
Ocular Manifestations in Patients Affected by p63-Associated Disorders: Ectrodactyly-Ectodermal Dysplasia-Clefting (EEC) and Ankyloblepharon-Ectodermal Defects-Cleft Lip Palate (AEC) Syndromes
 , , and
, , and
Abstract
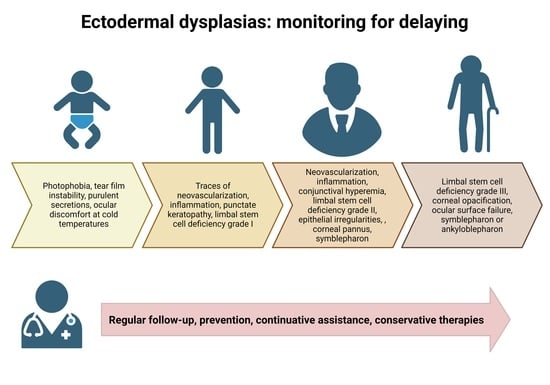
:
1. Introduction
2. Materials and Methods
3. Results
3.1. Case 1
3.2. Case 2
3.3. Case 3
3.4. Case 4
3.5. Case 5
3.6. Case 6
3.7. Case 7
3.8. Case 8
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 Homolog at 3q27–29, Encodes Multiple Products with Transactivating, Death-Inducing, and Dominant-Negative Activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Chakravarti, D.; Flores, E.R. p63 steps into the limelight: Crucial roles in the suppression of tumorigenesis and metastasis. Nat. Rev. Cancer 2013, 13, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Novelli, F.; Ganini, C.; Melino, G.; Nucci, C.; Han, Y.; Shi, Y.; Wang, Y.; Candi, E. p63 in corneal and epidermal differentiation. Biochem. Biophys. Res. Commun. 2022, 610, 15–22. [Google Scholar] [CrossRef]
- Soares, E.; Zhou, H. Master regulatory role of p63 in epidermal development and disease. Cell. Mol. Life Sci. 2018, 75, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Osterburg, C.; Osterburg, S.; Zhou, H.; Missero, C.; Dötsch, V. Isoform-Specific Roles of Mutant p63 in Human Diseases. Cancers 2021, 13, 536. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Memmi, E.M.; Pelicci, P.G.; Bernassola, F. Maintaining epithelial stemness with p63. Sci. Signal. 2015, 387, re9. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, E.; Barbaro, V.; Ruzza, A.; Ponzin, D.; Pellegrini, G.; De Luca, M. Isoforms of ΔNp63 and the migration of ocular limbal cells in human corneal regeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 9523–9528. [Google Scholar] [CrossRef]
- Candi, E.; Rufini, A.; Terrinoni, A.; Dinsdale, D.; Ranalli, M.; Paradisi, A.; De Laurenzi, V.; Spagnoli, L.G.; Catani, M.V.; Ramadan, S.; et al. Differential roles of p63 isoforms in epidermal development: Selective genetic complementation in p63 null mice. Cell Death Differ. 2006, 13, 1037–1047. [Google Scholar] [CrossRef]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef]
- Rinne, T.; Brunner, H.G.; Van Bokhoven, H. p63-Associated Disorders. Cell Cycle 2007, 6, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Brunner, H.G.; Hamel, B.C.J.; Van Bokhoven, H. The p63 gene in EEC and other syndromes. J. Med. Genet. 2002, 39, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Celli, J.; Duijf, P.; Hamel, B.C.; Bamshad, M.; Kramer, B.; Smits, A.P.; Newbury-Ecob, R.; Hennekam, R.C.; Van Buggenhout, G.; van Haeringen, A.; et al. Heterozygous Germline Mutations in the p53 Homolog p63 Are the Cause of EEC Syndrome. Cell 1999, 99, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, E.; Kaye, S.B.; Ponzin, D.; Barbaro, V.; Ferrari, S.; Böhm, E.; Nardiello, P.; Castaldo, G.; McGrath, J.A.; Willoughby, C.E. Limbal Stem Cell Deficiency and Ocular Phenotype in Ectrodactyly-Ectodermal Dysplasia-Clefting Syndrome Caused by p63 Mutations. Ophthalmology 2012, 119, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, V.; Nasti, A.A.; Del Vecchio, C.; Ferrari, S.; Migliorati, A.; Raffa, P.; Lariccia, V.; Nespeca, P.; Biasolo, M.; Willoughby, C.E.; et al. Correction of Mutant p63 in EEC Syndrome Using siRNA Mediated Allele-Specific Silencing Restores Defective Stem Cell Function. Stem Cells 2016, 34, 1588–1600. [Google Scholar] [CrossRef] [PubMed]
