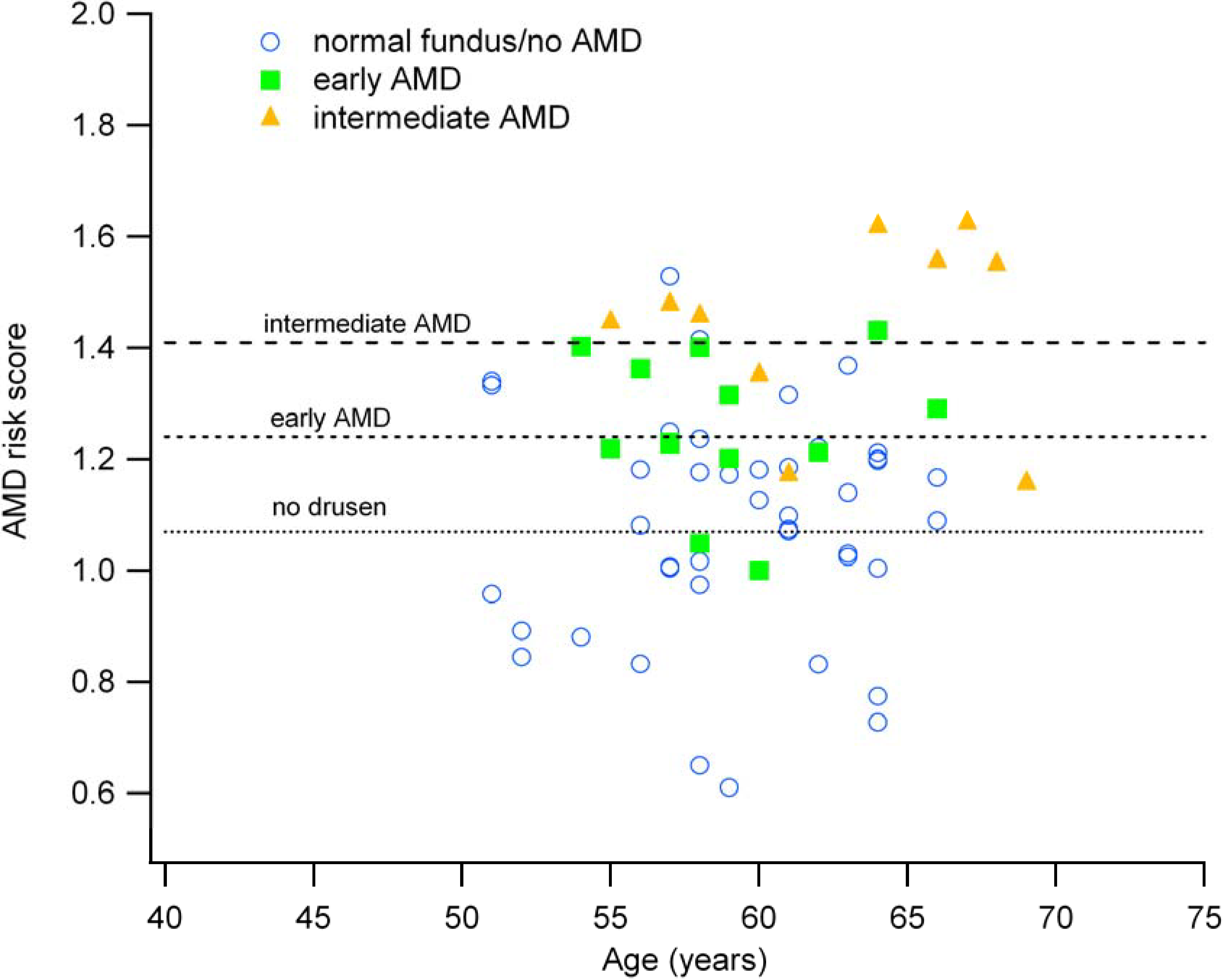
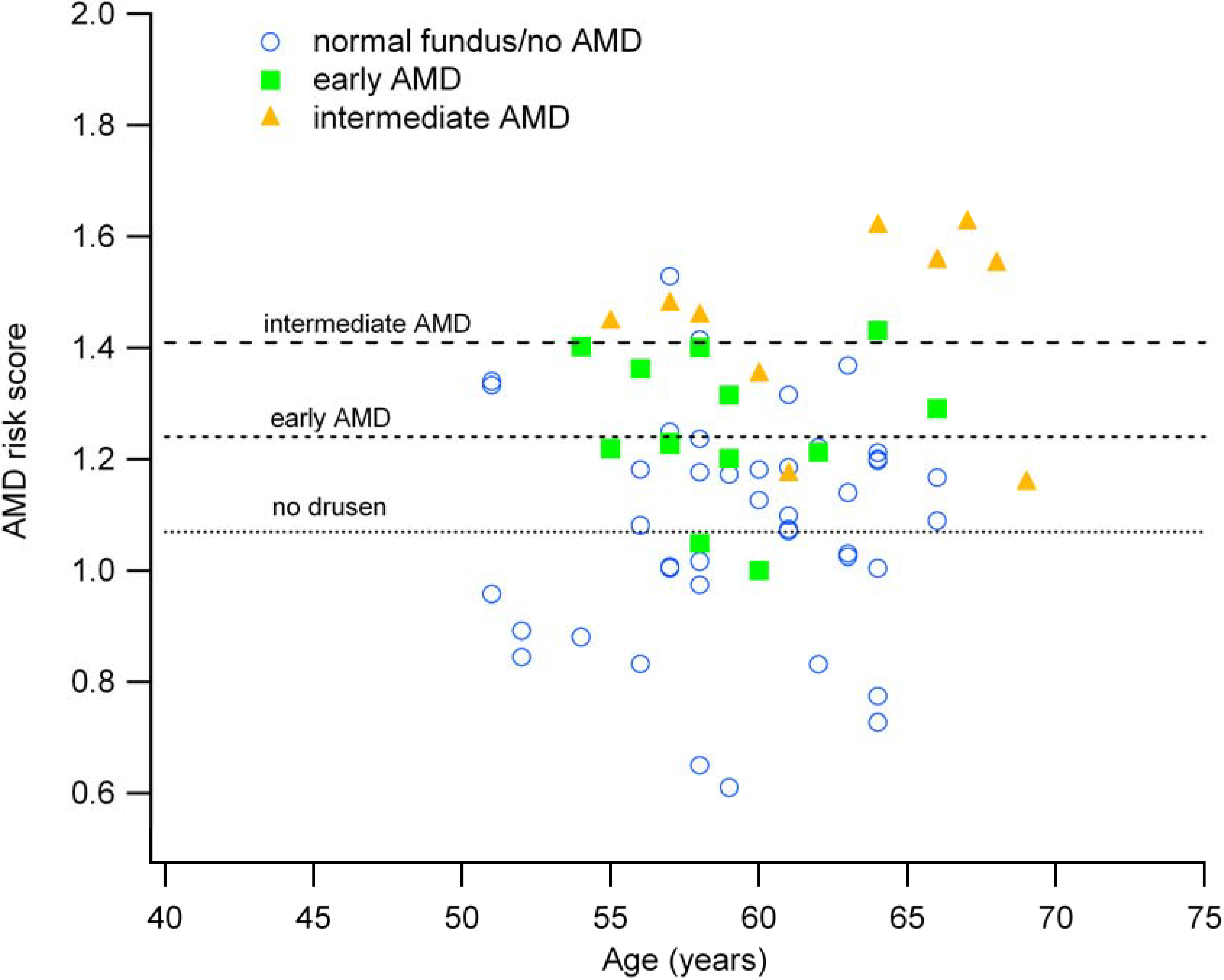
3.2. Genetic Risk Scores are Associated with Fundus Appearance
| Fundus Grading based on OCT Imaging | N | Mean Risk Score (±1 SD) |
|---|---|---|
| Normal (no drusen) | 43 | 1.07 (0.20) |
