The Roles of Epithelial-to-Mesenchymal Transition (EMT) and Mesenchymal-to-Epithelial Transition (MET) in Breast Cancer Bone Metastasis: Potential Targets for Prevention and Treatment

Abstract
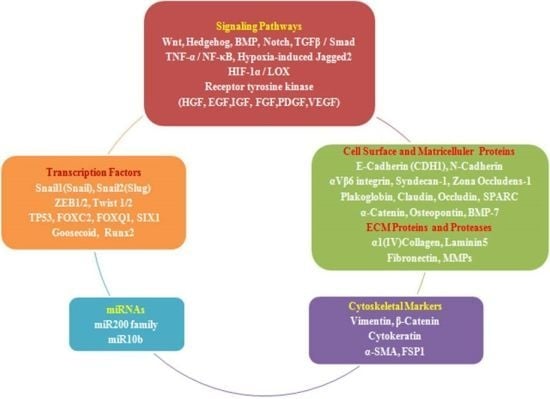
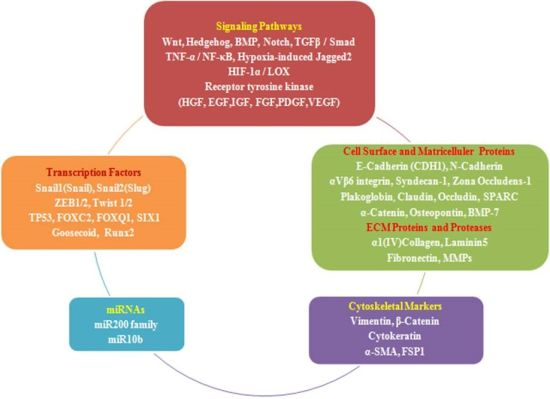
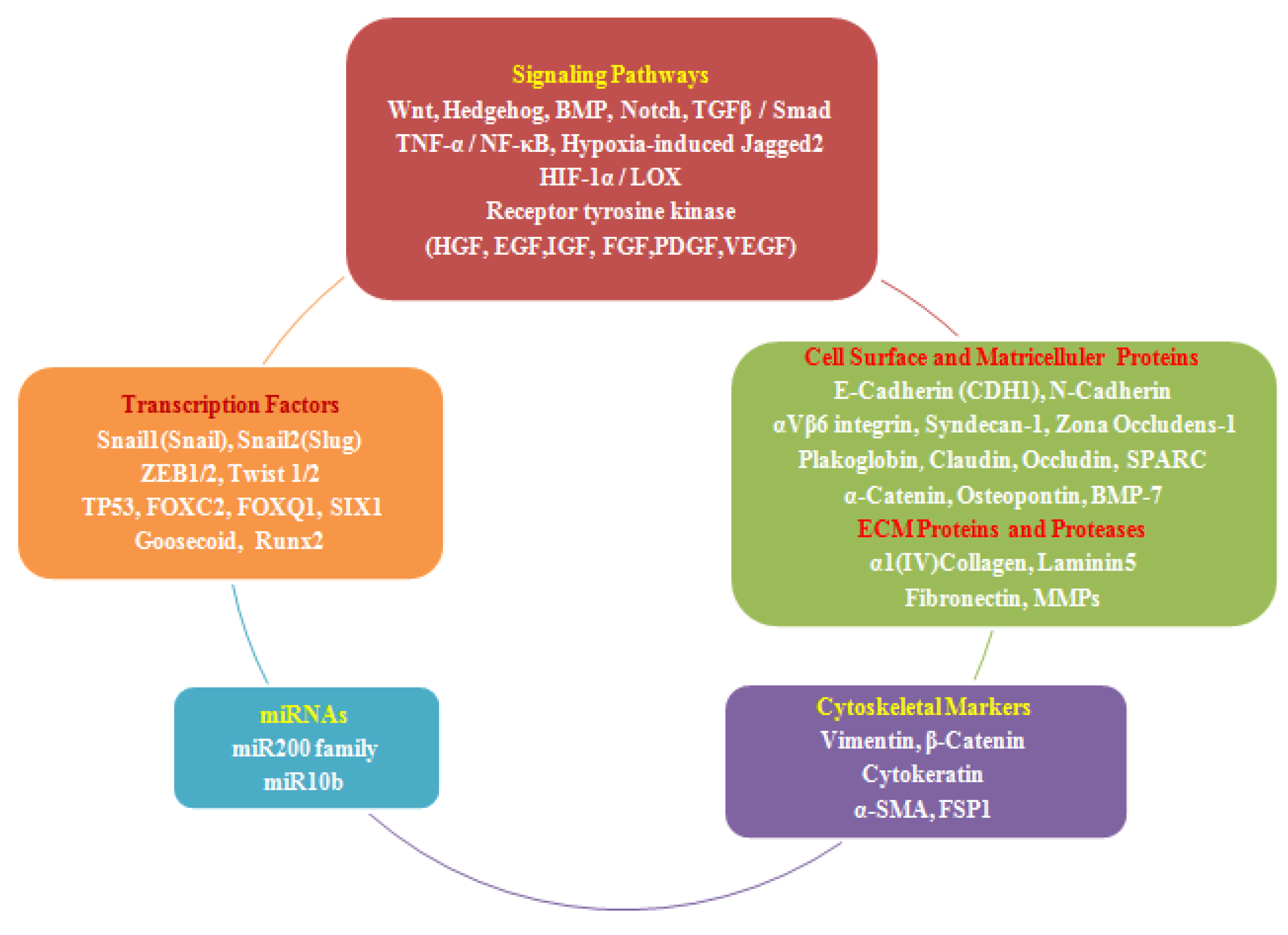
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Clues of Epithelial-Mesenchymal Transition
2.1. The Role of Peripheral Blood Circulating Tumor Cells and Bone Marrow Disseminated Tumor Cells
2.2. EMT Classification
2.3. Breast Cancer Stem Cells (BCSCs) and Mesenchymal Stem Cells (MSCs)
2.4. EMT Effectors and Signaling Pathways
3. The Clues of Mesenchymal-Epithelial Transition
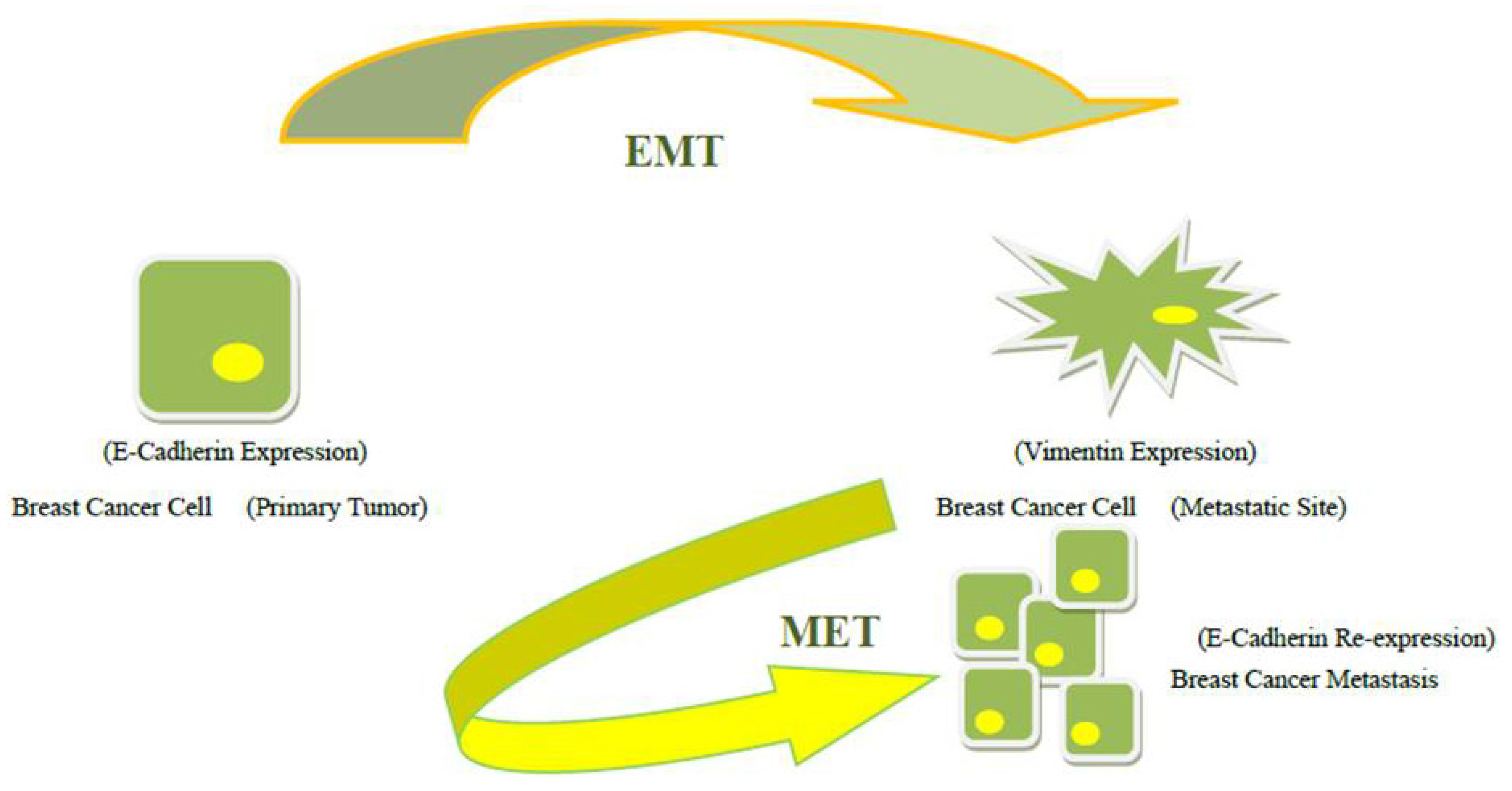
4. Clinical Implications of EMT and MET-Driven Growth of Metastases
5. Limitations
6. Conclusions
Conflicts of Interest
References
- Buijs, J.T.; van der Pluijm, G. Osteotropic cancers: From primary tumor to bone. Cancer Lett. 2009, 273, 177–193. [Google Scholar] [CrossRef]
- Sporn, M.B. The war on cancer. Lancet 1996, 347, 1377–1381. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Sainsbury, R. The development of endocrine therapy for women with breast cancer. Cancer Treat. Rev. 2013, 39, 507–517. [Google Scholar] [CrossRef]
- Rao, R.D.; Cobleigh, M.A. Adjuvant endocrine therapy for breast cancer. Oncology (Williston Park) 2012, 26, 541–547. [Google Scholar]
- Hernandez-Aya, L.F.; Gonzalez-Angulo, A.M. Adjuvant systemic therapies in breast cancer. Surg. Clin. North Am. 2013, 93, 473–491. [Google Scholar] [CrossRef]
- Vrbic, S.; Pejcic, I.; Filipovic, S.; Kocic, B.; Vrbic, M. Current and future anti-HER2 therapy in breast cancer. J. BUON 2013, 18, 4–16. [Google Scholar]
- Joerger, M.; Thürlimann, B. Chemotherapy regimens in early breast cancer: Major controversies and future outlook. Expert Rev. Anticancer Ther. 2013, 13, 165–178. [Google Scholar] [CrossRef]
- Andre, F.; Dieci, M.V.; Dubsky, P.; Sotiriou, C.; Curigliano, G.; Denkert, C.; Loi, S. Molecular pathways: Involvement of immune pathways in the therapeutic response and outcome in breast cancer. Clin. Cancer Res. 2013, 19, 28–33. [Google Scholar]
- Coleman, R.E. Skeletal complications of malignancy. Cancer 1997, 80, 1588–1594. [Google Scholar] [CrossRef]
- Steinman, R.A.; Brufsky, A.M.; Oesterreich, S. Zoledronic acid effectiveness against breast cancer metastases—A role for estrogen in the microenvironment? Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef]
- Lipton, A. Future treatment of bone metastases. Clin. Cancer Res. 2006, 12, 6305–6308. [Google Scholar] [CrossRef]
- Bouganim, N.; Dranitsaris, G.; Amir, E.; Clemons, M. Optimizing the use of bone-targeted agents in patients with metastatic cancers: A practical guide for medical oncologists. Support. Care Cancer 2011, 19, 1687–1696. [Google Scholar] [CrossRef]
- Rose, A.A.; Siegel, P.M. Emerging therapeutic targets in breast cancer bone metastases. Future Oncol. 2010, 6, 55–74. [Google Scholar] [CrossRef]