- Amparo, F.; Wang, H.; Yin, J.; Marmalidou, A.; Dana, R. Evaluating Corneal Fluorescein Staining Using a Novel Automated Method. Investig. Ophthalmol. Vis. Sci. 2017, 58, BIO168–BIO173. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Garg, N.K.; Lunde, E.; Han, K.Y.; Jain, S.; Azar, D.T. Corneal neovascularization: An anti-VEGF therapy review. Surv. Ophthalmol. 2012, 57, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.S.; Miller, P.E.; Bentley, E.; Thomasy, S.M.; Murphy, C.J. The SPOTS System: An Ocular Scoring System Optimized for Use in Modern Preclinical Drug Development and Toxicology. J. Ocul. Pharmacol. Ther. 2017, 33, 718–734. [Google Scholar] [CrossRef] [PubMed]
- Efron, N.; Morgan, P.B.; Katsara, S.S. Validation of grading scales for contact lens complications. Ophthalmic Physiol. Opt. 2001, 21, 17–29. [Google Scholar] [CrossRef]
- Almeida, S.F.; Solari, H.P. Ectodermal dysplasia, ectrodactyly and clefting syndrome: Ocular manifestations of this syndrome in a case report. Arq. Bras. Oftalkmol. 2007, 70, 125–128. [Google Scholar] [CrossRef]
- Gupta, S.; Fink, M.K.; Sinha, P.R.; Mohan, R.R. DELTA Scoring System: A Novel method for clinical grading of corneal neovascularization. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3527. [Google Scholar]
- Barbaro, V.; Nardiello, P.; Castaldo, G.; Willoughby, C.E.; Ferrari, S.; Ponzin, D.; Amato, F.; Bonifazi, E.; Parekh, M.; Calistri, A.; et al. A novel de novo missense mutation in TP63 underlying germline mosaicism in AEC syndrome:implications for recurrence risk and prenatal diagnosis. Am. J. Med. Genet. A 2012, 158A, 1957–1961. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kumar, C.; Bhalerao, S.; Pandita, A.; Shastri, S.; Sharma, P. Ectrodactyly, Ectodermal Dysplasia, Cleft Lip, and Palate (EEC Syndrome) with Tetralogy of Fallot: A Very Rare Combination. Front. Pediatr. 2015, 3, 51. [Google Scholar] [CrossRef]
- Gan, L.; van Setten, G.; Seregard, S.; Fagerholm, P. Proliferating cell nuclear antigen colocalization with corneal epithelial stem cells and involvement in physiological cell turnover. Acta Ophthalmol. Scand. 1995, 73, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Trinkaus-Randall, V. New insights in wound response and repair of epithelium. J. Cell. Physiol. 2013, 228, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Fete, M.; Vanbokhoven, H.; Clements, S.E.; McKeon, F.; Roop, D.R.; Koster, M.I.; Missero, C.; Attardi, L.D.; Lombillo, V.A.; Ratovitski, E.; et al. Conference Report: International Research Symposium on Ankyloblepharon-Ectodermal Defects-Cleft Lip and/or Palate (AEC) Syndrome. Am. J. Med. Genet. A 2009, 149A, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Elmann, S.; Hanson, S.A.; Bunce, C.N.; Shinder, R. Ectrodactyly Ectodermal Dysplasia Clefting (EEC) Syndrome: A Rare Cause of Congenital Lacrimal Anomalies. Ophthalmic Plast. Reconstr. Surg. 2015, 31, e35. [Google Scholar] [CrossRef] [PubMed]
- Batra, P.; Duggal, R.; Parkash, H. EEC syndrome-a case report. J. Indian Soc. Pedod. Prev. Dent. 2003, 21, 75–78. [Google Scholar]