| Early AMD (few small drusen) | 12 | 1.24 (0.12) |
| Intermediate AMD (multiple large drusen) | 14 | 1.41 (0.19) |
3.3. The AMD Risk Score and Age Correlate with Clinical Findings
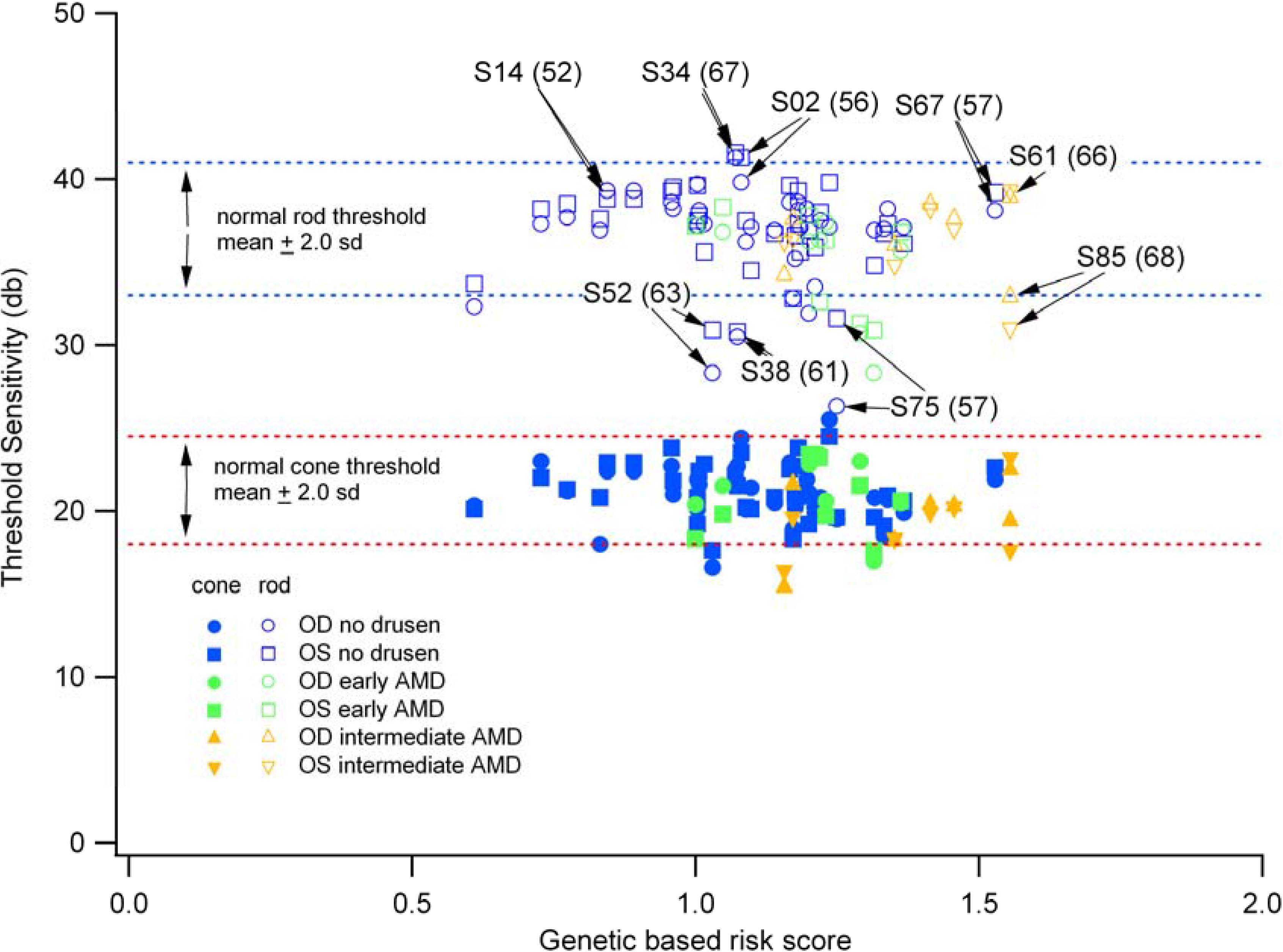