- Lee, B.L.; Higgins, M.J.; Goss, P.E. Denosumab and the current status of bone-modifying drugs in breast cancer. Acta Oncol. 2012, 51, 157–167. [Google Scholar] [CrossRef]
- Stopeck, A.T.; Lipton, A.; Body, J.J.; Steger, G.G.; Tonkin, K.; de Boer, R.H.; Lichinitser, M.; Fujiwara, Y.; Yardley, D.A.; Viniegra, M.; et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: A randomized, double-blind study. J. Clin. Oncol. 2010, 28, 5132–5139. [Google Scholar] [CrossRef]
- Young, R.J.; Coleman, R.E. Zoledronic acid to prevent and treat cancer metastasis: New prospects for an old drug. Future Oncol. 2013, 9, 633–643. [Google Scholar] [CrossRef]
- Casas, A.; Llombart, A.; Martín, M. Denosumab for the treatment of bone metastases in advanced breast cancer. Breast 2013, 22, 585–592. [Google Scholar] [CrossRef]
- Chang, J.C.; Wooten, E.C.; Tsimelzon, A.; Hilsenbeck, S.G.; Gutierrez, M.C.; Tham, Y.L.; Kalidas, M.; Elledge, R.; Mohsin, S.; Osborne, C.K.; et al. Patterns of resistance and incomplete response to docetaxel by gene expression profiling in breast cancer patients. J. Clin. Oncol. 2005, 23, 169–177. [Google Scholar]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Li, Q.Q.; Xu, J.D.; Wang, W.J.; Cao, X.X.; Chen, Q.; Tang, F.; Chen, Z.Q.; Liu, X.P.; Xu, Z.D. Twist1-mediated adriamycin-induced epithelial mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin. Cancer. Res. 2009, 15, 2657–2665. [Google Scholar] [CrossRef]
- Van der Pluijm, G. Epithelial plasticity, cancer stem cells and bone metastasis formation. Bone 2011, 48, 37–43. [Google Scholar] [CrossRef]
- Dave, B.; Mittal, V.; Tan, N.M.; Chang, J.C. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res. 2012, 14, 202. [Google Scholar]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Cardiff, R.D.; Couto, S.; Bolon, B. Three interrelated themes in current breast cancer research: Gene addiction, phenotypic plasticity, and cancer stem cells. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef]
- Takebe, N.; Warren, R.Q.; Ivy, S.P. Breast cancer growth and metastasis: İnterplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Weidner, N.; Folkman, J.; Pozza, F.; Bevilacqua, P.; Allred, E.N.; Moore, D.H.; Meli, S.; Gasparini, G. Tumor angiogenesis: A new significant and independent prognostic indicator in early-stage breast carcinoma. J. Natl. Cancer Inst. 1992, 84, 1875–1887. [Google Scholar] [CrossRef]
- Folkman, J.; Shing, Y. Angiogenesis. J. Biol. Chem. 1992, 267, 10931–10934. [Google Scholar]
- Folkman, J. The role of angiogenesis in tumor growth. Semin. Cancer Biol. 1992, 3, 65–71. [Google Scholar]
- Kiaris, H.; Chatzistamou, I.; Kalofoutis, C.; Koutselini, H.; Piperi, C.; Kalofoutis, A. Tumour-stroma interactions in carcinogenesis: Basic aspects and perspectives. Mol. Cell. Biochem. 2004, 261, 117–122. [Google Scholar] [CrossRef]
- Pupa, S.M.; Menard, S.; Forti, S.; Tagliabue, E. New insights into the role of extracellular matrix during tumor onset and progression. J. Cell. Physiol. 2002, 192, 259–267. [Google Scholar]
- Wells, A.; Chao, Y.L.; Grahovac, J.; Wu, Q.; Lauffenburger, D.A. Epithelial and mesenchymal phenotypic switchings modulate cell motility in metastasis. Front. Biosci. 2011, 16, 815–837. [Google Scholar] [CrossRef]