- McNab, A.A.; Potts, M.J.; Welham, R.A. The EEC syndrome and its ocular manifestations. Br. J. Ophthalmol. 1989, 73, 261–264. [Google Scholar] [CrossRef]
- Kennedy, D.P.; Chandler, J.W.; McCulley, J.P. Ocular surface involvements in ectrodactyly-ectodermal dysplasia-cleft syndrome. Contact Lens Anterior Eye 2015, 38, 228–231. [Google Scholar] [CrossRef]
- Augello, M.; Berg, B.-I.; Müller, A.A.; Schwenzer-Zimmerer, K. Two case reports with literature review of the EEC syndrome: Clinical presentation and management. Case Rep. Plast. Surg. Hand Surg. 2015, 2, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Garza-Leon, M.; León-Cachón, R.B.R.; Villafuerte-de la Cruz, R.; Martínez-Treviño, D.A. Infrared meibography and molecular assessment of p63 gene mutations in a Mexican patient with EEC syndrome. Arch. Soc. Española Oftalmol. 2018, 93, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Käsmann, B.; Ruprecht, K.W. Ocular manifestations in a father and son with EEC syndrome. Graefe’s Arch Clin. Exp. Ophthalmol. 1997, 235, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Majewski, F.; Küster, W. EEC syndrome sine sine? Clin. Genet. 1988, 33, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.K.; Marazita, M.L.; Pathak, R.; Beever, C.L.; Cooper, M.E.; Goldstein, T.; Shaw, D.F.; Field, L.L. TP63 mutation and clefting modifier genes in an EEC syndrome family. Clin. Genet. 2004, 66, 217–222. [Google Scholar] [CrossRef]
- Mawhorter, L.G.; Ruttum, M.S.; Koenig, S.B. Keratopathy in a Family with the Ectrodactyly-Ectodermal Dysplasia-clefting Syndrome. Ophthalmology 1985, 92, 1427–1431. [Google Scholar] [CrossRef] [PubMed]
- Bigatà, X.; Bielsa, I.; Artigas, M.; Azon, A.; Ribera, M.; Ferrandiz, C. The Ectrodactyly-Ectodermal Dysplasia-Clefting Syndrome (EEC): Report of Five Cases. Pediatr. Dermatol. 2003, 20, 113–118. [Google Scholar] [CrossRef]
- Barbaro, V.; Bonelli, F.; Ferrari, S.; La Vella, G.; Di Iorio, E. Innovative therapeutic approaches for the treatment of the ocular morbidities in patients with EEC syndrome. Cells 2023, 12, 495. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Clinical Examination | Analysis Method |
|---|---|
| Anterior segment | Slit lamp (CSO SL9900 LED c/PC) |
| Epithelial transparency | |
| Epithelial defects | |
| Corneal neovascularization | |
| Corneal opacity | |
| Inflammation | |
| Presence of palisades of Vogt | |
| Presence of Meibomian glands | |
| Fundus | |
| Corneal pannus extension | |
| Visual acuity | Snellen test |
| Intraocular pressure (IOP) | ICare tonometer |
| Epithelial integrity | Slit lamp + Fluorescein staining (Grading Oxford) |
| Ocular dryness | Schirmer’s test Tear Break-Up time (TBUT) test |
| Corneal thickness | Anterior Segment Optical Coherence Tomography (AS-OCT CSO MS39) |
| Ocular Parameter | Clinical Significance | Assessment Method | Grading Scale |
|---|---|---|---|
| Epithelial Transparency | Decreased transparency may indicate limbal stem cell deficiency | Slit lamp examination (cobalt blue filter) with fluorescein dye | Grade 0: Normal, clear corneal epithelium. Grade 1: Mild epithelial haze with minimal symptoms and no or mild corneal staining. Grade 2: Moderate epithelial haze with moderate symptoms and mild-to-moderate corneal staining. Grade 3: Severe epithelial haze with severe symptoms and significant corneal staining or vision loss [16]. |