3.4. Individuals with High-Risk Genetic Profiles Do Not Necessarily Exhibit Reductions in Scotopic and Photopic Sensitivity

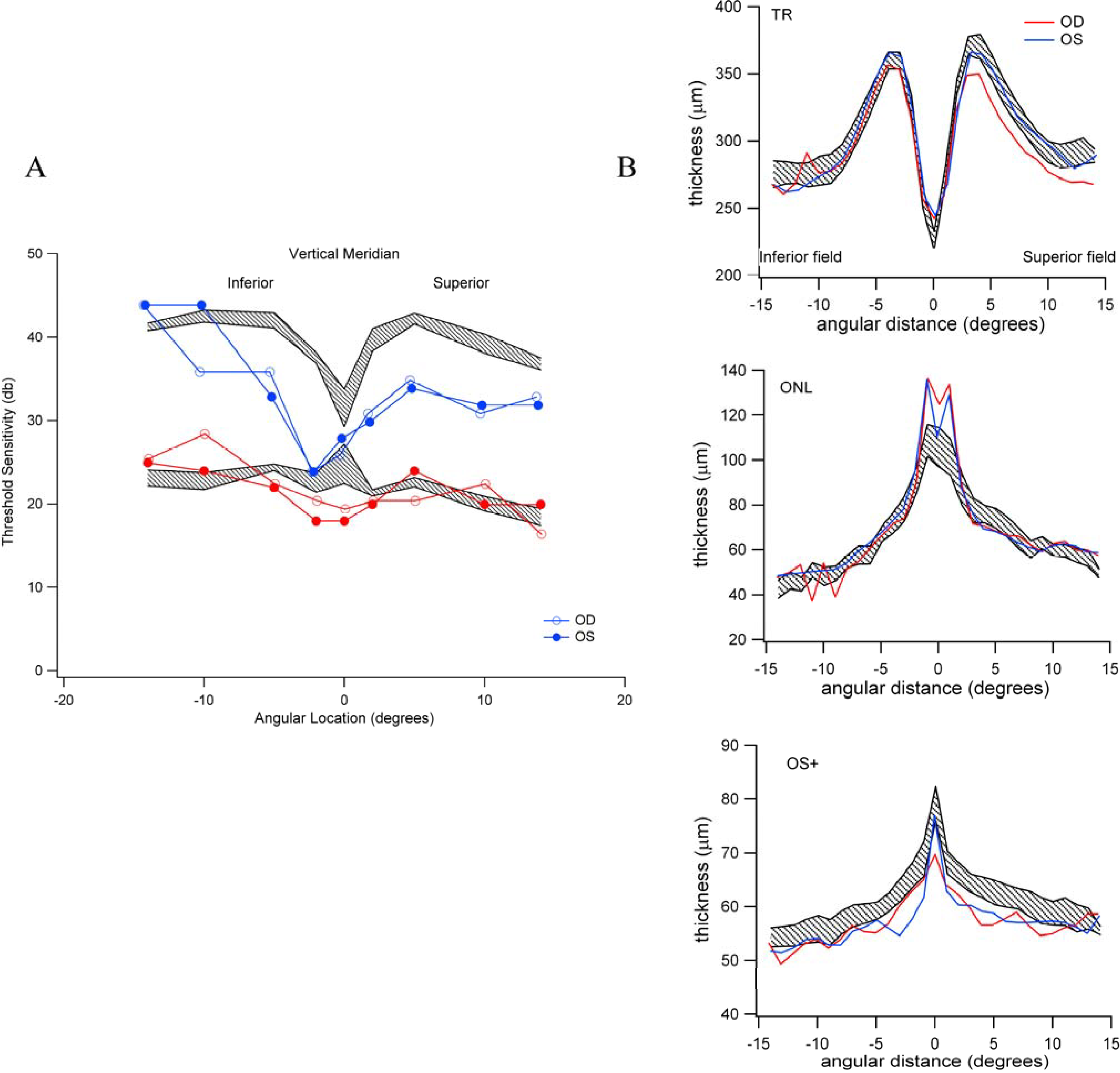
3.5. Reduced Rod-Mediated Threshold Sensitivity in Patients with Normal Fundus Is Not Clearly Accompanied by