Abstract
Currently, two-thirds of American adults are overweight or obese. This high prevalence of overweight/obesity negatively affects the health of the population, as obese individuals tend to develop several chronic diseases, such as type 2 diabetes and cardiovascular diseases. Due to obesity’s impact on health, medical costs, and longevity, the rise in the number of obese people has become a public health concern. Both genetic and environmental/dietary factors play a role in the development of metabolic diseases. Intuitively, it seems to be obvious to link over-nutrition to the development of obesity and other metabolic diseases. However, the underlying mechanisms are still unclear. Dietary nutrients not only provide energy derived from macronutrients, but also factors such as micronutrients with regulatory roles. How micronutrients, such as vitamin A (VA; retinol), regulate macronutrient homeostasis is still an ongoing research topic. As an essential micronutrient, VA plays a key role in the general health of an individual. This review summarizes recent research progress regarding VA’s role in carbohydrate, lipid, and protein metabolism. Due to the large amount of information regarding VA functions, this review focusses on metabolism in metabolic active organs and tissues. Additionally, some perspectives for future studies will be provided.
1. Introduction
The prevalence of overweight and obesity has become a public health concern around the world [1]. According to the World Health Organization’s 2010 report, 35% of adults aged 20 years and older worldwide were clinically overweight (body mass index, BMI ≥ 25 kg/m2) based on data from 2008. More than half a billion adults in the same category were obese (BMI ≥ 30 kg/m2) [2]. Moreover, 2.8 million deaths are linked to raised BMI each year [2]. Overweight and obesity are associated with an increased risk of developing an assortment of metabolic diseases, which mainly include hypertension, type 2 diabetes mellitus, non-alcoholic fatty liver disease, coronary heart disease, and stroke [1,3]. The implications of all those health conditions have brought gloomy biomedical and socioeconomic consequences to the well-being of the global population [3].
Both genetic and nutritional/environmental factors are instrumental to the development of obesity and its related metabolic diseases [4,5]. Mutations in the appetite-regulating hormones (e.g., leptin and proopiomelanocortin) and their corresponding receptors (e.g., leptin receptor and melanocortin 4 receptor, respectively) result in early onset of obesity and obesity-associated metabolic dysregulation in both humans and experimental rodent models [6]. Given the increasing numbers of the monogenic contributors, the majority of the obesity cases in the human population are believed to be polygenic [6]. On the other hand, overnutrition related to unhealthy eating habits probably drives the rising global prevalence of overweight and obesity [7,8]. In spite of the self-evident link between overnutrition and obesity, the underlying mechanisms that cause obesity and its related metabolic diseases due to excessive intake of nutrients have not been fully understood.
The utilization of dietary macronutrients requires coordinative regulation of the metabolic pathways in different organs and tissues of the body. In a fed state, excessive dietary macronutrients are conserved in the form of glycogen and triglycerides (TG) for future energy needs. During fasting, glycogen and TG are, respectively, broken down into glucose and fatty acids (FAs) to meet the energy needs of different organs and tissues in the body. These processes are coordinately regulated by both hormonal and nutritional stimuli in response to feeding and energy states so that the body reaches energy homeostasis [4,5]. Dysregulated hormonal balance and nutrient metabolism not only disrupt the energy homeostasis, but also predispose individuals to developing obesity and related metabolic diseases [5].
Insulin, secreted from pancreatic β-cells in response to physiological stimuli, is an important peptide hormone in anabolism [9]. Upon secretion, it binds to the insulin receptor on cell membranes and exerts its action through a signal transduction cascade that is mainly composed of kinases and phosphatases. The binding causes conformational changes of the insulin receptor and transphosphorylation of the β-subunit tyrosine kinase [10,11], which initiates the signal transduction cascade [12]. The signal is transduced by multiple components in a complex network containing multiple kinases and phosphatases [13,14]. The activation of the insulin signaling pathway results in changes in the activities and/or the expression levels of enzymes involved in nutrient metabolism. These subsequently alter the metabolic state of tissues and organs in the body. For example, insulin promotes glycolysis, glycogenesis, and lipogenesis, and, at the same time, suppresses gluconeogenesis in the liver [14]. However, in overweight and obese individuals, a certain dose of insulin only produces subnormal physiological responses. This observation is regarded as insulin resistance, which occurs concurrently with profound alterations in glucose and lipid metabolism [9,15,16]. Even though the molecular mechanisms leading to insulin resistance have not been fully understood, it is postulated that systemic inflammation, dysregulated lipid metabolism, and gastrointestinal dysbiosis, triggered by overnutrition, interplay with each other and result in the impairment of insulin action [16].
Overnutrition not only provides excessive energy from macronutrients, but also superfluous amounts of vitamins and essential factors, which have regulatory roles. How micronutrients, such as vitamin A (VA; retinol), regulate the homeostasis of macronutrients is still an ongoing research topic. As an essential and lipophilic micronutrient, VA plays a key role in the general health of an individual [5]. This review summarizes recent research progresses regarding the role of VA in carbohydrate, lipid and protein metabolism in the liver, pancreas, skeletal muscle, and adipose tissues, which are major active players in glucose and FA metabolism.
2. Overview of VA and Its Metabolism
VA is important for a myriad of physiological functions, including vision formation, immune response, cell differentiation and proliferation, embryonic development and metabolism [17]. The investigation of its pleiotropic effect on animals resulted in the discovery of the retinoid signaling pathway by which VA exerts its function by modulating gene expression in target cells [18,19]. Dietary molecules with VA activities exist in two forms: provitamin A (carotenoids and cryptoxanthins) and preformed VA (retinol or retinyl esters). Provitamin A molecules are mainly found in plants. Herbivorous and omnivorous animals convert provitamin A molecules into retinol before storing it as retinyl esters in different organs and tissues, depending on the species [20]. Retinol and retinyl esters are then introduced into the food chain and transferred to carnivorous and omnivorous animals at the higher tiers of the food chain [21].
After consumption (Figure 1), dietary retinol, retinyl esters, and carotenoids are released from the digested food matrix in the gastrointestinal tract. In the intestinal lumen, the brush-border retinyl ester hydrolase hydrolyzes retinyl esters into retinol and free FAs [22,23]. Due to their lipophilic characteristic, these molecules are incorporated into micelles in the intestinal lumen prior to absorption. Carotenoids enter enterocyte (intestinal mucosal cell) via passive diffusion, which neither requires cell membrane transporters nor energy [24,25]. Retinol is thought to be taken up into enterocytes by a brush border membrane transporter via facilitated diffusion [26].
Inside the enterocytes, the majority of β-carotene (major dietary carotenoid) will be symmetrically cleaved by 15,15′-dioxygenase to generate retinal [27]. Residual intact β-carotene can be delivered to the liver, where the same enzyme in hepatocytes catalyzes the cleavage reaction [25]. Aside from the symmetrical cleavage, β-carotene can also be asymmetrically cleaved at the 9′,10′ double bond by a specific enzyme in the enterocyte [28,29]. The resulting retinal from the cleavage of the β-carotene is readily reduced to retinol, which converges into the retinol repertoire from the hydrolysis of retinyl esters [30]. The majority of the retinol in the enterocyte is re-esterified into retinyl ester by lecithin:retinol acyltransferase [31] (LRAT) or acyl-CoA:retinol acyltransferase [32] (ARAT), and then incorporated into chylomicrons for delivery [30].
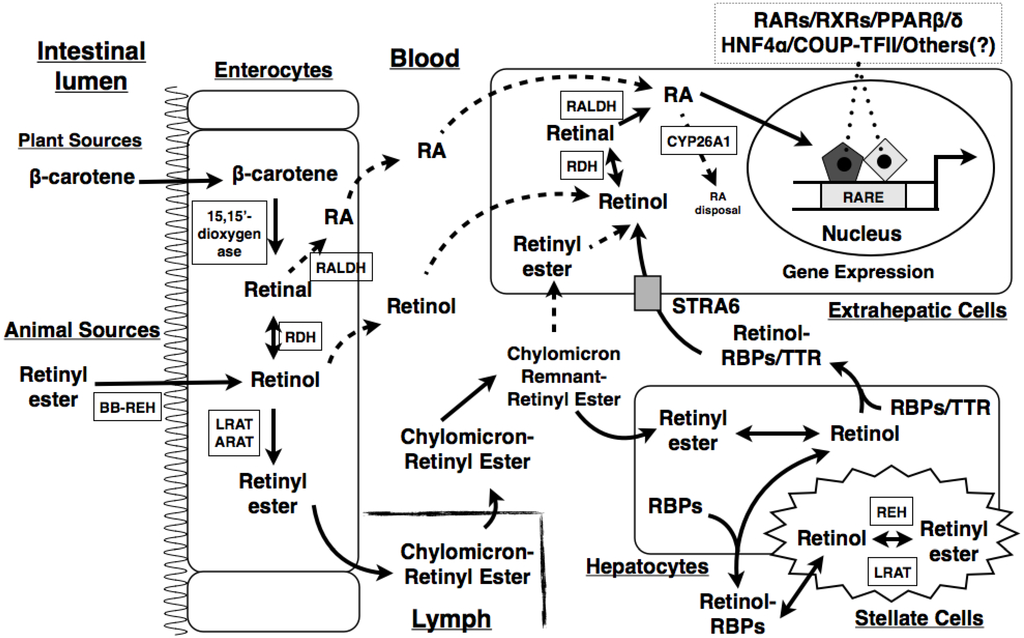
Figure 1.
Schematic representation of vitamin A (VA) digestion, absorption, transport, and metabolism in the body. Dietary VA from plant and animal sources is digested in the intestinal lumen and absorbed by enterocytes via different mechanisms. Within the enterocytes, the dietary VA is converted into retinyl esters, which are packaged into chylomicrons for the secretion into the lymph and eventually enter the circulation. The lipoprotein lipase hydrolyzes triglycerides (TGs) on the chylomicrons to produce chylomicron remnants. The retinyl ester-containing chylomicron remnants are eventually taken up by hepatocytes, where the retinyl esters are again hydrolyzed into retinol. The released retinol can be transported to target cells and catabolized into retinal, retinoic acid (RA), or other metabolites. Excessive retinol is re-esterified into retinyl esters, which are stored in stellate cells. BB-REH, brush-border retinyl ester hydrolase; LRAT, lecithin:retinol acyltransferase; ARAT, acyl-CoA:retinol acyltransferase; RBP, retinol binding protein; RDH, retinol dehydrogenase; RALDH, retinal dehydrogenase; REH, retinol ester hydrolase; STRA6, stimulated by retinoic acid 6; RA, retinoic acid; RARE, retinoic acid responsive element; RAR, retinoic acid receptor; RXR, retinoid X receptor; TTR, transthyretin; PPAR, peroxisome proliferator-activated receptor; HNF4α, hepatocyte nuclear factor 4α; COUP-TFII, chicken ovalbumin up-stream transcription factor II.
In the circulation, retinyl ester-containing chylomicrons interact with the lipoprotein lipase at the inner lining of endothelial cells of the vascular system, which gradually hydrolyzes the TGs on the chylomicrons to free FAs. This process decreases the size of the chylomicrons and converts them into chylomicron remnants. The retinyl ester-containing chylomicron remnants are eventually taken up by hepatocytes, where retinyl esters are again hydrolyzed into retinol. The released retinol can be transported to target cells and catabolized into retinal, retinoic acid (RA), or other metabolites for the different physiological functions. Excessive retinol is re-esterified into retinyl esters, which are stored in stellate cells inside the liver [5]. A combination of retinol and RA is shown to increase retinol uptake and esterification in the lung [33]. A portion of retinyl esters is absorbed by extrahepatic tissues [34].