| Recurrent epithelial defects | Corneal epithelial defects can indicate limbal stem cell deficiency | Slit lamp examination (cobalt blue filter) with fluorescein dye | Grade 0: No recurrent epithelial defects. Grade 1: Superficial punctate keratitis or minor epithelial defects with minimal symptoms and no or mild corneal staining. Grade 2: Moderate recurrent epithelial defects with moderate symptoms and mild-to-moderate corneal staining. Grade 3: Severe recurrent epithelial defects with severe symptoms and significant corneal staining or erosion [16]. |
| Corneal neovascularization | Limbal stem cell deficiency can lead to corneal neovascularization | Slit lamp examination with white light | Grade 0: No neovascularization. Grade 1: Superficial neovascularization limited to the peripheral cornea (≤2 mm from the limbus). Grade 2: Superficial neovascularization extending beyond the peripheral cornea (>2 mm from the limbus) or deep stromal neovascularization limited to the peripheral cornea. Grade 3: Superficial neovascularization extending to the central cornea or deep stromal neovascularization extending beyond the peripheral cornea. Grade 4: Total corneal neovascularization, with or without associated corneal opacity [17]. |
| Corneal opacity | Limbal stem cell deficiency can lead to corneal opacification | Slit lamp examination with white light | McDonald–Shadduck grading system: This grading system is based on the amount of corneal surface area covered by the opacity and ranges from 0 to 4. The grading is as follows: Grade 0: No corneal opacity. Grade 1: Less than 25% of the cornea covered by opacity. Grade 2: 25–50% of the cornea covered by opacity. Grade 3: 50–75% of the cornea covered by opacity. Grade 4: More than 75% of the cornea covered by opacity [18]. |
| Inflammation | Inflammation can indicate limbal stem cell deficiency | Slit lamp examination with white light | This parameter can be evaluated using a scale from 0 to 4, with 0 being no inflammation and 4 being severe inflammation. |
| Bulbar hyperemia | Increased blood flow to the eye can indicate limbal stem cell deficiency | Slit lamp examination with white light | Efron Grading Scale Grade 0: No hyperemia. Grade 1: Slight hyperemia with some injection visible. Grade 2: Mild hyperemia with injection easily visible. Grade 3: Moderate hyperemia with pronounced injection. Grade 4: Severe hyperemia with very pronounced injection [19]. |
| Palpebral rim abnormalities | Abnormalities in the eyelid can indicate limbal stem cell deficiency | External examination | Grading for palpebral rim abnormalities based on the severity of the abnormality and the extent of the lid involvement: Grade 0: Normal palpebral rim with no abnormalities. Grade 1: Mild palpebral rim abnormalities, involving less than half of the lid margin, including minimal notching, mild entropion, and minor lash abnormalities. Grade 2: Moderate palpebral rim abnormalities, involving more than half of the lid margin; including moderate notching, moderate entropion, and moderate lash abnormalities. Grade 3: Severe palpebral rim abnormalities, involving the entire lid margin; including severe notching, severe entropion, and severe lash abnormalities [20]. |