Retinal Structural Changes in the Absence of Drusen
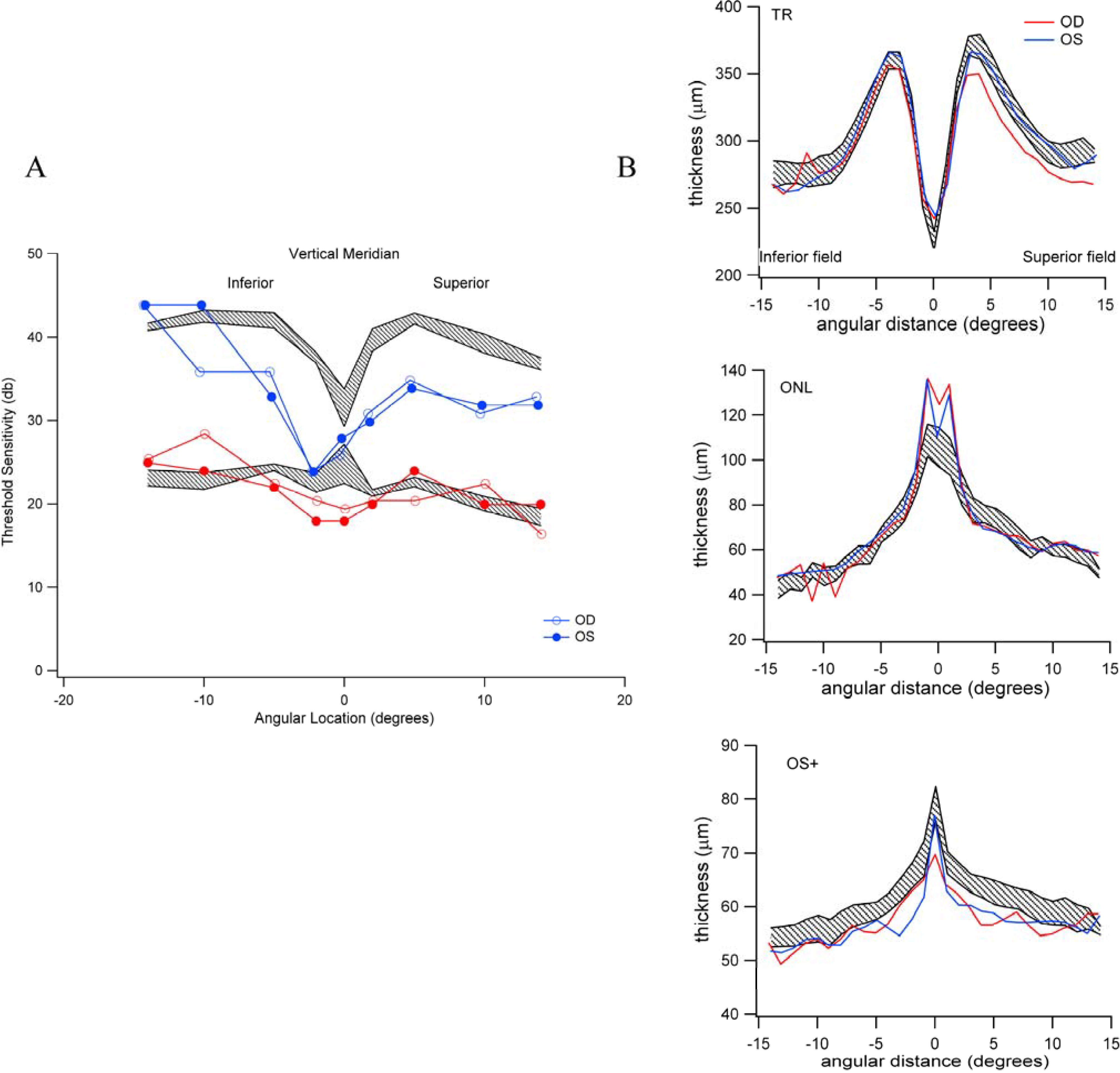
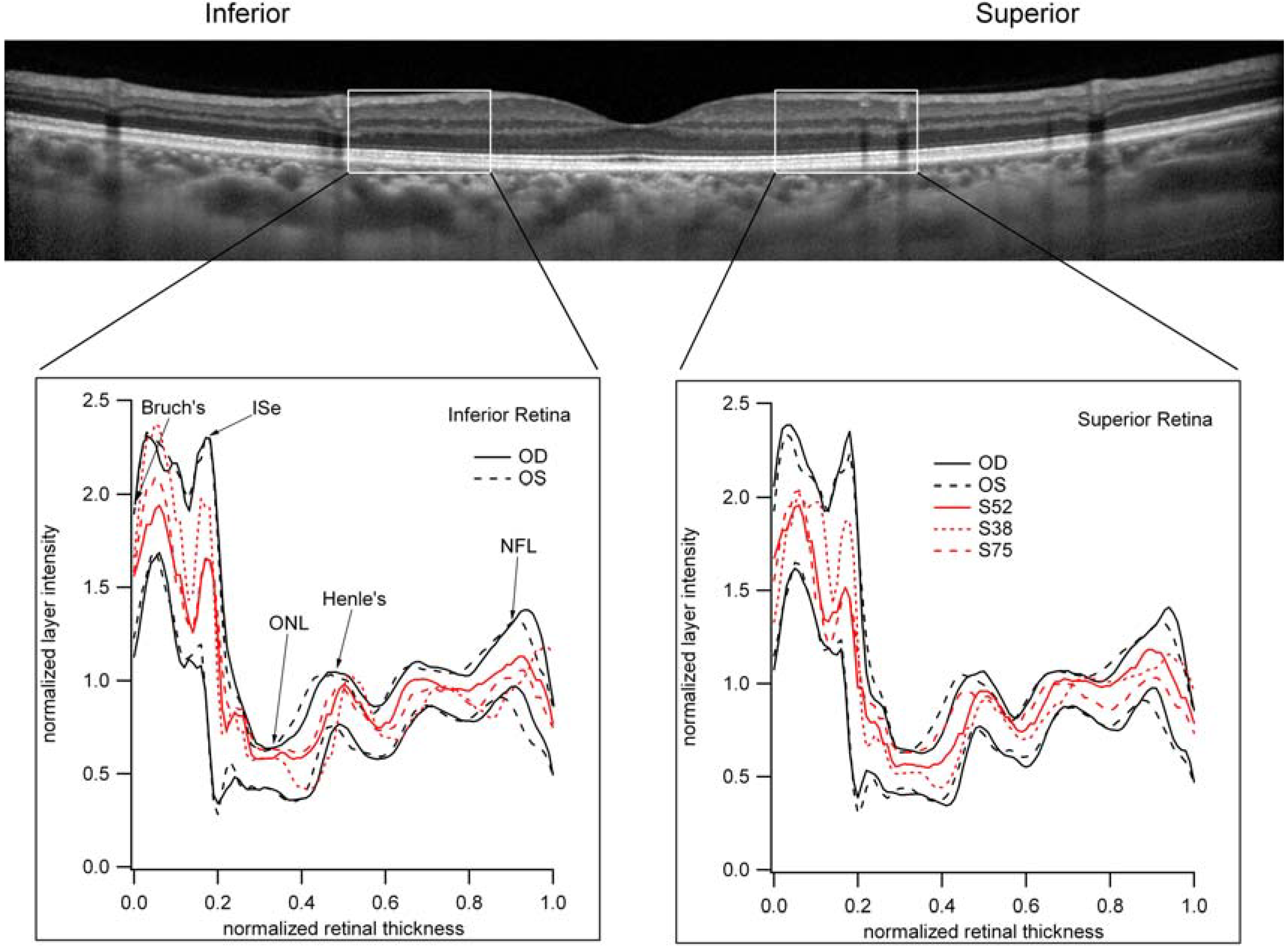
3.6. The Relative Brightness of the Bands Seen on OCT in Those Individuals with Reduced Scotopic Sensitivity Is Not Reduced?