Retinol derived from the liver is associated with retinol binding protein 4 (RBP4) and transthyretin (TTR) as a complex for the delivery to extrahepatic tissues. The uptake of retinol in the holo-RBP4-TTR complex is supposedly mediated by a receptor on the cell’s membrane. Stimulated by retinoic acid gene 6 (STRA6), a membrane receptor protein, is identified from bovine retinal pigment epithelium cells due to its high affinity to bind to RBPs [35]. Mutations in the human STRA6 gene cause Matthew-Wood Syndrome [36,37]. However, the deletion of the Stra6 gene in mice does not cause any apparent developmental or growth defect [38,39,40]. Interestingly, other than retinal pigment endothelium abnormalities [38,39,40], the levels of retinol and retinyl esters in the plasma and other tissues in Stra6−/− mice are similar to those in wild type control mice [38]. Stra6−/− mice have normal glucose tolerance [38,39]. When Stra6−/− mice were fed a VA deficient diet for 5 weeks, they were shown to have less retinol and retinyl ester levels in the white adipose tissue, kidney, heart, and testis than wild type control mice, indicating that STRA6 is responsible for part of the retinol uptake in these tissues [38]. The deletion of Stra6 in mice reduces insulin resistance mediated by the injection of holo-RBP and feeding of a high-fat diet, suggesting that STRA6 functions as a receptor to mediate RBP-induced insulin resistance [38]. These results indicate that STRA6 is not the only pathway for retinol transport, and other transporters or mechanisms may exist for retinol uptake in cells.
Many of VA’s physiological functions are mediated by RA. The generation and disposal of RA are highly regulated processes involving complex retinoid binding proteins and metabolic enzymes [41,42]. In essence, retinol can be reversibly oxidized into retinal by retinol dehydrogenases (RDHs), and further irreversibly oxidized into RA by retinal dehydrogenases (RALDHs), as reviewed in [41]. In vitro biochemical experiments have so far identified almost a dozen enzymes with retinol oxidation capabilities, but only RDH1, RDH10, and dehydrogenase/reductase member 9 (DHRS9) are verified and supported by in vivo evidence [41]. Comparatively, four RALDHs have been identified, which are expressed and regulated in a tissue specific manner [41]. RALDH1 is universally expressed and accounts for over 90% of retinal oxidation capability in rat liver and kidney [43]. Despite its ubiquitous expression profile, RALDH1 knockout mice do not manifest developmental differences compared to their wild type counterparts [44]. Interestingly, RALDH1 knockout mice are resistant against high-fat diet induced glucose intolerance and obesity [45,46]. In contrast to RALDH1, RALDH2 knockout mice are embryonically lethal. This suggests that RALDH2 is responsible for the local production of RA during embryo development [47]. RALDH3 does not recognize 9-cis retinal as a substrate, whereas RALDH4 only recognizes 9-cis retinal as a substrate [48,49]. The selective recognition of substrates suggests distinctly different functions of the two enzymes in vivo. However, more research is necessary to elucidate the underlying mechanisms [48,49]. The catabolic cytochrome P450 (CYP) family of enzymes partially contributes to RA homeostasis. These enzymes require NADPH and dioxygen to convert RA into a myriad of end products, including 4-hydroxy-RA, 4-oxo-RA, and 18-hydroxy-RA [50]. A representative member of the family, CYP26A1, was cloned and investigated for its role in the regulation of RA homeostasis in animals [51,52,53]. CYP26A1 levels are low in VA deficient animals, but CYP26A1 is substantially induced by RA or a VA sufficient diet [52]. CYP26A1 null mice are embryonically lethal, showing phenotypes similar to wild type mice treated with excessive RA [54]. These data implicate an autoregulatory mechanism by which CYP26A1 controls RA catabolism to maintain RA levels in vivo.
After RA molecule synthesis, they translocate to the nucleus, where they bind to and activate two subfamilies of nuclear receptors known as the retinoid acid receptors (RARs) and retinoid X receptors (RXRs). Nuclear receptors are ligand-activated transcription factors involved in a variety of physiological processes [55]. RARs and RXRs each contain three isoforms that have different tissue and developmental expression profiles [55]. It has been thought that all-trans RA is an exclusive ligand for RARs, whereas 9-cis RA is a common ligand for both RARs and RXRs [56]. Transcription factors mediating RA activity can recognize short consensus sequences, which are termed retinoic acid response elements (RAREs), located in the promoter region of the RA responsive genes. Depending on the specific gene, RXR can bind to RARE as a homodimer or a heterodimer with RAR or other nuclear receptors [19]. As shown in Figure 1, in addition to RARs and RXRs, RA signals have been suggested to be mediated by other nuclear receptors, such as peroxisome proliferator-activated receptor (PPAR) β/δ, hepatocyte nuclear factor 4α, and chicken ovalbumin up-stream transcription factor II [5,57]. The binding of their specific ligands to the nuclear receptors will help recruit corresponding transcriptional coactivators and dissociate transcriptional corepressors in the general transcription machinery, leading to the regulation of RA-targeted gene expression.
3. VA and Growth, Appetite, Taste and Olfaction
VA was first recognized as an essential factor for animal growth. Following the withdrawal of VA from the diet, growth retardation is the earliest and most reproducible sign of deficiency in many experimental animals [58]. Rats fed on a VA deficient diet upon weaning (about three weeks old), manifest cessation of growth and significant weight loss later on [59,60]. The reduction of body mass can be prevented by RA supplementation. However, once RA is removed from those rats fed on the VA deficient diet, the weight loss will reappear in a couple of days [61,62]. These results demonstrate that VA is important for normal growth and development of animals.
In comparison to rats, mice are more resistant to the development of VA deficiency. A normal growth curve is usually observed in weaning mice with sufficient hepatic VA storage, despite being sustained on a VA deficient diet [63,64]. In one study, a significant weight loss was achieved in mice fed on a VA deficient diet only when the VA storage was pre-depleted prior to weaning [63]. This suggests that mice might have special mechanisms to maintain VA homeostasis or they might use VA more efficiently. As a result, experimental conclusions obtained in mice cannot be directly extrapolated to rats or humans in terms of VA and its effect on growth and metabolism.
VA deficient animals exhibit decreased appetite and food intake during the period of growth cessation [59,60]. The underlying molecular mechanisms by which VA regulates energy intake remain elusive. Since leptin, an adipocyte-derived peptide hormone, exerts its action in the brain to regulate appetite, energy expenditure, and metabolism [65], some researchers investigated the effects of VA on the expression of leptin in rodents. The results showed that both RA treatment and VA supplementation were able to decrease leptin mRNA in adipocytes, which, in turn, decreased serum leptin levels [66,67]. Interestingly, the energy intake of these rodents did not change during the course of VA supplementation [68]. These data suggest that the VA-induced decrease in leptin levels does not correlate with food intake in these animals [68].
VA has also been implicated in the normal functions of the taste and olfactory system. VA depletion causes the loss of both preference to sodium chloride and aversion to quinine in rats, and these abnormal responses could be restored by VA supplementation [69]. One study also shows that the response to sweetness in VA deficient rats is impaired [70]. Furthermore, VA supplementation can improve the impairment of taste and olfaction in patients with cirrhosis [71]. Despite these many observations, the mechanism is not clear. Keratin infiltration into the taste buds is proposed to be a mechanism by which VA deficiency affects taste in rats [69]. But the physical change could not be confirmed in all VA deficient rodent models [70].
4. VA and Plasma Parameters: Clinical Evidence
The association between VA and metabolism was discovered from clinical observations in human subjects with type 2 diabetes. Biopsies of the liver of diabetic patients showed that the hepatic VA content was twice as much compared with healthy individuals [72]. Independent studies carried out more than eighty years ago also found that about 85% of adult patients with type 2 diabetes had elevated plasma carotene levels, and more than 10% were clinically diagnosed with xanthosis [73,74]. Similar results were obtained to a lesser degree in type 2 diabetic children, but their plasma VA levels were found to be subnormal [75]. Additionally, reduction of serum retinol and RBP levels was found in type 1 diabetic patients [76]. These data collectively suggest that VA plays important roles in metabolic homeostasis.
Recently, RBP4, a serum retinol transporter, was shown to be elevated in insulin resistant and type 2 diabetic subjects [77,78,79]. Multiple single nucleotide polymorphisms (SNP) of RBP4 were also discovered in the human genome, which predict the susceptibility to insulin resistance, obesity, and type 2 diabetes [80,81]. Despite suggesting a positive association, several clinical studies did not identify significant associations between changes in RBP4 levels and metabolic diseases [82,83,84]. It is worth to note that elevation of plasma RBP4 levels reduced insulin sensitivity in mice, and RBP4 knockout mice had improved insulin sensitivity [85]. When fenretinide, a synthetic retinoid derivative, was used to disrupt the association of RBP4 and TTR, insulin sensitivity in diet-induced obese mice was improved [85]. However, when a specific compound was developed to disrupt the RBP4 and TTR interaction and successfully reduced the plasma RBP4 level, insulin sensitivity in the insulin resistant mice was not improved, suggesting that another mechanism may be responsible for the fenretinide-mediated improvement of insulin sensitivity in mice [86]. Moreover, the improvement of insulin sensitivity in RBP4 knockout mice could not be observed in this study [86]. Further studies are needed to understand the role of VA in insulin resistance.
The association between VA and plasma lipid metabolism was also observed in patients taking retinoid drugs. Medical administration of isotretinoin (13-cis RA) results in hypertriglyceridemia in human subjects with acne [87]. Treatment of patients with acute promyelocytic leukemia with all-trans RA leads to weight gain and elevation of plasma TG and cholesterol levels [88,89]. It is postulated that the dysregulated plasma lipid levels are caused by the RA-induced apolipoprotein CIII expression [90]. Apolipoprotein CIII is regarded as an inhibitor of lipoprotein lipase activity [91]. On the contrary, long-term administration of fenretinide, a synthetic retinoid drug currently in phase II clinical trial evaluation, could prevent diet-induced obesity, insulin resistance, and hepatosteatosis [92,93]. As more and more retinoid drugs are synthesized and tested, it would be interesting to see how they affect human metabolism in any given health condition.
5. VA and Metabolism in the Liver
5.1. VA and Hepatic Carbohydrate Metabolism
The liver plays a leading role in the regulation of carbohydrate metabolism in response to the feeding and fasting statuses. Experiments in rodents show that both hypervitaminosis A and hypovitaminosis A affect carbohydrate metabolism in the liver [4].
Glycogen is a crucial polysaccharide in which intracellular glucose is stored. Hepatic glycogen synthesis and degradation contributes to plasma glucose homeostasis. In VA deficient rats, the liver glycogen content is abolished due to decreased glycogenesis from acetate, lactate, and glycerol, rather than directly from glucose [94,95]. The decreased glycogenesis can be recovered by the administration of glucocorticoid hormones [95]. Since VA deficient animals have lower adrenal steroid production, it is possible that VA deficiency partially affects glycogen metabolism via decreasing glucocorticoid hormone synthesis [95]. In hypervitaminotic A rats, the liver glycogen deposition in the fed state is almost the same or slightly lower than in rats fed on a chow diet [96,97]. However, the hepatic glycogenesis of hypervitaminotic A rats after fasted for 18 to 20 h is significantly higher than in rats fed chow diet [97]. This suggests that excessive VA intake for a short-term can prevent the hepatic glycogenolysis under the fasting condition. Additionally, excessive VA can also enhance the hepatic glycogenesis from glucose after refeeding in normal rats, but not in adrenalectomized rats [98]. This suggests that adrenal hormones are involved in the VA-regulated hepatic glycogen metabolism [98].