- Mathias, R.A.; Gopala, S.K.; Simpson, R.J. Contribution of cells undergoing epithelial-mesenchymal transition to the tumour microenvironment. J. Proteomics 2013, 78, 545–557. [Google Scholar] [CrossRef]
- Gunasinghe, N.P.A.D.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 2009, 119, 1429–1437. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Blick, T.; Hugo, H.; Widodo, E.; Waltham, M.; Pinto, C.; Mani, S.A.; Weinberg, R.A.; Neve, R.M.; Lenburg, M.E.; Thompson, E.W. Epithelial mesenchymal transition traits in human breast cancer cell lines paralel the CD44 (hi)/CD24 (lo/−) stem cell phenotype in human breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 235–252. [Google Scholar] [CrossRef]
- Beug, H. Breast cancer stem cells: Eradication by differentiation therapy? Cell 2009, 138, 623–625. [Google Scholar] [CrossRef]
- Woelfle, U.; Cloos, J.; Sauter, G.; Riethdorf, L.; Janicke, F.; van Diest, P.; Brakenhoff, R.; Pantel, K. Molecular signature associated with bone marrow micrometastasis in human breast cancer. Cancer Res. 2003, 63, 5679–5684. [Google Scholar]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal transition in breast cancer progression and metastasis. Chin. J. Cancer 2011, 30, 603–611. [Google Scholar] [CrossRef]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 2008, 3, e2888. [Google Scholar]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar]
- Pratap, J.; Lianb, J.B.; Steinb, G.S. Metastatic bone disease: Role of transcription factors and future targets. Bone 2011, 48, 30–36. [Google Scholar] [CrossRef]
- Vallone, V.B.F.; Hofer, E.L.; Choi, H.; Bordenave, R.H.; Batagelj, E.; Feldman, L.; La Russa, V.; Caramutti, D.; Dimase, F.; Labovsky, V.; et al. Behaviour of mesenchymal stem cells from bone marrow of untreated advanced breast and lung cancer patients without bone osteolytic metastasis. Clin. Exp. Metastasis 2013, 30, 317–332. [Google Scholar] [CrossRef]
- Josson, S.; Nomura, T.; Lin, J.-T.; Huang, W.-C.; Wu, D.; Zhau, H.E.; Zayzafoon, M.; Weizmann, M.N.; Gururajan, M.; Chung, L.W.K. Transition and confers cancer lethality and bone β2-microglobulin induces epithelial to mesenchymal metastasis in human cancer cells. Cancer Res. 2011, 71, 2600–2610. [Google Scholar] [CrossRef]
- Martin, F.T.; Dwyer, R.M.; Kelly, J.; Khan, S.; Murphy, J.M.; Curran, C.; Miller, N.; Hennessy, E.; Dockery, P.; Barry, F.P.; et al. Potential role of mesenchymal stem cells (MSCs) in the breast tumour microenvironment: Stimulation of epithelial to mesenchymal transition (EMT). Breast Cancer Res. Treat. 2010, 124, 317–326. [Google Scholar] [CrossRef]
- Jain, V.K.; Turner, N.C. Challenges and opportunities in the targeting of fibroblast growth factor receptors in breast cancer. Breast Cancer Res. 2012, 14, 208. [Google Scholar] [CrossRef]