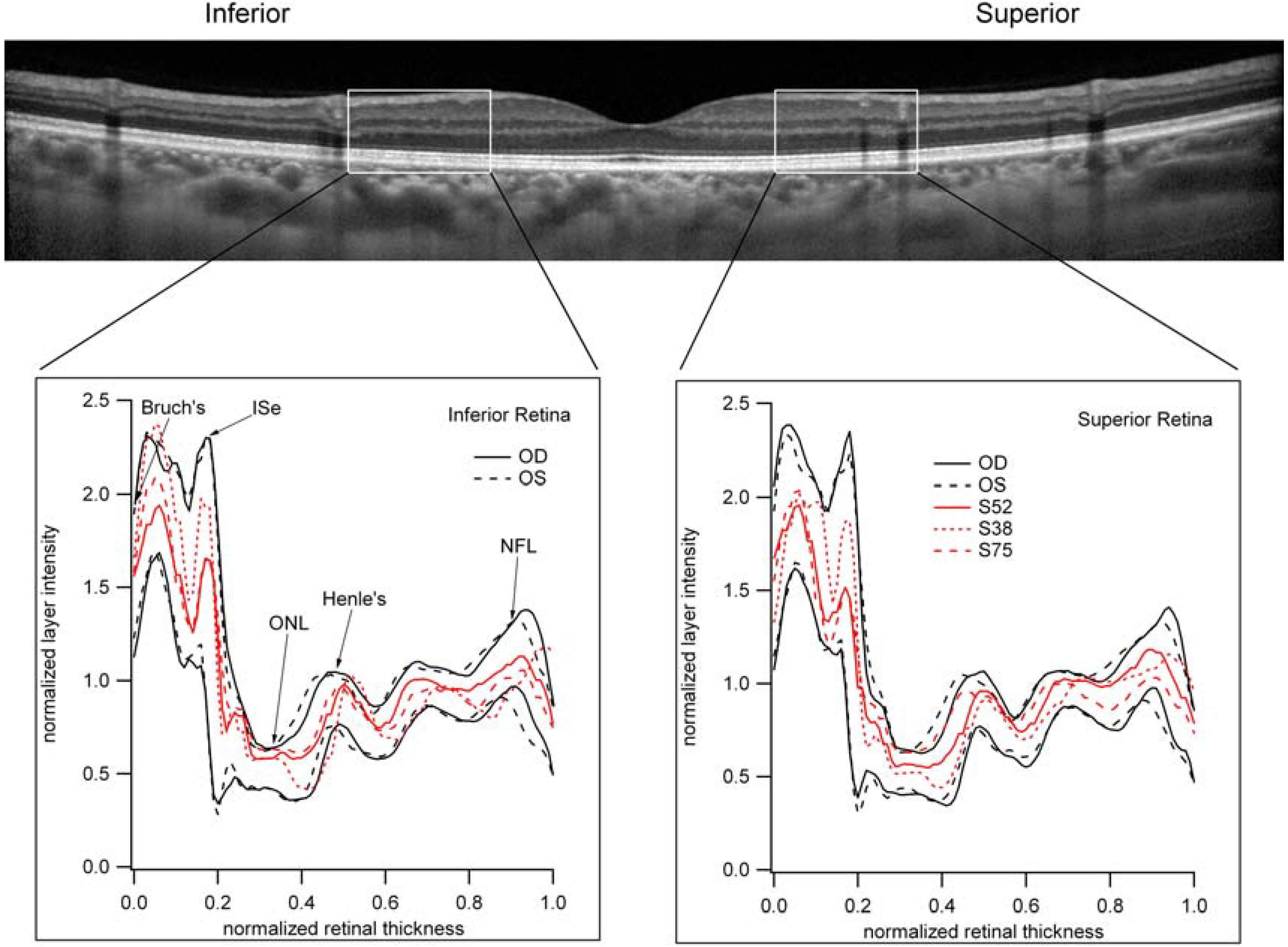
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Klaver, C.C.; Kliffen, M.; van Duijn, C.M.; Hofman, A.; Cruts, M.; Grobbee, D.E.; van Broeckhoven, C.; de Jong, P.T. Genetic association of apolipoprotein E with age-related macular degeneration. Am. J. Hum. Genet. 1998, 63, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Zareparsi, S.; Branham, K.E.; Li, M.; Shah, S.; Klein, R.J.; Ott, J.; Hoh, J.; Abecasis, G.R.; Swaroop, A. Strong association of the Y402H variant in complement factor H at 1q32 with susceptibility to age-related macular degeneration. Am. J. Hum. Genet. 2005, 77, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.; Spencer, K.L.; Kwan, S.Y.; Noureddine, M.; Gilbert, J.R.; et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.O.; Ritter, R., III; Abel, K.J.; Manning, A.; Panhuysen, C.; Farrer, L.A. Complement factor H polymorphism and age-related macular degeneration. Science 2005, 308, 421–424. [Google Scholar] [CrossRef]
- Rivera, A.; Fisher, S.A.; Fritsche, L.G.; Keilhauer, C.N.; Lichtner, P.; Meitinger, T.; Weber, B.H. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum. Mol. Genet. 2005, 14, 3227–3236. [Google Scholar] [CrossRef] [PubMed]
- Jakobsdottir, J.; Conley, Y.P.; Weeks, D.E.; Mah, T.S.; Ferrell, R.E.; Gorin, M.B. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am. J. Hum. Genet. 2005, 77, 389–407. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Dewan, A.; Liu, M.; Hartman, S.; Zhang, S.S.; Liu, D.T.; Zhao, C.; Tam, P.O.; Chan, W.M.; Lam, D.S.; Snyder, M.; et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science 2006, 314, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-Related Macular Degeneration: Genetics and Biology Coming Together. Annu. Rev. Genomics Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.O.; Fridley, B.L.; James, K.M.; Sharma, A.K.; Cunningham, J.M.; Tosakulwong, N. Evaluation of clustering and genotype distribution for replication in genome wide association studies: the age-related eye disease study. PloS One 2008, 3, e3813. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Glowacki, S.; Kauppinen, A.; Kaarniranta, K. Mitochondrial and nuclear DNA damage and repair in age-related macular degeneration. Int. J. Mol. Sci. 2013, 14, 2996–3010. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.M.; Manwaring, N.; Wang, J.J.; Rochtchina, E.; Mitchell, P.; Sue, C.M. Mitochondrial DNA haplogroups and age-related maculopathy. Arch. Ophthalmol. 2007, 125, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Kenney, M.C.; Chwa, M.; Atilano, S.R.; Pavlis, J.M.; Falatoonzadeh, P.; Ramirez, C.; Malik, D.; Hsu, T.; Woo, G.; Soe, K.; et al. Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: Implications for age-related macular degeneration. PloS One 2013, 8, e54339. [Google Scholar] [CrossRef] [PubMed]
- SanGiovanni, J.P.; Arking, D.E.; Iyengar, S.K.; Elashoff, M.; Clemons, T.E.; Reed, G.F.; Henning, A.K.; Sivakumaran, T.A.; Xu, X.; DeWan, A.; et al. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PloS One 2009, 4, e5508. [Google Scholar] [CrossRef] [PubMed]
- Tilleul, J.; Richard, F.; Puche, N.; Zerbib, J.; Leveziel, N.; Sahel, J.A.; Cohen, S.Y.; Korobelnik, J.F.; Feingold, J.; Munnich, A.; et al. Genetic association study of mitochondrial polymorphisms in neovascular age-related macular degeneration. Mol. Vis. 2013, 19, 1132–1140. [Google Scholar] [PubMed]
- Raychaudhuri, S.; Iartchouk, O.; Chin, K.; Tan, P.L.; Tai, A.K.; Ripke, S.; Gowrisankar, S.; Vemuri, S.; Montgomery, K.; Yu, Y.; et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011, 43, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Triebwasser, M.P.; Wong, E.K.; Schramm, E.C.; Thomas, B.; Reynolds, R.; Mardis, E.; Atkinson, J.P.; Daly, M.; Raychaudhuri, S.; et al. Whole-exome sequencing identifies rare, functional CFH variants in families with macular degeneration. Hum. Mol. Genet. 2014, 19, 5283–5293. [Google Scholar] [CrossRef]
- Duvvari, M.R.; Paun, C.C.; Buitendijk, G.H.; Saksens, N.T.; Volokhina, E.B.; Ristau, T.; Schoenmaker-Koller, F.E.; van de Ven, J.P.; Groenewoud, J.M.; van den Heuvel, L.P.; et al. Analysis of rare variants in the C3 gene in patients with age-related macular degeneration. PloS One 2014, 9, e94165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, J.M.; Yu, Y.; Miller, E.C.; Reynolds, R.; Tan, P.L.; Gowrisankar, S.; Goldstein, J.I.; Triebwasser, M.; Anderson, H.E.; Zerbib, J.; et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 2013, 45, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.D.; Cooke Bailey, J.N.; D’Aoust, L.N.; Cade, W.; Ayala-Haedo, J.; Fuzzell, M.D.; Laux, R.A.; Adams, L.; Reinhart-Mercer, L.; Caywood, L.; et al. Rare Complement Factor H Variant Associated with Age-related Macular Degeneration in the Amish. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4455–4460. [Google Scholar] [CrossRef]