Glycolysis is a series of enzymatic reactions, which break down glucose to produce pyruvate and generate ATP and NADH. This important carbohydrate metabolic pathway is influenced by VA status. In VA deficient hamsters, the hepatic generation of glucose-6-phosphate was decreased by up to 90% due to reduced glucokinase (gene Gck) activity [99]. Similarly, the generation of mannose-6-phosphate was also decreased due to impaired hexokinase activity [99]. On the contrary, hypervitaminosis A does not affect the enzymatic activities of hepatic glucokinase or hexokinase [100]. Instead, excessive VA can decrease the enzymatic activities of phosphofructokinase and pyruvate kinase [100]. Additionally, Gck mRNA levels in the liver of VA deficient rats are lower than in VA sufficient counterparts [60]. Retinoids not only increase hepatic Gck expression in rats, but also synergize with insulin to induce Gck expression in primary rat hepatocytes [101]. These observations suggest that VA is an important regulator of hepatic glycolytic enzymes.
Gluconeogenesis is a series of enzymatic reactions, which utilize non-carbohydrate substrates (e.g., lactate, pyruvate, amino acids) to generate glucose. This metabolic pathway is very important in maintaining fasting blood glucose levels in animals. Experiments with VA deficient rats show that the enzymatic activities of glucose-6-phosphatase and fructose-1,6-bisphosphatase were decreased in the liver, showing an inhibition of hepatic gluconeogenesis [102]. Comparably, hypervitaminotic A rats have elevated hepatic enzymatic activities of the cytosolic form of phosphoenolpyruvate carboxykinase (PEPCK, gene Pck1), glucose-6-phosphatase, and fructose-1,6-bisphosphatase [103,104]. In addition, the VA-mediated increase in gluconeogenesis is abolished in adrenalectomized rats [105], showing the requirement of adrenal hormones in the process. The regulation of gluconeogenesis by VA can also occur at the transcription level of Pck1 expression in the liver. RA not only induces Pck1 expression in primary rat hepatocytes, but also attenuates insulin-suppressed Pck1 expression [106]. In fact, insulin can inhibit RA-activated RXRs, but not RARs in the Pck1 promoter. This mechanism underlies the production of Pck1 mRNAs in the presence of RA, despite the insulin-suppressed state [106]. Additionally, a Pck1 transgenic mice model shows that all-trans and 9-cis RAs differentially regulate hepatic Pck1 expression in the periportal region of the liver acinus [107]. This retinoid-regulated Pck1 expression is purportedly mediated by different RAREs and nuclear receptors at its promoter [107,108].
5.2. VA and Hepatic Protein Metabolism
Ever since the increased nitrogenous metabolism was first described in VA deficient rats [109], many animal models were used to investigate the effects of VA on protein metabolism. In young male rats fed on a VA deficient diet, urinary nitrogen excretion is increased with a concurrent negative nitrogen balance [110]. It is in line with the observation of increased plasma urea in VA deficient adult rats [111]. Interestingly, these physiological changes cannot be obtained in VA deficient female rats, indicating a sex difference in the effects of VA on protein metabolism [112]. Additionally, VA deficiency increases both the mRNA levels and enzyme activities of most urea cycle enzymes in the rat liver [111]. These data suggest that VA deficiency increases protein catabolism by upregulating the enzymes of the urea cycle in both the liver and kidney.
The effect of VA deficiency on protein anabolism is controversial. The analysis of the incorporation rate of 14C-leucine into proteins in both VA deficient and chow-fed rats shows that VA deficiency does not adversely affect protein synthesis [113]. However, in vitro protein synthesis assay using isolated rat liver ribosomes show the opposite effect. Cell precipitates from the pH 5.1 fraction of the VA deficient rat liver homogenate exhibit enhanced protein synthesis capability [114]. The reason for these conflicting results is not known.
Very few studies looked at the effects of hypervitaminosis A on protein metabolism in animals. It is proposed in one study that 400 times the normal VA dose causes toxicity-induced weight loss and negative nitrogen balance [115]. However, the molecular mechanism by which the VA toxicity offsets the nitrogen balance in vivo is unknown.
5.3. VA and Hepatic Lipid Metabolism
Dysregulation in the metabolism of either VA or lipids could negatively affect the metabolism of the other. On the one hand, long-term retinoid drug users and people who take excessive amounts of VA supplements exhibit symptoms of hypercholesterolemia, hypertriglyceridemia, and high serum low density lipoprotein levels [116,117,118]. On the other hand, patients with severe type V hyperlipoproteinemia-associated hypertriglyceridemia have an increased risk for developing hypervitaminosis A [119]. The mechanisms underlying these phenomena have been actively investigated in rodent models. Oral administration of large doses of retinol leads to the accumulation of lipid droplets in the rat liver [120]. Overdose of retinol or retinyl palmitate also causes the elevation of hepatic cholesterol, FA, and TG contents in different strains of rats [121,122,123].
It has been shown that hypervitaminotic A rats have increased rates of hepatic FA oxidation and ketogenesis [123]. On the other hand, the hepatic TG synthesis rate is greatly enhanced in hypervitaminotic A rats. This is due to the increased incorporation rate of palmitate into TGs and the formation of glycerophosphate from glucose [122,123]. Interestingly, the rate of hepatic TG secretion is not changed in hypervitaminotic A rats [124]. These data suggest that VA increases hepatic lipid synthesis to an extent much greater than that it elevates hepatic lipid oxidation. The net result is the accumulation of lipids in the liver. A further investigation shows that the hyperlipidemic effect of excessive intake of VA cannot be observed in adrenalectomized rats, showing the involvement of adrenal hormones [122].
Contrary to the hyperlipidemic effect of hypervitaminosis A, VA deficiency causes a partial hypolipidemic effect in rodents. In general, VA deficient rats have decreased hepatic phospholipid content and decreased serum levels of TG, cholesterol, and high density lipoprotein [60,125]. Interestingly, these rats manifest unaltered hepatic contents of TG and cholesterol [123,126,127,128]. Depending on the severity of VA deficiency in the animals, not all of the above-mentioned symptoms can be observed. The partial hypolipidemic effect of VA deficiency in rodents may be caused by a decreased FA synthesis activity in the liver and impaired cholesterol synthesis from mevalonate [125,128]. However, it is worth mentioning that the VA deficiency-induced body mass loss and food intake drop may also contribute to the hypolipidemic symptoms. Recent evidence shows that the decrease in total percentage of body fat is similar between VA deficient rats and their counterparts pair-fed on a VA sufficient diet [129]. In drastic contrast to VA deficient rats, VA deficient mice show TG accumulation in the liver, resulting from reduced expression levels of mitochondrial FA oxidation genes [130]. The altered expression of these genes in the liver may be attributed to the downregulation of the hepatic PPARα gene [130]. Accordingly, RA treatments in mice increase hepatic FA oxidation, which has been reviewed in detail elsewhere [131]. Available data suggest that the dyslipidemia in VA deficient and hypervitaminotic A animals is a multicausal effect. The distinct responses of hepatic lipid metabolism to VA status in mice and rats deserve further investigation.
Hepatic FA synthesis is governed by an allotment of lipogenic genes, including acetyl-CoA carboxylase (ACC, gene Acc) and FA synthase (FAS, gene Fas). The transcription of hepatic Acc and Fas is controlled by a transcriptional factor termed sterol-responsive element binding protein 1c (SREBP-1c, gene Srebp-1c) [132]. In primary rat hepatocytes, insulin induces Srebp-1c expression through transcription factors associated with the liver X receptor elements in the Srebp-1c gene promoter [133]. Interestingly, the corresponding liver X receptor elements in the Srebp-1c promoter are also identified as RAREs [134]. This finding provides a possible mechanism by which RA synergizes with insulin to induce hepatic Srebp-1c expression [134]. It also showcases how nutritional and hormonal factors converge at the transcriptional level to regulate de novo FA synthesis in the liver.
5.4. VA and Mitochondrial Functions in Hepatocytes
Mitochondria are the powerhouses of the cell in which the tricarboxylic acid cycle (TCA cycle) and coupled-oxidative phosphorylation conjointly generate ATP for physiological events. The TCA cycle consists of a series of enzymatic reactions that generate reducing agents (NADH and FADH2) by utilizing acetyl-CoA derived from monosaccharides, amino acids, and FAs. The reducing agents ultimately provide electrons, which are consumed in the coupled oxidative phosphorylation process to reduce O2 and generate ATP.
In the rodent liver, both hypo- and hypervitaminosis A are implicated in the uncoupled oxidative phosphorylation in mitochondria [135,136,137]. On the one hand, the dioxygen consumption by liver homogenates of hypo- and hypervitaminotic A rats is significantly increased upon the supply of some TCA intermediates [135,136]. On the other hand, the capacity for oxidative phosphorylation is severely impaired in liver mitochondria of hypo- and hypervitaminotic A rats without affecting ATPase activity [137]. These data suggest that deficient or excessive VA status may increase basal energy metabolism in the liver.
6. VA and Islets of Langerhans in the Pancreas
Islets of Langerhans, which account for less than 2% of the total pancreas mass, produce and secrete peptide hormones from five different types of specialized cells, including α-, β-, polypeptide (PP-), δ-, and ε-cells [138,139]. Among all the hormones produced by islets, glucagon from α-cells and insulin from β-cells are of great clinical interest due to their concerted roles in the regulation of blood glucose levels. Glucagon promotes hepatic glucose production, glycogenolysis, and ketone production in response to hypoglycemia, whereas insulin promotes glucose disposal, glycogenesis, and lipogenesis in response to the increase of plasma glucose levels [140].
The secretion of glucagon is controlled by the autonomous nervous system, direct action of glucose on α-cells, and indirect effects of paracrine factors from non-α-cells on α-cells [141]. On the other hand, the secretion of insulin is mainly stimulated by the influx of glucose into β-cells. Glucose metabolism in β-cells leads to elevated ATP/ADP ratios, which in turn, inhibit the ATP-sensitive potassium channels on the cell membrane. The inhibition depolarizes the plasma membrane, which results in the secretion of insulin. In addition to glucose, amino acids and neural stimuli can also stimulate insulin secretion [142].
It has been shown that VA and its metabolites can affect the secretion of glucagon. In VA deficient rats, impaired glucagon secretion occurs from early stages of deficiency, and the impairment cannot be rescued by RA replenishment [143]. This demonstrates a critical physiologic role of VA in the normal function of α-cells. Interestingly, in the isolated intact rat islets and glucagon secreting cell lines, both retinol and RA inhibit glucagon secretion in a dose-dependent manner [144]. This result suggests that the acute effects of VA on glucagon secretion in vitro do not correlate with its physiological effects in vivo.
VA deficiency has been shown to decrease β-cell functions in rats in two ways. First, it reduces the β-cell mass in fetal islets by reducing fetal β-cell replication [145]. Second, it impairs glucose-stimulated insulin secretion (GSIS) from β-cells [146]. In comparison, the effects of VA and its metabolites on isolated islets and insulin secreting cells are both chemical- and dosage-dependent. For example, retinol at 10−7 M stimulates GSIS from isolated rat islets, which is in opposition to the inhibitory effect of retinol at 10−4‒10−5 M [147]. Additionally, all-trans RA potentiates GSIS [148,149,150], whereas 9-cis RA inhibits GSIS [151] from rodent islets and INS-1 cells. Cellular retinol-binding protein I knockout mice exhibit increased levels of 9-cis RA in the pancreas and reduced GSIS [152]. It is also observed that all-trans RA increases Gck mRNA levels and induces enzyme activity of glucokinase [150], whereas 9-cis RA treatment is associated with decreased GLUT2 and glucokinase activities in rodent islets [151]. Given the fact that retinoid receptors require preferential ligands to exert their full function, these results suggest that the production and balance of VA and its metabolites are critical for the normal functioning of β-cells.