| Corneal pannus | Limbal stem cell deficiency can lead to corneal pannus formation | Slit lamp examination with white light | Grading system for corneal pannus based on its location and extent: Grade 0: No pannus Grade 1: Peripheral pannus with 20–30% cornea affected. Grade 2: Mid- peripheral pannus with 30–50% cornea affected. Grade 3: Mid- peripheral pannus more than 50–70% cornea affected. Grade 4: Central pannus, more than 70–90% cornea affected [21]. |
| Case n | Age | Mutation | Eye | Palisades of Vogt | Meibomian/Lacrimal Glands | Epithelial Transparency [16] | Recurrent Epithelial Defects [16] | Corneal Neovascularization [17] | Corneal Opacity [18] | Inflammation [19] | Bulbar Hyperemia [20] | Palpebral Rim Abnormalities [21] | Corneal Pannus [22] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 6 | R279H | RE | + | Partial aplasia | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 |
| LE | + | Partial aplasia | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | |||
| 2 | 17 | L523P | RE | - | Absent | 1 | 1 | 2 | 1 | 2 | 2 | 3 | 1 |
| LE | - | Absent | 1 | 1 | 2 | 1 | 2 | 2 | 3 | 1 | |||
| 3 | 20 | L523P | RE | - | Absent | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 2 |
| LE | - | Absent | 2 | 2 | 2 | 2 | 2 | 3 | 3 | 2 | |||
| 4 | 31 | R279H | RE | - | Absent | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 |
| LE | - | Absent | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | |||
| 5 | 42 | R279H | RE | - | Partial aplasia | 2 | 2 | 3 | 3 | 3 | 4 | 1 | 3 |
| LE | - | Partial aplasia | 2 | 2 | 3 | 3 | 3 | 4 | 1 | 3 | |||
| 6 | 44 | R279C | RE | - | Absent | 2 | 2 | 3 | 2 | 1 | 1 | 2 | 2 |
| LE | - | Absent | 2 | 2 | 2 | 1 | 1 | 1 | 2 | 1 | |||
| 7 | 61 | R279H | RE | - | Absent | 3 | 3 | 4 | 4 | 1 | 1 | 3 | 4 |
| LE | - | Absent | 3 | 3 | 4 | 4 | 1 | 1 | 3 | 4 | |||
| 8 | 69 | R279C; R340G | RE * | - | Absent | 3 | 3 | 4 | 4 | 1 | 3 | 2 | 4 |
| LE | - | Absent | 2 | 2 | 1 | 1 | 1 | 1 | 2 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Iorio, E.; Bonelli, F.; Bievel-Radulescu, R.; Decastello, N.; Ferrari, S.; Barbaro, V.; Ponzin, D. Ocular Manifestations in Patients Affected by p63-Associated Disorders: Ectrodactyly-Ectodermal Dysplasia-Clefting (EEC) and Ankyloblepharon-Ectodermal Defects-Cleft Lip Palate (AEC) Syndromes. J. Clin. Med. 2023, 12, 7377. https://doi.org/10.3390/jcm12237377
Di Iorio E, Bonelli F, Bievel-Radulescu R, Decastello N, Ferrari S, Barbaro V, Ponzin D. Ocular Manifestations in Patients Affected by p63-Associated Disorders: Ectrodactyly-Ectodermal Dysplasia-Clefting (EEC) and Ankyloblepharon-Ectodermal Defects-Cleft Lip Palate (AEC) Syndromes. Journal of Clinical Medicine. 2023; 12(23):7377. https://doi.org/10.3390/jcm12237377
Chicago/Turabian StyleDi Iorio, Enzo, Filippo Bonelli, Raluca Bievel-Radulescu, Nicolò Decastello, Stefano Ferrari, Vanessa Barbaro, and Diego Ponzin. 2023. "Ocular Manifestations in Patients Affected by p63-Associated Disorders: Ectrodactyly-Ectodermal Dysplasia-Clefting (EEC) and Ankyloblepharon-Ectodermal Defects-Cleft Lip Palate (AEC) Syndromes" Journal of Clinical Medicine 12, no. 23: 7377. https://doi.org/10.3390/jcm12237377