- Ahluwalia, N.; Blacher, J.; Szabo de Edelenyi, F.; Faure, P.; Julia, C.; Hercberg, S.; Galan, P. Prognostic value of multiple emerging biomarkers in cardiovascular risk prediction in patients with stable cardiovascular disease. Atherosclerosis 2013, 228, 478–484. [Google Scholar] [CrossRef]
- The Emerging Risk Factors Collaboration. C-Reactive protein, fibrinogen, and cardiovascular disease prediction. New Engl. J. Med. 2012, 367, 1310–1320. [Google Scholar]
- Lenzenweger, M.F. Endophenotype, intermediate phenotype, biomarker: Definitions, concept comparisons, clarifications. Depress. Anxiety 2013, 30, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Quellec, G.; Russell, S.R.; Seddon, J.M.; Reynolds, R.; Scheetz, T.; Mahajan, V.B.; Stone, E.M.; Abramoff, M.D. Automated discovery and quantification of image-based complex phenotypes: A twin study of drusen phenotypes in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9195–9206. [Google Scholar] [CrossRef]
- Hageman, G.S.; Mullins, R.F. Molecular composition of drusen as related to substructural phenotype. Mol. Vis. 1999, 5, 28. [Google Scholar] [PubMed]
- Saade, C.; Smith, R.T. Reticular macular lesions: A review of the phenotypic hallmarks and their clinical significance. Clin. Exp. Ophthalmol. 2014. [Google Scholar] [CrossRef]
- Steinberg, J.S.; Auge, J.; Jaffe, G.J.; Fleckenstein, M.; Holz, F.G.; Schmitz-Valckenberg, S.; Group, G.A.P.S. Longitudinal analysis of reticular drusen associated with geographic atrophy in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4054–4060. [Google Scholar] [CrossRef]
- Garg, A.; Oll, M.; Yzer, S.; Chang, S.; Barile, G.R.; Merriam, J.C.; Tsang, S.H.; Bearelly, S. Reticular pseudodrusen in early age-related macular degeneration are associated with choroidal thinning. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7075–7081. [Google Scholar] [CrossRef]
- Schmitz-Valckenberg, S.; Alten, F.; Steinberg, J.S.; Jaffe, G.J.; Fleckenstein, M.; Mukesh, B.N.; Hohman, T.C.; Holz, F.G. The Geographic Atrophy Progression Study Group. Reticular drusen associated with geographic atrophy in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5009–5015. [Google Scholar] [CrossRef]
- Gottesman, I.I.; Gould, T.D. The endophenotype concept in psychiatry: Etymology and strategic intentions. Am. J. Psychiatry 2003, 160, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, L.; Jiang, F.; Liang, J.; Wu, D.Z. Scotopic sensitivity of central retina in early age-related macular degeneration. Yan Ke Xue Bao 2003, 19, 15–19. [Google Scholar] [PubMed]
- Dimitrov, P.N.; Robman, L.D.; Varsamidis, M.; Aung, K.Z.; Makeyeva, G.; Busija, L.; Vingrys, A.J.; Guymer, R.H. Relationship between clinical macular changes and retinal function in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5213–5220. [Google Scholar] [CrossRef]
- Haimovici, R.; Owens, S.L.; Fitzke, F.W.; Bird, A.C. Dark adaptation in age-related macular degeneration: Relationship to the fellow eye. Graefe’s Arch. Clin. Exp. Ophthalmol. 2002, 240, 90–95. [Google Scholar] [CrossRef]
- Jackson, G.R.; Edwards, J.G. A short-duration dark adaptation protocol for assessment of age-related maculopathy. J. Ocul. Biol. Dis. Inform. 2008, 1, 7–11. [Google Scholar] [CrossRef]
- Jackson, G.R.; Felix, T.; Owsley, C. The Scotopic Sensitivity Tester-1 and the detection of early age-related macular degeneration. Ophthalmic Physiolo. Opt. 2006, 26, 431–437. [Google Scholar] [CrossRef]
- Midena, E.; Degli Angeli, C.; Blarzino, M.C.; Valenti, M.; Segato, T. Macular function impairment in eyes with early age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 1997, 38, 469–477. [Google Scholar]
- Owsley, C.; Jackson, G.R.; White, M.; Feist, R.; Edwards, D. Delays in rod-mediated dark adaptation in early age-related maculopathy. Ophthalmology 2001, 108, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Owsley, C.; McGwin, G., Jr.; Jackson, G.R.; Kallies, K.; Clark, M. Cone- and rod-mediated dark adaptation impairment in age-related maculopathy. Ophthalmology 2007, 114, 1728–1735. [Google Scholar] [CrossRef] [PubMed]
- Do, C.B.; Hinds, D.A.; Francke, U.; Eriksson, N. Comparison of family history and SNPs for predicting risk of complex disease. PLoS Genet. 2012, 8, e1002973. [Google Scholar] [CrossRef] [PubMed]
- Aiyar, L.; Shuman, C.; Hayeems, R.; Dupuis, A.; Pu, S.; Wodak, S.; Chitayat, D.; Velsher, L.; Davies, J. Risk estimates for complex disorders: Comparing personal genome testing and family history. Genet. Med. 2014, 16, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Charbel Issa, P.; Barnard, A.R.; Singh, M.S.; Carter, E.; Jiang, Z.; Radu, R.A.; Schraermeyer, U.; MacLaren, R.E. Fundus autofluorescence in the Abca4(−/−) mouse model of Stargardt disease—Correlation with accumulation of A2E, retinal function, and histology. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5602–5612. [Google Scholar]