7. VA and Metabolism in Adipose Tissues
7.1. VA and Metabolism in White Adipose Tissue
White adipose tissue not only stores excessive energy as TGs, but also secretes important adipocyte-derived hormones and cytokines to regulate whole body energy metabolism. VA and its metabolites have determinant effects on the metabolic homeostasis in white adipose tissue.
First, VA status has been shown to affect adiposity in animals. In VA deficient rats, the loss of body fat deposit mass is reflected in the decreased total body mass [110]. Since VA deficient rats have significantly lower body mass compared to their VA sufficient diet pair-fed counterparts, it suggests that the reduction of fat mass in VA deficient rats cannot be fully attributed to the VA deficiency associated with the reduction of food intake [110]. In Zucker lean rats, VA deficiency decreases the epididymal fat mass and its fat mass/body mass ratio [60]. In Zucker fatty rats, a genetic rat model of obesity, VA deficiency retards the development of obesity [60]. Additionally, hypervitaminotic A rats have decreased release of FAs from white adipocytes, whereas VA deficient rats exhibit increased rates of lipolysis in white adipose tissue [153]. Despite the fact that these findings support VA’s role in maintaining adiposity, other studies have reported different observations. Mice on a VA deficient diet for 10 weeks show increased adiposity [154]. More importantly, in humans, low plasma VA status has been associated with overweight and obesity [155,156]. These divergent data warrant further research into the relationship between VA status and adiposity in animals.
Second, it has been reported that RA treatment induces lipolysis and depletes lipid storage in mature white adipocytes, which leads to weight loss in diet-induced obese mice [157,158]. All-trans RA treatment also decreases white adipose tissue mass in healthy lean mice [159]. The anti-obesogenic effect of all-trans RA may be mediated through the activation of PPARβ/δ and RARs, which are important factors in the upregulation of energy dissipation [157,158]. It is also hypothesized that all-trans RA promotes the acquisition of brown adipose tissue-like properties in white adipocytes [159]. Indeed, all-trans RA induces the expression of uncoupling protein 1 (UCP1), a mediator of adipose thermogenesis (Figure 2), in mouse white adipocytes, possibly via the activation of PPARβ/δ and RARs [160,161,162]. However, all-trans RA has no effect, or even inhibits the expression of UCP1 in human white adipocytes [160]. These conflicting results suggest that other retinoids besides RA may regulate energy metabolism in white adipose tissue. Interestingly, Raldh1 deficiency has been shown to induce a brown adipose tissue-like transcriptional program in white adipose tissue [163]. Furthermore, retinal can induce the expression of Ucp1 mRNA and UCP1 protein in white adipose tissue by activating RAR [163]. These data show that both retinal and RALDH1 are regulators of adaptive thermogenesis in white adipose tissue.
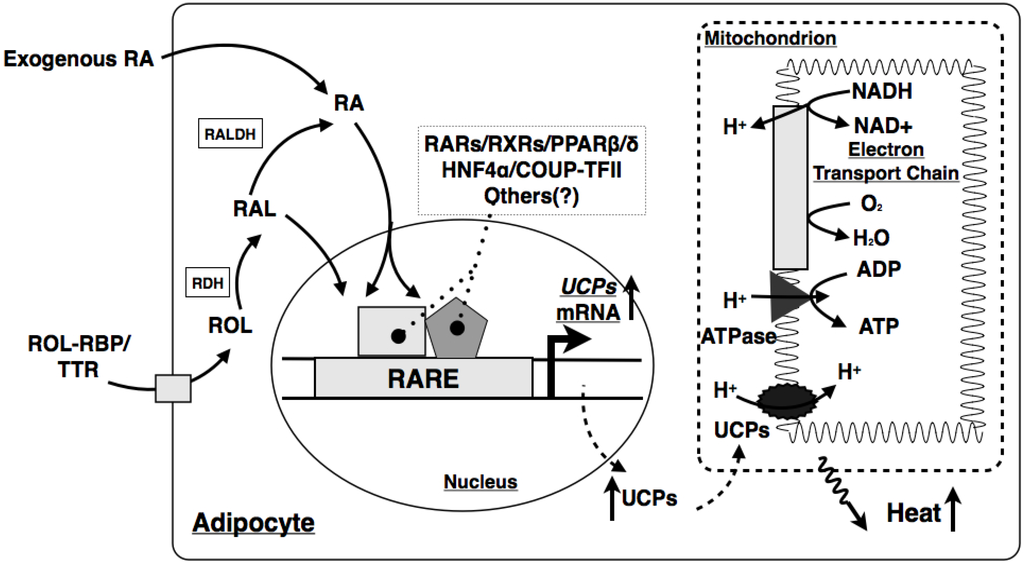
Figure 2.
Mechanism by which RA induces thermogenesis in adipocytes. Stepwise enzymatic reactions convert retinol into retinal, and then retinal into RA in the adipocytes. RA enters the nucleus, where it modulates the expression of UCP genes by activating the corresponding nuclear receptors in the promoters of the genes. Increased UCP proteins uncouple oxidative phosphorylation from ATP production. The reduced H+ gradient across the mitochondrial membranes leads to the increase of thermogenesis in the adipocytes. ROL, retinol; RBP, retinol binding protein; TTR, transthyretin; RAL, retinal; RDH, retinol dehydrogenase; RALDH, retinal dehydrogenase; RA, retinoic acid; RARE, retinoic acid responsive element; RAR, retinoic acid receptor; RXR, retinoid X receptor; UCP, uncoupling protein; PPAR, peroxisome proliferator activated receptor; HNF4α, hepatocyte nuclear factor 4α; COUP-TFII, chicken ovalbumin up-stream transcription factor II.
Third, RA distinctly influences different stages of adipocyte differentiation. In several mouse preadipocyte cell lines and 3T3-L1 cells, RA can block preadipocyte differentiation through inhibiting the induction of PPARγ and CCAAT-enhancer-binding protein α (C/EBPα) [164]. It has been shown that cellular retinoic-acid binding protein II (CRABP II) mediates the effect of RA [165]. Interestingly, the inhibitory effect of RA on differentiation cannot be observed in the late stage of adipocyte differentiation due to reduced RAR expression levels [166]. Additionally, low doses of all-trans RA potentiated the differentiation of Ob17 mouse pre-adipocytes [167].
7.2. VA and Metabolism in Brown Adipose Tissue
Brown adipose tissue is engaged in thermogenesis in mammals [168]. It is derived from a cell lineage with a myogenic gene expression signature, which differs from the origin of white adipose tissue [169]. A zinc finger protein, PRD1-BF-1-RIZ1 homologous domain containing protein 16 (PRDM16), induces the expression of a variety of marker genes, which determines brown adipocyte identity and promotes the differentiation of brown preadipocytes into brown adipocytes [170,171]. On the other hand, UCP1 expression levels can be induced in cultured white adipocytes, while the expression of white adipocyte marker genes are retained [172]. Furthermore, beige adipocytes, which are a distinct type of adipocytes with characteristics between white and brown adipocytes, are identified in mouse and human white adipose tissue [173,174,175]. In the human neck region, the adipocytes are aligned in a gradient fashion with the classical brown adipocytes at the inside, beige/brite adipocytes in the middle, and white adipocytes on the outside, which corresponds to the production of heat and usage of fuels [176].
Feeding a VA deficient diet to mice reduced the expression of UCP genes in their brown adipose tissue [177]. In contrast, retinol supplementation induced the expression of Ucp1 mRNA in rat brown adipocytes [66]. Additionally, in both cultured brown adipocytes and rodent brown adipose tissue, RA treatment stimulated thermogenesis by upregulating the expression of Ucp1 [177,178,179]. This regulation is mediated through the RARE in the promoter of Ucp1 gene [180]. It has been shown that all-trans RA treatment also reduces RARα and RXRα protein levels in mouse brown adipose tissue [181]. Collectively, these data provide an interesting mechanism by which VA and its metabolites regulate brown adipose tissue thermogenesis.
8. VA and Metabolism in the Skeletal Muscle
The skeletal muscle is the largest organ in the body, which plays a critical role in the regulation of energy metabolism [182]. However, very limited research has been performed to investigate the effects of VA on carbohydrate and protein metabolism in the skeletal muscle. In avian species, VA deficiency depletes the glycogen content in the pectoralis major muscle [183]. On the other hand, RA treatment of mouse myoblast C2C12 cells leads to increased glucose uptake, possibly through the activation of AMP-activated protein kinase [184]. In addition, VA deficient rats have lowered protein synthesis and increased proteolysis rates in the skeletal muscle [111,185]. This change in protein metabolism partly contributes to the body mass loss in VA deficient animals. Interestingly, acute VA toxicity accelerates myofibrillar protein breakdown without affecting the rate of protein synthesis, which may also lead to muscle wasting [186]. These data, though sparing, collectively demonstrate the involvement of VA in the regulation of glucose and protein metabolism in the skeletal muscle.
In terms of lipid metabolism, VA deficient rats do not show significant changes in the FA oxidation capacity in the skeletal muscle [123]. In contrast, RA treatment of mouse skeletal muscle dose-dependently increases the transcripts of genes that are involved in FA oxidation and thermogenesis [187]. Indeed, RA treatment and retinyl palmitate supplementation induce the expression of UCP3 in the mouse skeletal muscle [68,157]. Despite the different results observed between rats and mice, these data suggest that VA may increase the energy dissipation in the skeletal muscle.
9. Conclusions
VA is a crucial regulator of carbohydrate, protein, and lipid metabolism in all of the major metabolically active organs. The manifold of VA effects may be attributed to the roles of RA (or other VA metabolites) in regulating the expression of critical genes in different metabolic pathways. A lot of effort and progress has been made in understanding the actions of VA (RA) in different organs. We think that further investigations are warranted in the following areas: (1) the molecular mechanisms by which VA regulates the transcription of critical metabolic genes in different organs and tissues, such as Gck, Pck1, and Srebp-1c; (2) the interplay between VA and other hormones in the regulation of metabolic homeostasis; (3) the roles of VA in the regulation of macronutrient metabolism in various disease states, especially obesity, type 2 diabetes, and other metabolic diseases. The findings in the abovementioned areas will not only help understand the effects of micronutrients on energy metabolism, but also help to develop new pharmaceutical strategies to combat metabolic diseases.
Acknowledgments
Guoxun Chen is supported by research grants from Allen Foundation Inc., start-up fund from the University of Tennessee at Knoxville, and Scientist Development Grant from American Heart Association (09SDG2140003). Wei Chen received financial support from the China Scholarship Council.