- Martorana, A.M.; Zheng, G.; Crowe, T.C.; O’Grady, R.L.; Lyons, J.G. Epithelial cells up-regulate matrix metalloproteinases in cells within the same mammary carcinoma that have undergone an epithelial-mesenchymal transition. Cancer Res. 1998, 58, 4970–4979. [Google Scholar]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Guise, T.A.; Chirgwin, J.M. Transforming growth factor-β in osteolytic breast cancer bone metastases. Clin. Orthop. 2003, 415, 32–38. [Google Scholar] [CrossRef]
- Buijs, T.J.; Stayrook, K.R.; Guise, T.A. TGF-β in the bone microenvironment: Role in breast cancer metastases. Cancer Microenviron. 2011, 4, 261–281. [Google Scholar] [CrossRef]
- Deckers, M.; van Dinther, M.; Buijs, J.; Que, I.; Lowik, C.; van der Pluijm, G.; ten Dijke, P. The tumor suppressor Smad4 is required for transforming growth factor B-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res. 2006, 66, 2202–2209. [Google Scholar] [CrossRef]
- Giehl, K.; Imamichib, Y.; Menke, A. Smad4-independent TGF-β signaling in tumor cell migration. Cells Tissues Organs 2007, 185, 123–130. [Google Scholar] [CrossRef]
- Sundqvist, A.; ten Dijke, P.; van Dam, H. Key signaling nodes in mammary gland development and cancer: Smad signal integration in epithelial cell plasticity. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef]
- Gal, A.; Sjoblom, T.; Fedorova, L.; Imreh, S.; Beug, H.; Moustakas, A. Sustained TGF beta exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 2008, 27, 1218–1230. [Google Scholar]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor β in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef]
- Ten Dijke, P.; Goumans, M.-J.; Itoh, F.; Itoh, S. Regulation of cell proliferation by Smad proteins. J. Cell. Physiol. 2002, 191, 1–16. [Google Scholar] [CrossRef]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massagué, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Invest. 1999, 103, 197–206. [Google Scholar] [CrossRef]
- Tanos, T.; Rojo, L.J.; Echeverria, P.; Brisken, C. ER and PR signaling nodes during mammary gland development. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef] [Green Version]
- Beleut, M.; Rajaram, R.D.; Caikovski, M.; Ayyanan, A.; Germano, D.; Choi, Y.; Schneider, P.; Brisken, C. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc. Natl. Acad. Sci. USA 2009, 107, 2989–2994. [Google Scholar]
- Joshi, P.A.; Jackson, H.W.; Beristain, A.G.; di Grappa, M.A.; Mote, P.A.; Clarke, C.L.; Stingl, J.; Waterhouse, P.D.; Khokha, R. Progesterone induces adult mammary stem cell expansion. Nature 2010, 465, 803–807. [Google Scholar] [CrossRef]
- Asselin-Labat, M.L.; Vaillant, F.; Sheridan, J.M.; Pal, B.; Wu, D.; Simpson, E.R.; Yasuda, H.; Smyth, G.K.; Martin, T.J.; Lindeman, G.J.; et al. Control of mammary stem cell function by steroid hormone signalling. Nature 2010, 465, 798–802. [Google Scholar] [CrossRef]
- Hinck, L.; Silberstein, G.B. Key stages in mammary gland development: The mammary end bud as a motile organ. Breast Cancer Res. 2005, 7, 245–251. [Google Scholar] [CrossRef]