- Hunter, J.J.; Masella, B.; Dubra, A.; Sharma, R.; Yin, L.; Merigan, W.H.; Palczewska, G.; Palczewski, K.; Williams, D.R. Images of photoreceptors in living primate eyes using adaptive optics two-photon ophthalmoscopy. Biomed. Opt. Expr. 2010, 2, 139–148. [Google Scholar] [CrossRef]
- Bernstein, P.S.; Ahmed, F.; Liu, A.; Allman, S.; Sheng, X.; Sharifzadeh, M.; Ermakov, I.; Gellermann, W. Macular pigment imaging in AREDS2 participants: An ancillary study of AREDS2 subjects enrolled at the Moran Eye Center. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6178–6186. [Google Scholar] [CrossRef]
- Frandsen, J.E.; Llop, S.; Digre, K.B.; Bernstein, P.S.; Sharifzadeh, M.; Warner, J.E.; Gellerman, W.; Katz, B.J. Quantification of macular carotenoids using autofluorescence imaging in patients with photosensitive migraine and benign essential blepharospasm. Arch. Ophthalmol. 2012, 130, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Masters, B.R. Noninvasive redox fluorometry: How light can be used to monitor alterations of corneal mitochondrial function. Curr. Eye Res. 1984, 3, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.E.; Cope, M.; Springett, R.; Amess, P.N.; Penrice, J.; Tyszczuk, L.; Punwani, S.; Ordidge, R.; Wyatt, J.; Delpy, D.T. Use of mitochondrial inhibitors to demonstrate that cytochrome oxidase near-infrared spectroscopy can measure mitochondrial dysfunction noninvasively in the brain. J. Cereb. Blood Flow Metab. 1999, 19, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P.; Zrenner, E. Electrophysiology in the investigation of acquired retinal disorders. Surv. Ophthalmol. 2000, 45, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Feigl, B.; Brown, B.; Lovie-Kitchin, J.; Swann, P. The rod-mediated multifocal electroretinogram in aging and in early age-related maculopathy. Curr. Eye Res. 2006, 31, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.R.; Owsley, C. Scotopic sensitivity during adulthood. Vis. Res. 2000, 40, 2467–2473. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.R.; Owsley, C.; Cordle, E.P.; Finley, C.D. Aging and scotopic sensitivity. Vis. Res. 1998, 38, 3655–3662. [Google Scholar] [CrossRef] [PubMed]
- Sturr, J.F.; Zhang, L.; Taub, H.A.; Hannon, D.J.; Jackowski, M.M. Psychophysical evidence for losses in rod sensitivity in the aging visual system. Vis. Res. 1997, 37, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Feigl, B.; Brown, B.; Lovie-Kitchin, J.; Swann, P. Monitoring retinal function in early age-related maculopathy: Visual performance after 1 year. Eye 2005, 19, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Owsley, C.; Jackson, G.R.; Cideciyan, A.V.; Huang, Y.; Fine, S.L.; Ho, A.C.; Maguire, M.G.; Lolley, V.; Jacobson, S.G. Psychophysical evidence for rod vulnerability in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2000, 41, 267–273. [Google Scholar]
- Brown, B.; Adams, A.J.; Coletta, N.J.; Haegerstrom-Portnoy, G. Dark adaptation in age-related maculopathy. Ophthalmic physiol. Opt. 1986, 6, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, R.L.; Polkinghorne, P.C.; Fitzke, F.W.; Kemp, C.M.; Bird, A.C. Abnormal dark adaptation and rhodopsin kinetics in Sorsby’s fundus dystrophy. Investig. Ophthalmol. Vis. Sci. 1992, 33, 1633–1636. [Google Scholar]
- Dimitrov, P.N.; Guymer, R.H.; Zele, A.J.; Anderson, A.J.; Vingrys, A.J. Measuring rod and cone dynamics in age-related maculopathy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Gaffney, A.J.; Binns, A.M.; Margrain, T.H. The effect of pre-adapting light intensity on dark adaptation in early age-related macular degeneration. Doc. Ophthalmol. 2013, 127, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Vujosevic, S.; Smolek, M.K.; Lebow, K.A.; Notaroberto, N.; Pallikaris, A.; Casciano, M. Detection of macular function changes in early (AREDS 2) and intermediate (AREDS 3) age-related macular degeneration. Ophthalmologica 2011, 225, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Schnell-Inderst, P.; Schwarzer, R.; Gohler, A.; Grandi, N.; Grabein, K.; Stollenwerk, B.; Klauss, V.; Wasem, J.; Siebert, U. Prognostic value, clinical effectiveness and cost-effectiveness of high sensitivity C-reactive protein as a marker in primary prevention of major cardiac events. GMS Health Technol. Assess. 2009, 5. [Google Scholar] [CrossRef]
- Wilson, P.W.; Nam, B.H.; Pencina, M.; D’Agostino, R.B., Sr.; Benjamin, E.J.; O’Donnell, C.J. C-reactive protein and risk of cardiovascular disease in men and women from the Framingham Heart Study. Arch. Intern. Med. 2005, 165, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, N.; Almasy, L.; Schroeder, M.; Mulvihill, W.P.; Weeks, D.E. Mega2: Data-handling for facilitating genetic linkage and association analyses. Bioinformatics 2005, 21, 2556–2557. [Google Scholar] [CrossRef] [PubMed]