Author Contributions
Wei Chen and Guoxun Chen wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. Consultation, Obesity: Preventing and Managing the Global Epidemic. In World Health Organization Technical Report Series; WHO: Geneva, Switzerland, 2000. [Google Scholar]
- WHO. Burden: Mortality, Morbidity and Risk Factors. In Global Status Report on Noncommunicable Diseases 2010—Description of the Global Burden of NCDs, Their Risk Factors and Determinants; World Health Organization: Geneva, Switzerland, 2011; pp. 1–23. [Google Scholar]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Loria, C.M.; Ard, J.D.; Millen, B.E.; Comuzzie, A.G.; Nonas, C.A.; Donato, K.A.; Pi-Sunyer, F.X.; et al. 2013 AHA/ACC/TOS Guideline for the Management of Overweight and Obesity in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J. Am. Coll. Cardiol. 2013. [Google Scholar] [CrossRef]
- Zhao, S.; Li, R.; Li, Y.; Chen, W.; Zhang, Y.; Chen, G. Roles of vitamin A status and retinoids in glucose and fatty acid metabolism. Biochem. Cell Biol. 2012, 90, 142–152. [Google Scholar] [CrossRef]
- Chen, G. Roles of Vitamin A Metabolism in the Development of Hepatic Insulin Resistance. ISRN Hepatol. 2013, 2013, 1–21. [Google Scholar]
- Fall, T.; Ingelsson, E. Genome-wide association studies of obesity and metabolic syndrome. Mol. Cell. Endocrinol. 2014, 382, 740–757. [Google Scholar] [CrossRef]
- Kopelman, P. Symposium 1: Overnutrition: Consequences and solutions. Foresight Report: The obesity challenge ahead. Proc. Nutr. Soc. 2010, 69, 80–85. [Google Scholar] [CrossRef]
- Key, T.J.; Spencer, E.A.; Reeves, G.K. Symposium 1: Overnutrition: Consequences and solutions. Obesity and cancer risk. Proc. Nutr. Soc. 2010, 69, 86–90. [Google Scholar] [CrossRef]
- White, M.F. Insulin signaling in health and disease. Science 2003, 302, 1710–1711. [Google Scholar] [CrossRef]
- Kasuga, M.; Karlsson, F.A.; Kahn, C.R. Insulin stimulates the phosphorylation of the 95,000-dalton subunit of its own receptor. Science 1982, 215, 185–187. [Google Scholar]
- Petruzzelli, L.M.; Ganguly, S.; Smith, C.J.; Cobb, M.H.; Rubin, C.S.; Rosen, O.M. Insulin activates a tyrosine-specific protein kinase in extracts of 3T3-L1 adipocytes and human placenta. Proc. Natl. Acad. Sci. USA 1982, 79, 6792–6796. [Google Scholar] [CrossRef]
- McKern, N.M.; Lawrence, M.C.; Streltsov, V.A.; Lou, M.-Z.; Adams, T.E.; Lovrecz, G.O.; Elleman, T.C.; Richards, K.M.; Bentley, J.D.; Pilling, P.A.; et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 2006, 443, 218–221. [Google Scholar] [CrossRef]
- Cohen, P. The twentieth century struggle to decipher insulin signalling. Nat. Rev. Mol. Cell Biol. 2006, 7, 867–873. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef]
- Johnson, A.M.F.; Olefsky, J.M. The Origins and Drivers of Insulin Resistance. Cell 2013, 152, 673–684. [Google Scholar] [CrossRef]
- IARC. IARC Handbooks of Cancer Prevention; IARC Scientific Publications: Lyon, France, 1999. [Google Scholar]
- Chambon, P. The retinoid signaling pathway: Molecular and genetic analyses. Semin. Cell Biol. 1994, 5, 115–125. [Google Scholar] [CrossRef]
- Lefebvre, P.; Martin, P.J.; Flajollet, S.; Dedieu, S.; Billaut, X.; Lefebvre, B. Transcriptional activities of retinoic acid receptors. Vitam. Horm. 2005, 70, 199–264. [Google Scholar] [CrossRef]
- Moore, T. Vitamin A and carotene: The absence of the liver oil vitamin A from carotene. VI. The conversion of carotene to vitamin A in vivo. Biochem. J. 1930, 24, 692–702. [Google Scholar]
- Moore, T. Vitamin A; Elsevier Publishing Company: Amsterdam, The Netherlands, 1957. [Google Scholar]
- Rigtrup, K.M.; Ong, D.E. A retinyl ester hydrolase activity intrinsic to the brush border membrane of rat small intestine. Biochemistry 1992, 31, 2920–2926. [Google Scholar] [CrossRef]
- Rigtrup, K.M.; Kakkad, B.; Ong, D.E. Purification and partial characterization of a retinyl ester hydrolase from the brush border of rat small intestine mucosa: Probable identity with brush border phospholipase B. Biochemistry 1994, 33, 2661–2666. [Google Scholar] [CrossRef]
- Erdman, J.W.; Bierer, T.L.; Gugger, E.T. Absorption and transport of carotenoids. Ann. N. Y. Acad. Sci. 1993, 691, 76–85. [Google Scholar] [CrossRef]
- Parker, R.S. Absorption, metabolism, and transport of carotenoids. FASEB J. 1996, 10, 542–551. [Google Scholar]
- Dew, S.E.; Ong, D.E. Specificity of the retinol transporter of the rat small intestine brush border. Biochemistry 1994, 33, 12340–12345. [Google Scholar] [CrossRef]
- Goodman, D.S.; Huang, A.S. Biosynthesis of vitamin A with rat intestinal enzymes. Science 1965, 149, 879–880. [Google Scholar]
- Wang, X.D.; Russell, R.M.; Liu, C.; Stickel, F.; Smith, D.E.; Krinsky, N.I. β-Oxidation in rabbit liver in vitro and in the perfused ferret liver contributes to retinoic acid biosynthesis from β-apocarotenoic acids. J. Biol. Chem. 1996, 271, 26490–26498. [Google Scholar] [CrossRef]
- Kiefer, C.; Hessel, S.; Lampert, J.M.; Vogt, K.; Lederer, M.O.; Breithaupt, D.E.; Lintig, J. Identification and characterization of a mammalian enzyme catalyzing the asymmetric oxidative cleavage of provitamin A. J. Biol. Chem. 2001, 276, 14110–14116. [Google Scholar]
- Harrison, E.H. Mechanisms of digestion and absorption of dietary vitamin A. Annu. Rev. Nutr. 2005, 25, 87–103. [Google Scholar] [CrossRef]
- MacDonald, P.N.; Ong, D.E. Evidence for a lecithin-retinol acyltransferase activity in the rat small intestine. J. Biol. Chem. 1988, 263, 12478–12482. [Google Scholar]
- Helgerud, P.; Petersen, L.B.; Norum, K.R. Acyl CoA: Retinol acyltransferase in rat small intestine: Its activity and some properties of the enzymic reaction. J. Lipid Res. 1982, 23, 609–618. [Google Scholar]
- Ross, A.C.; Li, N.-Q. Retinol combined with retinoic acid increases retinol uptake and esterification in the lungs of young adult rats when delivered by the intramuscular as well as oral routes. J. Nutr. 2007, 137, 2371–2376. [Google Scholar]
- Blomhoff, R.; Green, M.H.; Berg, T.; Norum, K.R. Transport and storage of vitamin A. Science 1990, 250, 399–404. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef]
- Chassaing, N.; Golzio, C.; Odent, S.; Lequeux, L.; Vigouroux, A.; Martinovic-Bouriel, J.; Tiziano, F.D.; Masini, L.; Piro, F.; Maragliano, G.; et al. Phenotypic spectrum of STRA6 mutations: From Matthew-Wood syndrome to non-lethal anophthalmia. Hum. Mutat. 2009, 30, E673–E681. [Google Scholar] [CrossRef]
- Golzio, C.; Martinovic-Bouriel, J.; Thomas, S.; Mougou-Zrelli, S.; Grattagliano-Bessieres, B.; Bonniere, M.; Delahaye, S.; Munnich, A.; Encha-Razavi, F.; Lyonnet, S.; et al. Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6. Am. J. Hum. Genet. 2007, 80, 1179–1187. [Google Scholar] [CrossRef]
- Berry, D.C.; Jacobs, H.; Marwarha, G.; Gely-Pernot, A.; O’Byrne, S.M.; DeSantis, D.; Klopfenstein, M.; Feret, B.; Dennefeld, C.; Blaner, W.S.; et al. The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J. Biol. Chem. 2013, 288, 24528–24539. [Google Scholar] [CrossRef]
- Ruiz, A.; Mark, M.; Jacobs, H.; Klopfenstein, M.; Hu, J.; Lloyd, M.; Habib, S.; Tosha, C.; Radu, R.A.; Ghyselinck, N.B.; et al. Retinoid content, visual responses, and ocular morphology are compromised in the retinas of mice lacking the retinol-binding protein receptor, STRA6. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3027–3039. [Google Scholar] [CrossRef]
- Terra, R.; Wang, X.; Hu, Y.; Charpentier, T.; Lamarre, A.; Zhong, M.; Sun, H.; Mao, J.; Qi, S.; Luo, H.; et al. To investigate the necessity of STRA6 upregulation in T cells during T cell immune responses. PLoS One 2013, 8, e82808. [Google Scholar] [CrossRef]
- Napoli, J.L. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta 2012, 1821, 152–167. [Google Scholar]
- Wolf, G. Retinoic acid homeostasis: Retinoic acid regulates liver retinol esterification as well as its own catabolic oxidation in liver. Nutr. Rev. 2001, 59, 391–394. [Google Scholar] [CrossRef]
- Akawi, Z.; Napoli, J.L. Rat liver cytosolic retinal dehydrogenase: Comparison of 13-cis-, 9-cis-, and all-trans-retinal as substrates and effects of cellular retinoid-binding proteins and retinoic acid on activity. Biochemistry 1994, 33, 1938–1943. [Google Scholar] [CrossRef]
- Matt, N.; Dupé, V.; Garnier, J.-M.; Dennefeld, C.; Chambon, P.; Mark, M.; Ghyselinck, N.B. Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 2005, 132, 4789–4800. [Google Scholar] [CrossRef]
- Ziouzenkova, O.; Orasanu, G.; Sharlach, M.; Akiyama, T.E.; Berger, J.P.; Viereck, J.; Hamilton, J.A.; Tang, G.; Dolnikowski, G.G.; Vogel, S.; et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat. Med. 2007, 13, 695–702. [Google Scholar] [CrossRef]
- Reichert, B.; Yasmeen, R.; Jeyakumar, S.M.; Yang, F.; Thomou, T.; Alder, H.; Duester, G.; Maiseyeu, A.; Mihai, G.; Harrison, E.H.; et al. Concerted action of aldehyde dehydrogenases influences depot-specific fat formation. Mol. Endocrinol. 2011, 25, 799–809. [Google Scholar] [CrossRef]
- Zhai, Y.; Sperkova, Z.; Napoli, J.L. Cellular expression of retinal dehydrogenase types 1 and 2: Effects of vitamin A status on testis mRNA. J. Cell. Physiol. 2001, 186, 220–232. [Google Scholar] [CrossRef]
- Lin, M.; Zhang, M.; Abraham, M.; Smith, S.M.; Napoli, J.L. Mouse retinal dehydrogenase 4 (RALDH4), molecular cloning, cellular expression, and activity in 9-cis-retinoic acid biosynthesis in intact cells. J. Biol. Chem. 2003, 278, 9856–9861. [Google Scholar] [CrossRef]