- Tanos, T.; Sflomos, G.; Echeverria, P.C.; Ayyanan, A.; Gutierrez, M.; Delaloye, J.F.; Raffoul, W.; Fiche, M.; Dougall, W.; Schneider, P.;et al. Progesterone/RANKL is a major regulatory axis in the human breast. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Ismail, P.M.; DeMayo, F.J.; Amato, P.; Lydon, J.P. Progesterone induction of calcitonin expression in the murine mammary gland. J. Endocrinol. 2004, 180, 287–295. [Google Scholar] [CrossRef]
- Andrews, J.L.; Kim, A.C.; Hens, J.R. The role and function of cadherins in the mammary gland. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef]
- Kowalski, P.J.; Rubin, M.A.; Kleer, C.G. E-Cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 2003, 5, 217–222. [Google Scholar] [CrossRef]
- Berx, G.; Staes, K.; van Hengel, J.; Molemans, F.; Bussemakers, M.J.; van Bokhoven, A.; van Roy, F. Cloning and characterization of the human invasion suppressor gene E cadherin (CDH1). Genomics 1995, 26, 281–289. [Google Scholar] [CrossRef]
- Pecina-Slaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3. [Google Scholar] [CrossRef] [Green Version]
- Perl, A.K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef]
- Wells, A.; Yates, C.; Shepard, C.R. E-Cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin. Exp. Metastasis 2008, 25, 621–628. [Google Scholar] [CrossRef]
- Saha, B.; Chaiwun, B.; Imam, S.S.; Tsao-Wei, D.D.; Groshen, S.; Naritoku, W.Y.; Imam, S.A. Overexpression of E-cadherin protein in metastatic breast cancer cells in bone. Anticancer Res. 2007, 27, 3903–3908. [Google Scholar]
- Lehtinen, L.; Ketola, K.; Mäkelä, R.; Mpindi, J.P.; Viitala, M.; Kallioniemi, O.; Iljin, K. High-throughput RNAi screening for novel modulators of vimentin expression identifies MTHFD2 as a regulator of breast cancer cell migration and invasion. Oncotarget 2013, 4, 48–63. [Google Scholar]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef]
- Bullock, M.D.; Sayan, A.E.; Packham, G.K.; Mirnezami, A.H. MicroRNAs: Critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression. Biol. Cell 2012, 104, 3–12. [Google Scholar] [CrossRef]
- Schwarzenbacher, D.; Balic, M.; Pichler, M. The role of microRNAs in breast cancer stem cells. Int. J. Mol. Sci. 2013, 14, 14712–14723. [Google Scholar] [CrossRef]
- Hwang, M.S.; Yu, N.; Stinson, S.Y.; Yue, P.; Newman, R.J.; Allan, B.B.; Dornan, D. miR-221/222 targets adiponectin receptor 1 to promote the epithelial-to-mesenchymal transition in breast cancer. PLoS One 2013, 8, e66502. [Google Scholar]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef]
- De Herreros, A.G.; Peiro, S.; Nassour, M.; Savagner, P. Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 135–147. [Google Scholar]
- Maeda, M.; Johnson, K.R.; Wheelock, M.J. Cadherin switching: Essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J. Cell Sci. 2005, 118, 873–887. [Google Scholar] [CrossRef]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- El-Haibia, C.P.; Bellb, G.W.; Zhangc, J.; Collmanna, A.Y.; Woodd, D.; Scherbere, C.M.; Csizmadiaf, E.; Marianig, O.; Zhua, C.; Campagne, A.; et al. Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy. PNAS 2012, 109, 17460–17465. [Google Scholar] [CrossRef]