- Renteria, M.E.; Cortes, A.; Medland, S.E. Using PLINK for Genome-Wide Association Studies (GWAS) and data analysis. Meth. Mol. Biol. 2013, 1019, 193–213. [Google Scholar]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birch, D.G.; Wen, Y.; Locke, K.; Hood, D.C. Rod sensitivity, cone sensitivity, and photoreceptor layer thickness in retinal degenerative diseases. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7141–7147. [Google Scholar] [CrossRef]
- Hood, D.C.; Zhang, X.; Ramachandran, R.; Talamini, C.L.; Raza, A.; Greenberg, J.P.; Sherman, J.; Tsang, S.H.; Birch, D.G. The inner segment/outer segment border seen on optical coherence tomography is less intense in patients with diminished cone function. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9703–9709. [Google Scholar] [CrossRef]
- Rangaswamy, N.V.; Patel, H.M.; Locke, K.G.; Hood, D.C.; Birch, D.G. A comparison of visual field sensitivity to photoreceptor thickness in retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4213–4219. [Google Scholar] [CrossRef]
- Wen, Y.; Klein, M.; Hood, D.C.; Birch, D.G. Relationships among multifocal electroretinogram amplitude, visual field sensitivity, and SD-OCT receptor layer thicknesses in patients with retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2012, 53, 833–840. [Google Scholar] [CrossRef]
- Sadigh, S.; Cideciyan, A.V.; Sumaroka, A.; Huang, W.C.; Luo, X.; Swider, M.; Steinberg, J.D.; Stambolian, D.; Jacobson, S.G. Abnormal thickening as well as thinning of the photoreceptor layer in intermediate age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1603–1612. [Google Scholar] [CrossRef]
- Spaide, R.F.; Curcio, C.A. Anatomical correlates to the bands seen in the outer retina by optical coherence tomography: Literature review and model. Retina 2011, 31, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Cobos, E.; Arias, L.; Ruiz-Moreno, J.; Rubio, M.; Garcia-Bru, P.; Caminal, J.; Catala-Mora, J.; Arruga, J. Preoperative study of the inner segment/outer segment junction of photoreceptors by spectral-domain optical coherence tomography as a prognostic factor in patients with epiretinal membranes. Clin. Ophthalmol. 2013, 7, 1467–1470. [Google Scholar] [CrossRef]
- Scholda, C.; Wirtitsch, M.; Hermann, B.; Unterhuber, A.; Ergun, E.; Sattmann, H.; Ko, T.H.; Fujimoto, J.G.; Fercher, A.F.; Stur, M.; et al. Ultrahigh resolution optical coherence tomography of macular holes. Retina 2006, 26, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Spaide, R.F.; Koizumi, H.; Freund, K.B. Photoreceptor outer segment abnormalities as a cause of blind spot enlargement in acute zonal occult outer retinopathy-complex diseases. Am. J. Ophthalmol. 2008, 146, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Morita, S.; Watanabe, Y.; Kaneko, T.; Yamane, S.; Kobayashi, S.; Arakawa, A.; Kadonosono, K. Inner segment/outer segment junction assessed by spectral-domain optical coherence tomography in patients with idiopathic epiretinal membrane. Am. J. Ophthalmol. 2010, 150, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ayton, L.N.; Luu, C.D.; Guymer, R.H. Relationship between Retinal Microstructures on Optical Coherence Tomography and Microperimetry in Age-Related Macular Degeneration. Ophthalmology 2014, 121, 1445–1452. [Google Scholar] [CrossRef]
- Wu, Z.; Ayton, L.N.; Guymer, R.H.; Luu, C.D. Relationship between the second reflective band on optical coherence tomography and multifocal electroretinography in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2800–2806. [Google Scholar] [CrossRef]
- Spaide, R.F. Outer retinal atrophy after regression of subretinal drusenoid deposits as a newly recognized form of late age-related macular degeneration. Retina 2013, 33, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Acton, J.H.; Smith, R.T.; Hood, D.C.; Greenstein, V.C. Relationship between retinal layer thickness and the visual field in early age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7618–7624. [Google Scholar] [CrossRef]
- Feigl, B.; Morris, C.P. The challenge of predicting macular degeneration. Curr. Med. Res. Opin. 2011, 27, 1745–1748. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorin, M.B.; Weeks, D.E.; Baron, R.V.; Conley, Y.P.; Ortube, M.C.; Nusinowitz, S. Endophenotypes for Age-Related Macular Degeneration: Extending Our Reach into the Preclinical Stages of Disease. J. Clin. Med. 2014, 3, 1335-1356. https://doi.org/10.3390/jcm3041335
Gorin MB, Weeks DE, Baron RV, Conley YP, Ortube MC, Nusinowitz S. Endophenotypes for Age-Related Macular Degeneration: Extending Our Reach into the Preclinical Stages of Disease. Journal of Clinical Medicine. 2014; 3(4):1335-1356. https://doi.org/10.3390/jcm3041335
Chicago/Turabian StyleGorin, Michael B., Daniel E. Weeks, Robert V. Baron, Yvette P. Conley, Maria C. Ortube, and Steven Nusinowitz. 2014. "Endophenotypes for Age-Related Macular Degeneration: Extending Our Reach into the Preclinical Stages of Disease" Journal of Clinical Medicine 3, no. 4: 1335-1356. https://doi.org/10.3390/jcm3041335