- Sima, A.; Parisotto, M.; Mader, S.; Bhat, P.V. Kinetic characterization of recombinant mouse retinal dehydrogenase types 3 and 4 for retinal substrates. Biochim. Biophys. Acta 2009, 1790, 1660–1664. [Google Scholar]
- Roberts, A.B.; Nichols, M.D.; Newton, D.L.; Sporn, M.B. In vitro metabolism of retinoic acid in hamster intestine and liver. J. Biol. Chem. 1979, 254, 6296–6302. [Google Scholar]
- Fujii, H.; Sato, T.; Kaneko, S.; Gotoh, O.; Fujii-Kuriyama, Y.; Osawa, K.; Kato, S.; Hamada, H. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. EMBO J. 1997, 16, 4163–4173. [Google Scholar]
- Wang, Y.; Zolfaghari, R.; Ross, A.C. Cloning of rat cytochrome P450RAI (CYP26) cDNA and regulation of its gene expression by all-trans-retinoic acid in vivo. Arch. Biochem. Biophys. 2002, 401, 235–243. [Google Scholar] [CrossRef]
- Ray, W.J.; Bain, G.; Yao, M.; Gottlieb, D.I. CYP26, a novel mammalian cytochrome P450, is induced by retinoic acid and defines a new family. J. Biol. Chem. 1997, 272, 18702–18708. [Google Scholar]
- Abu-Abed, S.; Dollé, P.; Metzger, D.; Beckett, B.; Chambon, P.; Petkovich, M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001, 15, 226–240. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Dawson, M.I.; Hobbs, P.D.; Husmann, M.; Pfahl, M. Identification of retinoids with nuclear receptor subtype-selective activities. Cancer Res. 1991, 51, 4804–4809. [Google Scholar]
- Berry, D.C.; Noy, N. Is PPAR β/δ a Retinoid Receptor? PPAR Res. 2007, 2007. [Google Scholar] [CrossRef]
- Mayer, J.; Krehl, W.A. Influence of vitamin A deficiency on the gross efficiency of growth of rats. Yale J. Biol. Med. 1948, 20, 403–405. [Google Scholar]
- Sampson, M.M.; Korenchevsky, V. The influence of vitamin A deficiency on male rats in paired feeding experiments. Biochem. J. 1932, 26, 1322–1339. [Google Scholar]
- Zhang, Y.; Li, R.; Li, Y.; Chen, W.; Zhao, S.; Chen, G. Vitamin A status affects obesity development and hepatic expression of key genes for fuel metabolism in Zucker fatty rats. Biochem. Cell Biol. 2012, 90, 548–557. [Google Scholar] [CrossRef]
- Rogers, W.E.; Bieri, J.G. Vitamin A deficiency in the rat prior to weaning. Proc. Soc. Exp. Biol. Med. 1969, 132, 622–624. [Google Scholar] [CrossRef]
- Anzano, M.A.; Lamb, A.J.; Olson, J.A. Growth, appetite, sequence of pathological signs and survival following the induction of rapid, synchronous vitamin A deficiency in the rat. J. Nutr. 1979, 109, 1419–1431. [Google Scholar]
- McCarthy, P.T.; Cerecedo, L.R. Vitamin A deficiency in the mouse. J. Nutr. 1952, 46, 361–376. [Google Scholar]
- Sagazio, A.; Piantedosi, R.; Alba, M.; Blaner, W.S.; Salvatori, R. Vitamin A deficiency does not influence longitudinal growth in mice. Nutrition 2007, 23, 483–488. [Google Scholar] [CrossRef]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Kumar, M.V.; Sunvold, G.D.; Scarpace, P.J. Dietary vitamin A supplementation in rats: Suppression of leptin and induction of UCP1 mRNA. J. Lipid Res. 1999, 40, 824–829. [Google Scholar]
- Hollung, K.; Rise, C.P.; Drevon, C.A.; Reseland, J.E. Tissue-specific regulation of leptin expression and secretion by all-trans retinoic acid. J. Cell. Biochem. 2004, 92, 307–315. [Google Scholar] [CrossRef]
- Felipe, F.; Mercader, J.; Ribot, J.; Palou, A.; Bonet, M.L. Effects of retinoic acid administration and dietary vitamin A supplementation on leptin expression in mice: Lack of correlation with changes of adipose tissue mass and food intake. Biochim. Biophys. Acta 2005, 1740, 258–265. [Google Scholar]
- Bernard, R.A.; Halpern, B.P. Taste changes in vitamin A deficiency. J. Gen. Physiol. 1968, 52, 444–464. [Google Scholar] [CrossRef]
- Reifen, R.; Agami, O.; Weiser, H.; Biesalski, H.; Naim, M. Impaired responses to sweet taste in vitamin A-deficient rats. Metab. Clin. Exp. 1998, 47, 1–2. [Google Scholar]
- Garrett-Laster, M.; Russell, R.M.; Jacques, P.F. Impairment of taste and olfaction in patients with cirrhosis: The role of vitamin A. Hum. Nutr. Clin. Nutr. 1984, 38, 203–214. [Google Scholar]
- Moore, T. Vitamin A and carotene: The vitamin A reserve of the adult human being in health and disease. Biochem. J. 1937, 31, 155–164. [Google Scholar]
- Boeck, W.C.; Yater, W.M. Xanthemia and xanthosis (carotinemia): Clinical study. J. Lab. Clin. Med. 1929, 14, 1129–1143. [Google Scholar]
- Rabinowitch, I.M. Carotinemia and diabetes: II. The relationship between the sugar, cholesterol and carotin contents of blood plasma. Arch. Intern. Med. 1930, 45, 586–592. [Google Scholar] [CrossRef]
- Mosenthal, H.O.; Loughlin, W.C. Vitamins A, B and C in diabetic children. Arch. Intern. Med. 1944, 73, 391–396. [Google Scholar] [CrossRef]
- Basu, T.K.; Tze, W.J.; Leichter, J. Serum vitamin A and retinol-binding protein in patients with insulin-dependent diabetes mellitus. Am. J. Clin. Nutr. 1989, 50, 329–331. [Google Scholar]
- Graham, T.E.; Yang, Q.; Blüher, M.; Hammarstedt, A.; Ciaraldi, T.P.; Henry, R.R.; Wason, C.J.; Oberbach, A.; Jansson, P.-A.; Smith, U.; et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med. 2006, 354, 2552–2563. [Google Scholar] [CrossRef]
- Cho, Y.M.; Youn, B.-S.; Lee, H.; Lee, N.; Min, S.-S.; Kwak, S.H.; Lee, H.K.; Park, K.S. Plasma retinol-binding protein-4 concentrations are elevated in human subjects with impaired glucose tolerance and type 2 diabetes. Diabetes Care 2006, 29, 2457–2461. [Google Scholar]
- Wolf, G. Serum retinol-binding protein: A link between obesity, insulin resistance, and type 2 diabetes. Nutr. Rev. 2007, 65, 251–256. [Google Scholar] [CrossRef]
- Craig, R.L.; Chu, W.S.; Elbein, S.C. Retinol binding protein 4 as a candidate gene for type 2 diabetes and prediabetic intermediate traits. Mol. Genet. Metab. 2007, 90, 338–344. [Google Scholar] [CrossRef]
- Kovacs, P.; Geyer, M.; Berndt, J.; Klöting, N.; Graham, T.E.; Böttcher, Y.; Enigk, B.; Tönjes, A.; Schleinitz, D.; Schön, M.R.; et al. Effects of genetic variation in the human retinol binding protein-4 gene (RBP4) on insulin resistance and fat depot-specific mRNA expression. Diabetes 2007, 56, 3095–3100. [Google Scholar] [CrossRef]
- Janke, J.; Engeli, S.; Boschmann, M.; Adams, F.; Böhnke, J.; Luft, F.C.; Sharma, A.M.; Jordan, J. Retinol-binding protein 4 in human obesity. Diabetes 2006, 55, 2805–2810. [Google Scholar] [CrossRef]
- Ulgen, F.; Herder, C.; Kühn, M.C.; Willenberg, H.S.; Schott, M.; Scherbaum, W.A.; Schinner, S. Association of serum levels of retinol-binding protein 4 with male sex but not with insulin resistance in obese patients. Arch. Physiol. Biochem. 2010, 116, 57–62. [Google Scholar] [CrossRef]
- Kotnik, P.; Fischer-Posovszky, P.; Wabitsch, M. RBP4: A controversial adipokine. Eur. J. Endocrinol. 2011, 165, 703–711. [Google Scholar] [CrossRef]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef]
- Motani, A.; Wang, Z.; Conn, M.; Siegler, K.; Zhang, Y.; Liu, Q.; Johnstone, S.; Xu, H.; Thibault, S.; Wang, Y.; et al. Identification and characterization of a non-retinoid ligand for retinol-binding protein 4 which lowers serum retinol-binding protein 4 levels in vivo. J. Biol. Chem. 2009, 284, 7673–7680. [Google Scholar] [CrossRef]
- Bershad, S.; Rubinstein, A.; Paterniti, J.R.; Le, N.A.; Poliak, S.C.; Heller, B.; Ginsberg, H.N.; Fleischmajer, R.; Brown, W.V. Changes in plasma lipids and lipoproteins during isotretinoin therapy for acne. N. Engl. J. Med. 1985, 313, 981–985. [Google Scholar] [CrossRef]
- Tallman, M.S.; Kwaan, H.C. Reassessing the hemostatic disorder associated with acute promyelocytic leukemia. Blood 1992, 79, 543–553. [Google Scholar]
- Miller, V.A.; Rigas, J.R.; Muindi, J.R.; Tong, W.P.; Venkatraman, E.; Kris, M.G.; Warrell, R.P. Modulation of all-trans retinoic acid pharmacokinetics by liarozole. Cancer Chemother. Pharmacol. 1994, 34, 522–526. [Google Scholar] [CrossRef]
- Vu-Dac, N.; Gervois, P.; Torra, I.P.; Fruchart, J.C.; Kosykh, V.; Kooistra, T.; Princen, H.M.; Dallongeville, J.; Staels, B. Retinoids increase human apo C-III expression at the transcriptional level via the retinoid X receptor. Contribution to the hypertriglyceridemic action of retinoids. J. Clin. Investig. 1998, 102, 625–632. [Google Scholar] [CrossRef]
- Shachter, N.S. Apolipoproteins C-I and C-III as important modulators of lipoprotein metabolism. Curr. Opin. Lipidol. 2001, 12, 297–304. [Google Scholar] [CrossRef]
- Preitner, F.; Mody, N.; Graham, T.E.; Peroni, O.D.; Kahn, B.B. Long-term Fenretinide treatment prevents high-fat diet-induced obesity, insulin resistance, and hepatic steatosis. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1420–E1429. [Google Scholar] [CrossRef]
- Mcilroy, G.D.; Delibegovic, M.; Owen, C.; Stoney, P.N.; Shearer, K.D.; McCaffery, P.J.; Mody, N. Fenretinide treatment prevents diet-induced obesity in association with major alterations in retinoid homeostatic gene expression in adipose, liver, and hypothalamus. Diabetes 2013, 62, 825–836. [Google Scholar] [CrossRef]
- Wolf, G.; Lane, M.D.; Johnson, B.C. Studies on the function of vitamin A in metabolism. J. Biol. Chem. 1957, 225, 995–1008. [Google Scholar]
- Johnson, B.C.; Wolf, G. The function of vitamin A in carbohydrate metabolism. Its role in adrenocorticoid production. Vitam. Horm. 1960, 18, 457–483. [Google Scholar] [CrossRef]
- Ray, A.; Sadhu, D.P. Carbohydrate metabolism in hypervitaminosis A. Nature 1959, 184. [Google Scholar] [CrossRef]
- Singh, M.; Singh, V.N.; Venkitasubramanian, T.A. Early effects of feeding excess vitamin A: Hepatic glycogen, blood lactic acid, plasma NEFA and glucose tolerance in rats. Life Sci. 1968, 7, 239–247. [Google Scholar] [CrossRef]