- Korpal, M.; Ell, B.J.; Buffa, F.M.; Ibrahim, T.; Blanco, M.A.; Celià-Terrassa, T.; Mercatali, L.; Khan, Z.; Goodarzi, H.; Hua, Y.; et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat. Med. 2011, 17, 1101–1108. [Google Scholar] [CrossRef] [Green Version]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteomics 2010, 73, 1907–1920. [Google Scholar] [CrossRef]
- Janowska-Wieczorek, A.; Marquez-Curtis, L.A.; Wysoczynski, M.; Ratajczak, M.Z. Enhancing effect of platelet-derived microvesicles on the invasive potential of breast cancer cells. Transfusion 2006, 46, 1199–1209. [Google Scholar] [CrossRef]
- Friel, A.M.; Corcoran, C.; Crown, J.; O’Driscoll, L. Relevance of circulating tumor cells, extracellular nucleic acids, and exosomes in breast cancer. Breast Cancer Res. Treat. 2010, 123, 613–625. [Google Scholar]
- Simpson, R.J.; Jensen, S.S.; Lim, J.W. Proteomic profiling of exosomes: Current perspectives. Proteomics 2008, 8, 4083–4099. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef]
- Rappa, G.; Mercapide, J.; Lorico, A. Spontaneous formation of tumorigenic hybrids between breast cancer and multipotent stromal cells is a source of tumor heterogeneity. Am. J. Pathol. 2012, 180, 2504–2515. [Google Scholar] [CrossRef]
- Sabbah, M.; Emami, S.; Redeuilh, G.; Julien, S.; Prévost, G.; Zimber, A.; Ouelaa, R.; Bracke, M.; de Wever, O.; Gespach, C. Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist. Updat. 2008, 11, 123–151. [Google Scholar] [CrossRef]
- Shore, P.A. Role for Runx2 in normal mammary gland and breast cancer bone metastasis. J. Cell. Biochem. 2005, 96, 484–489. [Google Scholar] [CrossRef]
- Pratap, J.; Lian, J.B.; Javed, A.; Barnes, G.L.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev. 2006, 25, 589–600. [Google Scholar]
- Chimge, N.-O.; Baniwal, S.K.; Little, G.H.; Chen, Y.; Kahn, M.; Tripathy, D.; Borok, Z.; Frenkel, B. Regulation of breast cancer metastasis by Runx2 and estrogen signaling: The role of SNAI2. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef]
- Mendoza-Villanueva, D.; Zeef, L.; Shore, P. Metastatic breast cancer cells inhibit osteoblast differentiation through the Runx2/CBFb dependent expression of the Wnt antagonist, sclerostin. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA. Int. J. Mol. Med. 2008, 22, 271–275. [Google Scholar]
- Weinberg, R.A. Leaving home early: Reexamination of the canonical models of tumor progression. Cancer Cell 2008, 14, 283–284. [Google Scholar] [CrossRef]
- Scheel, C.; Weinberg, R.A. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Foroni, C.; Broggini, M.; Generali, D.; Damia, G. Epithelial-mesenchymal transition and breast cancer: Role, molecular mechanisms and clinical impact. Cancer Treat. Rev. 2012, 38, 689–697. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.S.; Fridlyand, J.; Sahin, A.; Agarwal, R.; Joy, C.; et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef]
- Howlett, A.R.; Bissell, M.J. The influence of tissue microenvironment (stroma and extracellular matrix) on the development and function of mammary epithelium. Epithel. Cell Biol. 1993, 2, 79–89. [Google Scholar]