- Singh, M.; Singh, V.N.; Venkitasubramanian, T.A. Early effects of excessive retinol intake on hepatic glycogen metabolism. Arch. Biochem. Biophys. 1976, 173, 93–99. [Google Scholar] [CrossRef]
- Shankar, S.; Creek, K.E.; de Luca, L.M. The effect of the progression of vitamin A deficiency on glucose, galactose and mannose incorporation into sugar phosphates and sugar nucleotides in hamster liver. J. Nutr. 1990, 120, 361–374. [Google Scholar]
- Singh, V.N.; Singh, M.; Dileepan, K.N. Early effects of vitamin A toxicity on hepatic glycolysis in rat. J. Nutr. 1978, 108, 1959–1962. [Google Scholar]
- Chen, G.; Zhang, Y.; Lu, D.; Li, N.-Q.; Ross, A.C. Retinoids synergize with insulin to induce hepatic Gck expression. Biochem. J. 2009, 419, 645–653. [Google Scholar]
- Dileepan, K.N.; Singh, V.N.; Ramachandran, C.K. Decreased hepatic gluconeogenesis in vitamin A-deficient rats. Proc. Soc. Exp. Biol. Med. 1981, 167, 248–253. [Google Scholar]
- Singh, M.; Ningh, V.N.; Venkitasubramanian, T.A. Role of adrenals in the vitamin A-mediated increase in the activities of gluconeogenic enzymes of rat liver. Life Sci. 1975, 17, 859–865. [Google Scholar] [CrossRef]
- Dileepan, K.N.; Singh, V.N.; Ramachandran, C.K. Early effects of hypervitaminosis A on gluconeogenic activity and amino acid metabolizing enzymes of rat liver. J. Nutr. 1977, 107, 1809–1815. [Google Scholar]
- Singh, M.; Singh, V.N.; Venkitasubramanian, T.A. Early effects of excessive retinol intake on gluconeogenesis. Involvement of adrenals in the increased activities on Gluconeogenic Enzymes of rat. Arch. Biochem. Biophys. 1976, 173, 82–92. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, R.; Chen, W.; Li, Y.; Chen, G. Retinoids induced Pck1 expression and attenuated insulin-mediated suppression of its expression via activation of retinoic acid receptor in primary rat hepatocytes. Mol. Cell. Biochem. 2011, 355, 1–8. [Google Scholar] [CrossRef]
- Shin, D.-J.; Odom, D.P.; Scribner, K.B.; Ghoshal, S.; McGrane, M.M. Retinoid regulation of the phosphoenolpyruvate carboxykinase gene in liver. Mol. Cell. Endocrinol. 2002, 195, 39–54. [Google Scholar] [CrossRef]
- Scribner, K.B.; Odom, D.P.; McGrane, M.M. Nuclear receptor binding to the retinoic acid response elements of the phosphoenolpyruvate carboxykinase gene in vivo: Effects of vitamin A deficiency. J. Nutr. Biochem. 2007, 18, 206–214. [Google Scholar] [CrossRef]
- Sampson, M.M.; Dennison, M.; Korenchevsky, V. The absorption of nitrogen and of fat from the alimentary canal of rats kept on a vitamin A-deficient diet. Biochem. J. 1932, 26, 1315–1321. [Google Scholar]
- Brown, E.F.; Morgan, A.F. The effect of vitamin A deficiency upon the nitrogen metabolism of the rat. J. Nutr. 1948, 35, 425–438. [Google Scholar]
- Esteban-Pretel, G.; Marín, M.P.; Cabezuelo, F.; Moreno, V.; Renau-Piqueras, J.; Timoneda, J.; Barber, T. Vitamin A deficiency increases protein catabolism and induces urea cycle enzymes in rats. J. Nutr. 2010, 140, 792–798. [Google Scholar] [CrossRef]
- John, A.; Sivakumar, B. Effect of vitamin A deficiency on nitrogen balance and hepatic urea cycle enzymes and intermediates in rats. J. Nutr. 1989, 119, 29–35. [Google Scholar]
- Rao, B.S.N. Effect of vitamin A deficiency on the incorporation of 14C-Leucine into tissue proteins of rats. Nature 1966, 210, 306–307. [Google Scholar]
- Tryfiates, G.P.; Krause, R.F. Effect of vitamin A deficiency on the protein synthetic activity of rat liver ribosomes. Proc. Soc. Exp. Biol. Med. 1971, 136, 946–948. [Google Scholar] [CrossRef]
- Nerurkar, M.K.; Sahasrabudhe, M.B. Metabolism of calcium, phosphorus and nitrogen in hypervitaminosis A in young rats. Biochem. J. 1956, 63, 344–349. [Google Scholar]
- Gerber, L.E.; Erdman, J.W. Changes in lipid metabolism during retinoid administration. J. Am. Acad. Dermatol. 1982, 6, 664–674. [Google Scholar] [CrossRef]
- Ellis, C.N.; Kang, S.; Vinik, A.I.; Grekin, R.C.; Cunningham, W.J.; Voorhees, J.J. Glucose and insulin responses are improved in patients with psoriasis during therapy with etretinate. Arch. Dermatol. 1987, 123, 471–475. [Google Scholar] [CrossRef]
- Yehya, A.; Baer, J.T.; Smiley, W.; Dollar, A.; Sperling, L. Hypervitaminosis A altering the lipid profile in a hypercholesterolemic patient. J. Clin. Lipidol. 2009, 3, 205–207. [Google Scholar] [CrossRef]
- Ellis, J.K.; Russell, R.M.; Makrauer, F.L.; Schaefer, E.J. Increased risk for vitamin A toxicity in severe hypertriglyceridemia. Ann. Intern. Med. 1986, 105, 877–879. [Google Scholar] [CrossRef]
- Lettinga, K.D.; Gutter, W.; van Noorden, C.J.; Schellens, J.P.; Frederiks, W.M. Early effects of high doses of retinol (vitamin A) on the in situ cellular metabolism in rat liver. Liver 1996, 16, 1–11. [Google Scholar] [CrossRef]
- Misra, U.K. Hypervitaminosis A and tissue fatty acids. Can. J. Biochem. 1965, 43, 1885–1886. [Google Scholar] [CrossRef]
- Singh, V.N.; Singh, M.; Venkitasubramanian, T.A. Early effects of feeding excess vitamin A: Mechanism of fatty liver production in rats. J. Lipid Res. 1969, 10, 395–401. [Google Scholar]
- Ramachandran, C.K.; Dileepan, K.N.; Singh, V.; Venkitasubramanian, T.A. Effect of excess and deficiency of vitamin A on the utilization of FFA by liver and skeletal muscle. Environ. Physiol. Biochem. 1975, 5, 208–214. [Google Scholar]
- Singh, M.; Singh, V.N. Fatty liver in hypervitaminosis A: Synthesis and release of hepatic triglycerides. Am. J. Physiol. 1978, 234, E511–E514. [Google Scholar]
- Oliveros, L.B.; Domeniconi, M.A.; Vega, V.A.; Gatica, L.V.; Brigada, A.M.; Gimenez, M.S. Vitamin A deficiency modifies lipid metabolism in rat liver. Br. J. Nutr. 2007, 97, 263–272. [Google Scholar] [CrossRef]
- Khanna, A.; Reddy, T.S. Effect of undernutrition and vitamin A deficiency on the phospholipid composition of rat tissues at 21 days of age—I. Liver, spleen and kidney. Int. J. Vitam. Nutr. Res. 1983, 53, 3–8. [Google Scholar]
- Green, B.; Lowe, J.S.; Morton, R.A. The effect of vitamin A deficiency on the cholesterol levels of the plasma and liver of the rat. Biochem. J. 1955, 61, 447–453. [Google Scholar]
- Wiss, O.; Wiss, V. Alterations of the lipid metabolism of rat liver as early symptoms of vitamin A deficiency. Int. J. Vitam. Nutr. Res. 1980, 50, 233–237. [Google Scholar]
- Green, H.N. Fat metabolism in vitamin A deficiency: The utilisation of fat and the desaturation of fat in the liver. Biochem. J. 1934, 28, 25–30. [Google Scholar]
- Kang, H.W.; Bhimidi, G.R.; Odom, D.P.; Brun, P.-J.; Fernandez, M.-L.; McGrane, M.M. Altered lipid catabolism in the vitamin: A deficient liver. Mol. Cell. Endocrinol. 2007, 271, 18–27. [Google Scholar] [CrossRef]
- Bonet, M.L.; Ribot, J.; Palou, A. Lipid metabolism in mammalian tissues and its control by retinoic acid. Biochim. Biophys. Acta 2012, 1821, 177–189. [Google Scholar]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Chen, G.; Liang, G.; Ou, J.; Goldstein, J.L.; Brown, M.S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 11245–11250. [Google Scholar] [CrossRef]
- Li, R.; Chen, W.; Li, Y.; Zhang, Y.; Chen, G. Retinoids synergized with insulin to induce Srebp-1c expression and activated its promoter via the two liver X receptor binding sites that mediate insulin action. Biochem. Biophys. Res. Commun. 2011, 406, 268–272. [Google Scholar] [CrossRef]
- Deluca, H.F.; Manatt, M.R.; Madsen, N.; Olson, E.B. Action of vitamin A on liver homogenate oxidation of tricarboxylic acid cycle intermediates. J. Nutr. 1963, 81, 383–386. [Google Scholar]
- Seward, C.R.; Vaughan, G.; Hove, E.L. Respiratory activities of hypo- and hypervitaminotic A rat liver homogenates. Proc. Soc. Exp. Biol. Med. 1964, 117, 477–480. [Google Scholar] [CrossRef]
- Seward, C.R.; Vaughan, G.; Hove, E.L. Effect of vitamin A deficiency or excess on the oxidative phosphorylation by rat liver mitochondria. J. Biol. Chem. 1966, 241, 1229–1232. [Google Scholar]
- Brissova, M.; Fowler, M.J.; Nicholson, W.E.; Chu, A.; Hirshberg, B.; Harlan, D.M.; Powers, A.C. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J. Histochem. Cytochem. 2005, 53, 1087–1097. [Google Scholar] [CrossRef]
- Andralojc, K.M.; Mercalli, A.; Nowak, K.W.; Albarello, L.; Calcagno, R.; Luzi, L.; Bonifacio, E.; Doglioni, C.; Piemonti, L. Ghrelin-producing epsilon cells in the developing and adult human pancreas. Diabetologia 2009, 52, 486–493. [Google Scholar] [CrossRef]
- Unger, R.H.; Cherrington, A.D. Glucagonocentric restructuring of diabetes: A pathophysiologic and therapeutic makeover. J. Clin. Investig. 2012, 122, 4–12. [Google Scholar] [CrossRef]
- Gylfe, E.; Gilon, P. Glucose regulation of glucagon secretion. Diabetes Res. Clin. Pract. 2013, 103, 1–10. [Google Scholar] [CrossRef]
- Jensen, M.V.; Joseph, J.W.; Ronnebaum, S.M.; Burgess, S.C.; Sherry, A.D.; Newgard, C.B. Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1287–E1297. [Google Scholar] [CrossRef]
- Chertow, B.S.; Driscoll, H.K.; Blaner, W.S.; Meda, P.; Cordle, M.B.; Matthews, K.A. Effects of vitamin A deficiency and repletion on rat glucagon secretion. Pancreas 1994, 9, 475–484. [Google Scholar] [CrossRef]
- Chertow, B.S.; Driscoll, H.K.; Primerano, D.A.; Cordle, M.B.; Matthews, K.A. Retinoic acid receptor transcripts and effects of retinol and retinoic acid on glucagon secretion from rat islets and glucagon-secreting cell lines. Metab. Clin. Exp. 1996, 45, 300–305. [Google Scholar] [CrossRef]