- Bonnomet, A.; Syne, L.; Brysse, A.; Feyereisen, E.; Thompson, E.W.; Noel, A.; Foidart, J.M.; Birembaut, P.; Polette, M.; Gilles, C. A dynamic in vivo model of epithelial-to-mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene 2012, 31, 3741–3753. [Google Scholar]
- Creighton, C.J.; Chang, J.C.; Rosen, J.M. Epithelialmesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 253–260. [Google Scholar] [CrossRef]
- Jechlinger, M.; Grunert, S.; Beug, H. Mechanisms in epithelial plasticity and metastasis: Insights from 3D cultures and expression profiling. J. Mammary Gland Biol. Neoplasia 2002, 7, 415–432. [Google Scholar] [CrossRef]
- Hugo, H.; Ackland, M.L.; Blick, T. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J. Cell. Physiol. 2007, 213, 374–383. [Google Scholar] [CrossRef]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef]
- Patel, S.A.; Dave, M.A.; Murthy, R.G.; Helmy, K.Y.; Rameshwar, P. Metastatic breast cancer cells in the bone marrow microenvironment: Novel insights into oncoprotection. Oncol. Rev. 2011, 5, 93–102. [Google Scholar] [CrossRef]
- Kallergi, G.; Papadaki, M.A.; Politaki, E.; Mavroudis, D.; Georgoulias, V.; Agelaki, S. Epithelial to mesenchymal transition markers expressed in circulating tumour cells of early and metastatic breast cancer patients. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef]
- Lopes, N.; Paredes, J.; Costa, J.L.; Ylstra, B.; Schmitt, F. Vitamin D and the mammary gland: A review on its role in normal development and breast cancer. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef]
- Lopes, N.; Carvalho, J.; Duraes, C.; Sousa, B.; Gomes, M.; Costa, J.L.; Oliveira, C.; Paredes, J.; Schmitt, F. 1Alpha,25-dihydroxyvitamin D3 induces de novo E-cadherin expression in triple-negative breast cancer cells by CDH1-promoter demethylation. Anticancer Res. 2012, 32, 249–257. [Google Scholar]
- Chao, Y.; Wu, Q.; Acquafondata, M.; Dhir, R.; Wells, A. Partial mesenchymal to epithelial reverting transition in breast and prostate cancer metastases. Cancer Microenviron. 2012, 5, 19–28. [Google Scholar] [CrossRef]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Drasin, D.J.; Robin, T.P.; Ford, H.L. Breast cancer epithelial-to-mesenchymal transition: Examining the functional consequences of plasticity. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef]
- Tomaskovic-Crook, E.; Thompson, E.W.; Thiery, J.P. Epithelial to mesenchymal transition and breast cancer. Breast Cancer Res. 2009, 11. [Google Scholar] [CrossRef]
- Sas, L.; Lardon, F.; Vermeulen, P.B.; Hauspy, J.; van Dam, P.; Pauwels, P.; Dirix, L.Y.; van Laere, S.J. The interaction between ER and NFκB in resistance to endocrine therapy. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef]
- Pece, S.; Tosoni, D.; Confalonieri, S.; Mazzarol, G.; Vecchi, M.; Ronzoni, S.; Bernard, L.; Viale, G.; Pelicci, P.G.; di Fiore, P.P. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 2010, 140, 62–73. [Google Scholar] [CrossRef]
- Torsvik, A.; Bjerkvig, R. Mesenchymal stem cell signaling in cancer progression. Cancer Treat. Rev. 2013, 39, 180–188. [Google Scholar]
- Palena, C.; Hamilton, D.H.; Fernando, R.I. Influence of IL-8 on the epithelial-mesenchymal transition and the tumor microenvironment. Future Oncol. 2012, 8, 713–722. [Google Scholar] [CrossRef]
- Buijs, J.T.; Petersen, M.; van der Horst, G.; van der Pluijm, G. Bone morphogenetic proteins and its receptors; therapeutic targets in cancer progression and bone metastasis? Curr. Pharm. Des. 2010, 16, 1291–1300. [Google Scholar] [CrossRef]
- Buijs, J.T.; Henriquez, N.V.; van Overveld, P.G.M.; van der Horst, G.; Que, I.; Schwaninger, R.; Rentsch, C.; ten Dijke, P.; Cleton-Jansen, A.-M.; Driouch, K.; et al. Bone morphogenetic protein 7 in the development and treatment of bone metastases from breast cancer. Cancer Res. 2007, 67, 8742–8751. [Google Scholar] [CrossRef]
- Fuxea, J.; Karlsson, M.C.I. TGF-β-induced epithelial-mesenchymal transition: A link between cancer and inflammation. Semin. Cancer Biol. 2012, 22, 455–461. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhou, H.; Dunstan, C.R.; Sutherland, R.L.; Seibel, M.J. The role of the bone microenvironment in skeletal metastasis. J. Bone Oncol. 2013, 2, 47–57. [Google Scholar]
- Chirgwin, J.M.; Mohammad, K.S.; Guise, T.A. Tumor-bone cellular interactions in skeletal metastases. J. Musculoskel. Neuron Interact. 2004, 4, 308–318. [Google Scholar]
- Kozlow, W.; Guise, T.A. Breast cancer metastasis to bone: Mechanisms of osteolysis and implications for therapy. J. Mammary Gland Biol. Neoplasia 2005, 10, 169–180. [Google Scholar] [CrossRef]
- Kakonen, S.M.; Mundy, G.R. Mechanisms of osteolytic bone metastases in breast carcinoma. Cancer 2003, 97, 834–839. [Google Scholar] [CrossRef]
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.-H.; Moustakas, A. TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 2005, 16, 1987–2002. [Google Scholar] [CrossRef]
- Kang, Y.; He, W.; Tulley, S.; Gupta, G.P.; Serganova, I.; Chen, C.R.; Manova-Todorova, K.; Blasberg, R.; Gerald, W.L.; Massagué, J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13909–13914. [Google Scholar]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Sethi, N.; Kang, Y. Notch signaling: Mediator and therapeutic target of bone metastasis. BoneKey Rep. 2012, 1. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Demirkan, B. The Roles of Epithelial-to-Mesenchymal Transition (EMT) and Mesenchymal-to-Epithelial Transition (MET) in Breast Cancer Bone Metastasis: Potential Targets for Prevention and Treatment. J. Clin. Med. 2013, 2, 264-282. https://doi.org/10.3390/jcm2040264
Demirkan B. The Roles of Epithelial-to-Mesenchymal Transition (EMT) and Mesenchymal-to-Epithelial Transition (MET) in Breast Cancer Bone Metastasis: Potential Targets for Prevention and Treatment. Journal of Clinical Medicine. 2013; 2(4):264-282. https://doi.org/10.3390/jcm2040264
Chicago/Turabian StyleDemirkan, Binnaz. 2013. "The Roles of Epithelial-to-Mesenchymal Transition (EMT) and Mesenchymal-to-Epithelial Transition (MET) in Breast Cancer Bone Metastasis: Potential Targets for Prevention and Treatment" Journal of Clinical Medicine 2, no. 4: 264-282. https://doi.org/10.3390/jcm2040264