- Matthews, K.A.; Rhoten, W.B.; Driscoll, H.K.; Chertow, B.S. Vitamin A deficiency impairs fetal islet development and causes subsequent glucose intolerance in adult rats. J. Nutr. 2004, 134, 1958–1963. [Google Scholar]
- Chertow, B.S.; Blaner, W.S.; Baranetsky, N.G.; Sivitz, W.I.; Cordle, M.B.; Thompson, D.; Meda, P. Effects of vitamin A deficiency and repletion on rat insulin secretion in vivo and in vitro from isolated islets. J. Clin. Investig. 1987, 79, 163–169. [Google Scholar] [CrossRef]
- Chertow, B.S.; Baker, G.R. The effects of vitamin A on insulin release and glucose oxidation in isolated rat islets. Endocrinology 1978, 103, 1562–1572. [Google Scholar] [CrossRef]
- Chertow, B.S.; Goking, N.Q.; Driscoll, H.K.; Primerano, D.A.; Matthews, K.A. Effects of all-trans-retinoic acid (ATRA) and retinoic acid receptor (RAR) expression on secretion, growth, and apoptosis of insulin-secreting RINm5F cells. Pancreas 1997, 15, 122–131. [Google Scholar] [CrossRef]
- Driscoll, H.K.; Adkins, C.D.; Chertow, T.E.; Cordle, M.B.; Matthews, K.A.; Chertow, B.S. Vitamin A stimulation of insulin secretion: Effects on transglutaminase mRNA and activity using rat islets and insulin-secreting cells. Pancreas 1997, 15, 69–77. [Google Scholar] [CrossRef]
- Cabrera-Valladares, G.; German, M.S.; Matschinsky, F.M.; Wang, J.; Fernandez-Mejia, C. Effect of retinoic acid on glucokinase activity and gene expression and on insulin secretion in primary cultures of pancreatic islets. Endocrinology 1999, 140, 3091–3096. [Google Scholar]
- Kane, M.A.; Folias, A.E.; Pingitore, A.; Perri, M.; Obrochta, K.M.; Krois, C.R.; Cione, E.; Ryu, J.Y.; Napoli, J.L. Identification of 9-cis-retinoic acid as a pancreas-specific autacoid that attenuates glucose-stimulated insulin secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 21884–21889. [Google Scholar] [CrossRef]
- Kane, M.A.; Folias, A.E.; Pingitore, A.; Perri, M.; Krois, C.R.; Ryu, J.Y.; Cione, E.; Napoli, J.L. CrbpI modulates glucose homeostasis and pancreas 9-cis-retinoic acid concentrations. Mol. Cell. Biol. 2011, 31, 3277–3285. [Google Scholar] [CrossRef]
- Ramachandran, C.K.; Dileepan, K.N.; Singh, V.N.; Venkitasubramanian, T.A. Metabolic potential of the adipose tissue of rats during hyper- and hypovitaminosis A. Proc. Soc. Exp. Biol. Med. 1986, 182, 73–78. [Google Scholar]
- Ribot, J.; Felipe, F.; Bonet, M.L.; Palou, A. Changes of Adiposity in Response to Vitamin A Status Correlate with Changes of PPARγ2 Expression. Obesity 2001, 9, 500–509. [Google Scholar] [CrossRef]
- De Souza Valente da Silva, L.; Valeria da Veiga, G.; Ramalho, R.A. Association of serum concentrations of retinol and carotenoids with overweight in children and adolescents. Nutrition 2007, 23, 392–397. [Google Scholar] [CrossRef]
- Zulet, M.A.; Puchau, B.; Hermsdorff, H.H.M.; Navarro, C.; Martínez, J.A. Vitamin A intake is inversely related with adiposity in healthy young adults. J. Nutr. Sci. Vitaminol. 2008, 54, 347–352. [Google Scholar] [CrossRef]
- Berry, D.C.; Noy, N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor β/δ and retinoic acid receptor. Mol. Cell. Biol. 2009, 29, 3286–3296. [Google Scholar] [CrossRef]
- Berry, D.C.; DeSantis, D.; Soltanian, H.; Croniger, C.M.; Noy, N. Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes 2012, 61, 1112–1121. [Google Scholar] [CrossRef]
- Mercader, J.; Ribot, J.; Murano, I.; Felipe, F.; Cinti, S.; Bonet, M.L.; Palou, A. Remodeling of white adipose tissue after retinoic acid administration in mice. Endocrinology 2006, 147, 5325–5332. [Google Scholar] [CrossRef]
- Murholm, M.; Isidor, M.S.; Basse, A.L.; Winther, S.; Sørensen, C.; Skovgaard-Petersen, J.; Nielsen, M.M.; Hansen, A.S.; Quistorff, B.; Hansen, J.B. Retinoic acid has different effects on UCP1 expression in mouse and human adipocytes. BMC Cell Biol. 2013, 14. [Google Scholar] [CrossRef]
- Schug, T.T.; Berry, D.C.; Shaw, N.S.; Travis, S.N.; Noy, N. Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 2007, 129, 723–733. [Google Scholar] [CrossRef]
- Shaw, N.; Elholm, M.; Noy, N. Retinoic acid is a high affinity selective ligand for the peroxisome proliferator-activated receptor β/δ. J. Biol. Chem. 2003, 278, 41589–41592. [Google Scholar] [CrossRef]
- Kiefer, F.W.; Vernochet, C.; O’Brien, P.; Spoerl, S.; Brown, J.D.; Nallamshetty, S.; Zeyda, M.; Stulnig, T.M.; Cohen, D.E.; Kahn, C.R.; et al. Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat. Med. 2012, 18, 918–925. [Google Scholar] [CrossRef]
- Dimaculangan, D.D.; Chawla, A.; Boak, A.; Kagan, H.M.; Lazar, M.A. Retinoic acid prevents downregulation of ras recision gene/lysyl oxidase early in adipocyte differentiation. Differentiation 1994, 58, 47–52. [Google Scholar] [CrossRef]
- Berry, D.C.; Soltanian, H.; Noy, N. Repression of cellular retinoic acid-binding protein II during adipocyte differentiation. J. Biol. Chem. 2010, 285, 15324–15332. [Google Scholar] [CrossRef]
- Xue, J.C.; Schwarz, E.J.; Chawla, A.; Lazar, M.A. Distinct stages in adipogenesis revealed by retinoid inhibition of differentiation after induction of PPARγ. Mol. Cell. Biol. 1996, 16, 1567–1575. [Google Scholar]
- Safonova, I.; Darimont, C.; Amri, E.Z.; Grimaldi, P.; Ailhaud, G.; Reichert, U.; Shroot, B. Retinoids are positive effectors of adipose cell differentiation. Mol. Cell. Endocrinol. 1994, 104, 201–211. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef]
- Timmons, J.A.; Wennmalm, K.; Larsson, O.; Walden, T.B.; Lassmann, T.; Petrovic, N.; Hamilton, D.L.; Gimeno, R.E.; Wahlestedt, C.; Baar, K.; et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc. Natl. Acad. Sci. USA 2007, 104, 4401–4406. [Google Scholar] [CrossRef]
- Seale, P.; Kajimura, S.; Yang, W.; Chin, S.; Rohas, L.M.; Uldry, M.; Tavernier, G.; Langin, D.; Spiegelman, B.M. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007, 6, 38–54. [Google Scholar] [CrossRef]
- Wolf, G. Brown adipose tissue: The molecular mechanism of its formation. Nutr. Rev. 2009, 67, 167–171. [Google Scholar] [CrossRef]
- Petrovic, N.; Walden, T.B.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic peroxisome proliferator-activated receptor gamma (PPARγ) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem. 2010, 285, 7153–7164. [Google Scholar]
- Wu, J.; Boström, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.-H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef]
- Cypess, A.M.; White, A.P.; Vernochet, C.; Schulz, T.J.; Xue, R.; Sass, C.A.; Huang, T.L.; Roberts-Toler, C.; Weiner, L.S.; Sze, C.; et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 2013, 19, 635–639. [Google Scholar] [CrossRef]
- Lidell, M.E.; Betz, M.J.; Dahlqvist Leinhard, O.; Heglind, M.; Elander, L.; Slawik, M.; Mussack, T.; Nilsson, D.; Romu, T.; Nuutila, P.; et al. Evidence for two types of brown adipose tissue in humans. Nat. Med. 2013, 19, 631–634. [Google Scholar] [CrossRef]
- Nedergaard, J.; Cannon, B. How brown is brown fat? It depends where you look. Nat. Med. 2013, 19, 540–541. [Google Scholar] [CrossRef]
- Bonet, M.L.; Oliver, J.; Picó, C.; Felipe, F.; Ribot, J.; Cinti, S.; Palou, A. Opposite effects of feeding a vitamin A-deficient diet and retinoic acid treatment on brown adipose tissue uncoupling protein 1 (UCP1), UCP2 and leptin expression. J. Endocrinol. 2000, 166, 511–517. [Google Scholar] [CrossRef]
- Puigserver, P.; Vázquez, F.; Bonet, M.L.; Picó, C.; Palou, A. In vitro and in vivo induction of brown adipocyte uncoupling protein (thermogenin) by retinoic acid. Biochem. J. 1996, 317, 827–833. [Google Scholar]
- Alvarez, R.; de Andrés, J.; Yubero, P.; Viñas, O.; Mampel, T.; Iglesias, R.; Giralt, M.; Villarroya, F. A novel regulatory pathway of brown fat thermogenesis. Retinoic acid is a transcriptional activator of the mitochondrial uncoupling protein gene. J. Biol. Chem. 1995, 270, 5666–5673. [Google Scholar] [CrossRef]
- Larose, M.; Cassard-Doulcier, A.M.; Fleury, C.; Serra, F.; Champigny, O.; Bouillaud, F.; Ricquier, D. Essential cis-acting elements in rat uncoupling protein gene are in an enhancer containing a complex retinoic acid response domain. J. Biol. Chem. 1996, 271, 31533–31542. [Google Scholar] [CrossRef]
- Ribot, J.; Felipe, F.; Bonet, M.L.; Palou, A. Retinoic acid administration and vitamin A status modulate retinoid X receptor alpha and retinoic acid receptor alpha levels in mouse brown adipose tissue. Mol. Cell. Biochem. 2004, 266, 25–30. [Google Scholar] [CrossRef]
- Pedersen, B.K. Muscles and their myokines. J. Exp. Biol. 2011, 214, 337–346. [Google Scholar] [CrossRef]
- Sundeen, G.; Richards, J.F.; Bragg, D.B. The effect of vitamin A deficiency on some postmortem parameters of avian muscle. Poult. Sci. 1980, 59, 2225–2236. [Google Scholar] [CrossRef]
- Lee, Y.M.; Lee, J.O.; Jung, J.-H.; Kim, J.H.; Park, S.-H.; Park, J.M.; Kim, E.-K.; Suh, P.-G.; Kim, H.S. Retinoic acid leads to cytoskeletal rearrangement through AMPK-Rac1 and stimulates glucose uptake through AMPK-p38 MAPK in skeletal muscle cells. J. Biol. Chem. 2008, 283, 33969–33974. [Google Scholar] [CrossRef]
- Narbonne, J.F.; Daubeze, M.; Bonmort, F. Protein metabolism in vitamin A deficient rats. II. Protein synthesis in striated muscle. Ann. Nutr. Aliment 1978, 32, 59–75. [Google Scholar]
- Hillgartner, F.B.; Morin, D.; Hansen, R.J. Effect of excessive vitamin A intake on muscle protein turnover in the rat. Biochem. J. 1982, 202, 499–508. [Google Scholar]
- Amengual, J.; Ribot, J.; Bonet, M.L.; Palou, A. Retinoic acid treatment increases lipid oxidation capacity in skeletal muscle of mice. Obesity 2008, 16, 585–591. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).