
Molecular Genetic Approach and Evaluation of Cardiovascular Events in Patients with Clinical Familial Hypercholesterolemia Phenotype from Romania

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Clinical and Biological Evaluation of FH Patients
2.3. Evaluation of the New Cardiovascular Events
- electrocardiogram (ECG) for ischemic changes assessment;
- ankle–brachial index (ABI) measurement with a sphygmomanometer and a portable ultrasonography device for determining sounds that detect systolic blood pressure in the lower limbs; the reference ABI values were between 0.9 and 1.3;
- echocardiography (Siemens Acuson CV70 Cardiac Vascular Ultrasound Machine), highlighting left ventricular (LV) wall motion abnormalities and ejection fraction values, important predictors of left ventricular systolic dysfunction;
- measurement of carotid intima–media thickness (cIMT) (at the levels of the carotid bifurcation, internal, external, right and left carotid arteries) by using Siemens Acuson CV70 Cardiac Vascular Ultrasound Machine, B-mode and color Doppler ultrasound (5–10 MHz). The average of the cIMT (the average of the six quantified segments) was also recorded. The reference cIMT values were under 0.9 mm [26,27].
2.4. Evaluation of the Mutations in the LDLR, APOB, and PCSK9 Genes
2.4.1. DNA Genomic Extraction
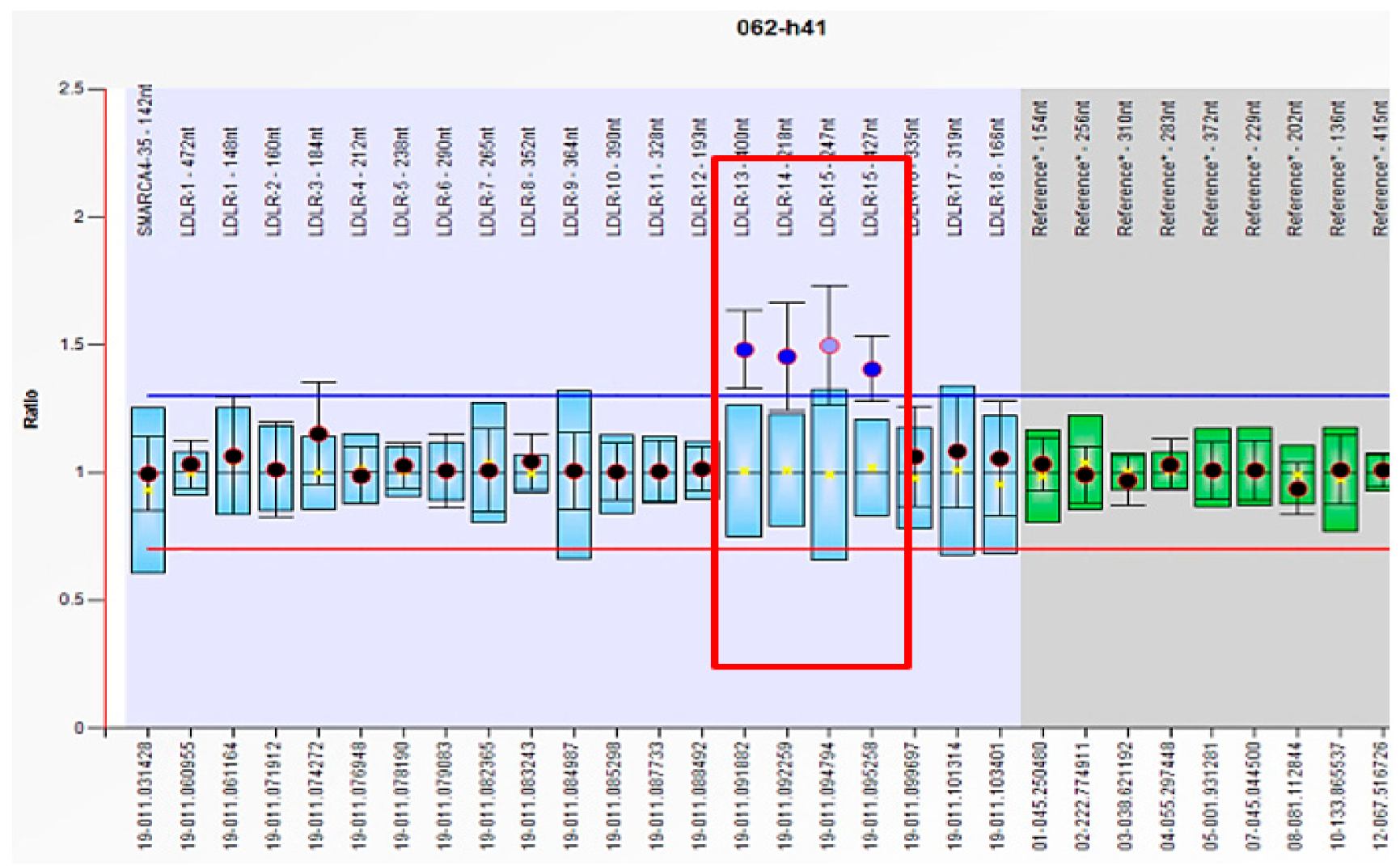
2.4.2. MLPA (P062, LDLR MLPA Kit, MRC Holland, Amsterdam, Netherlands)
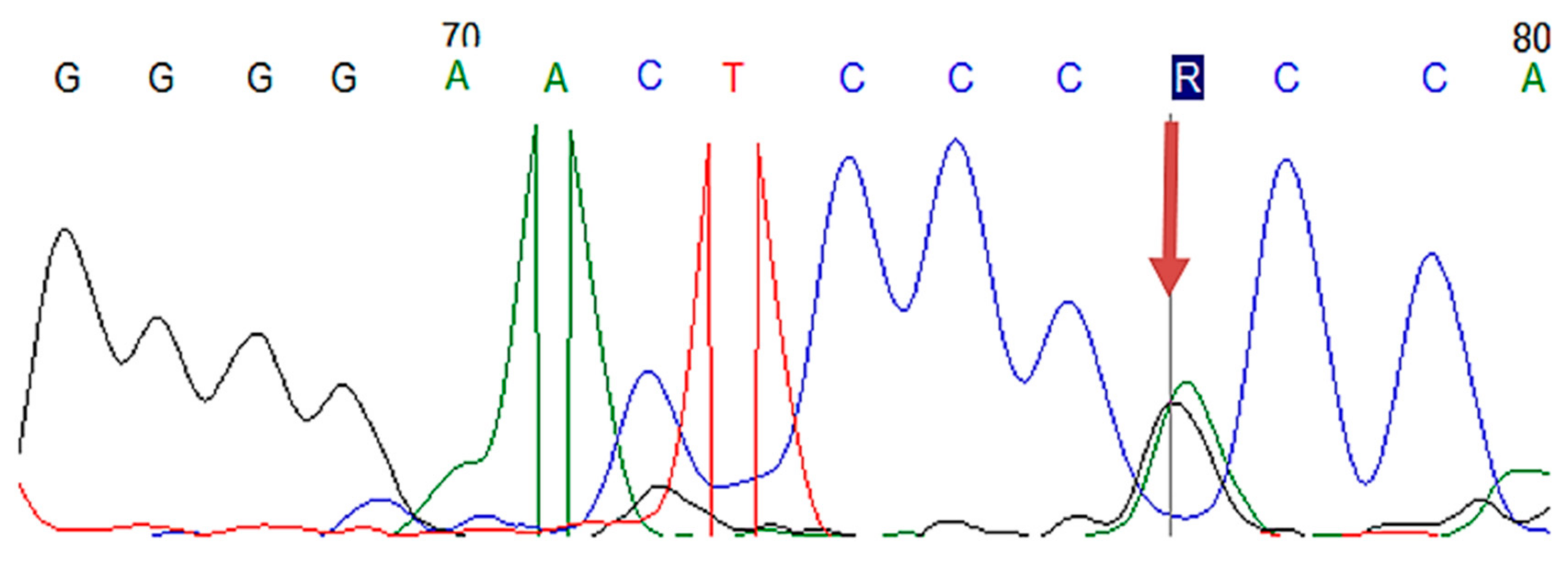
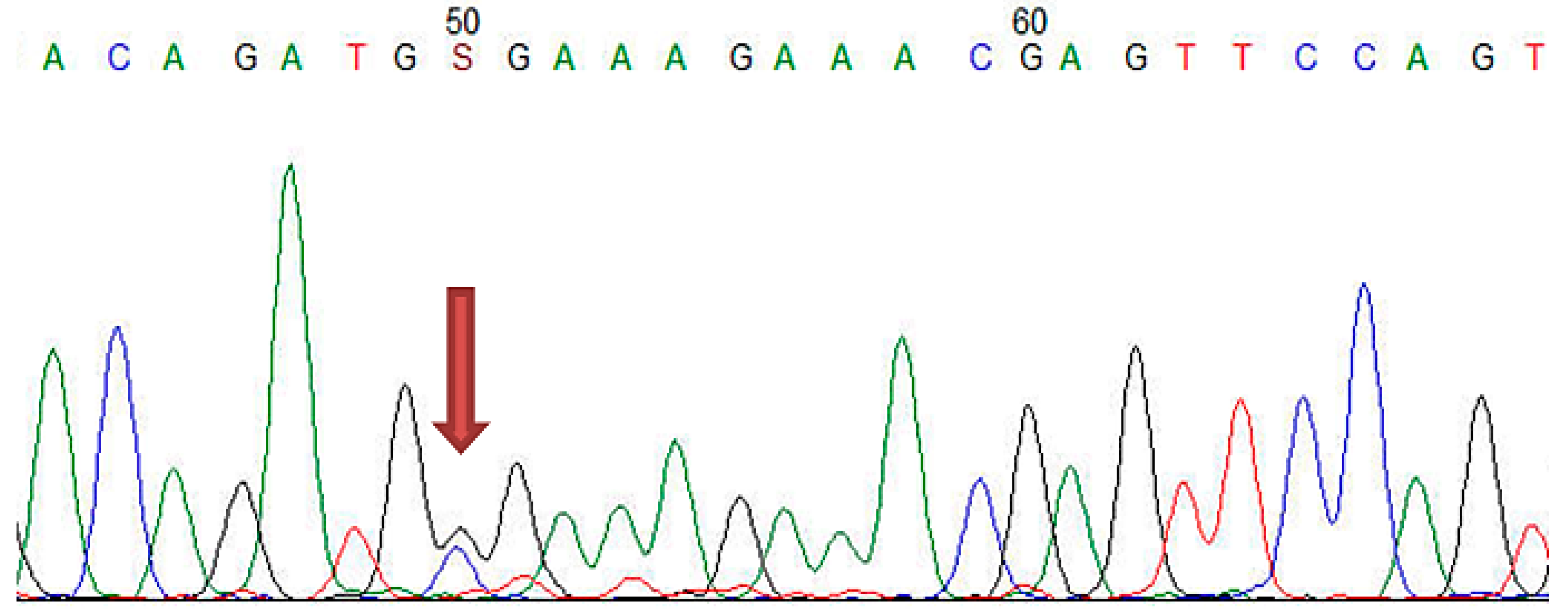
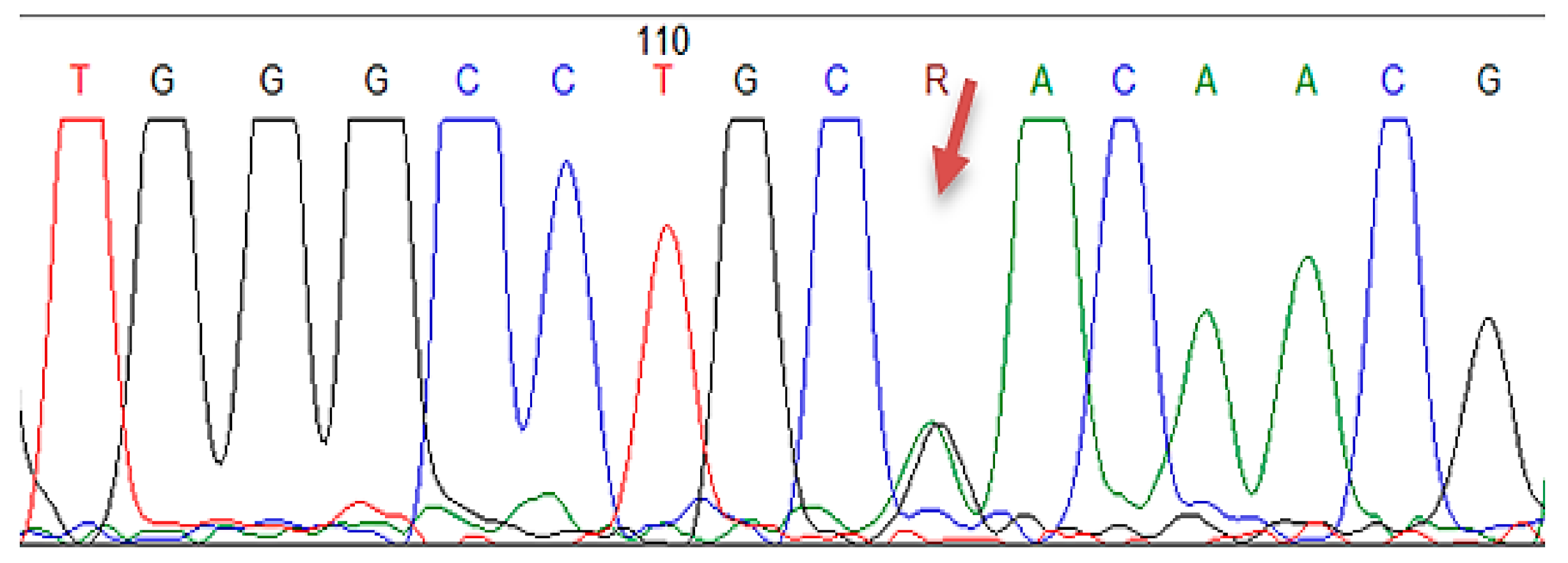
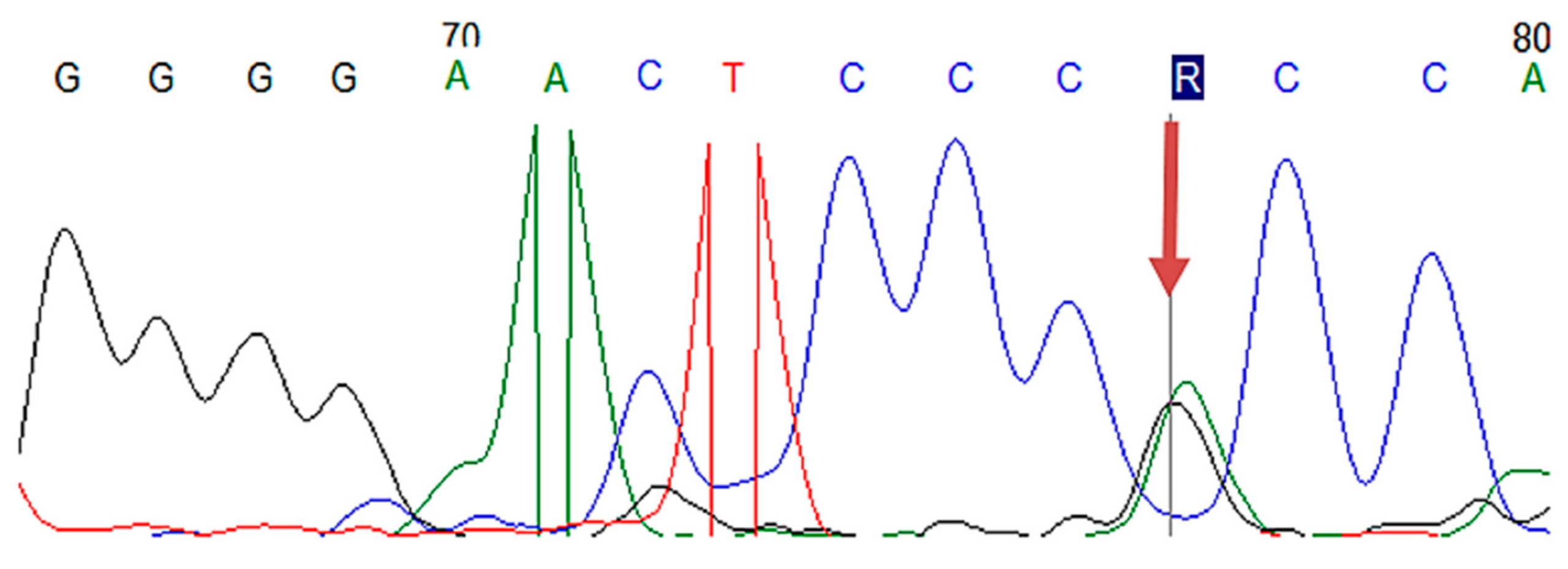
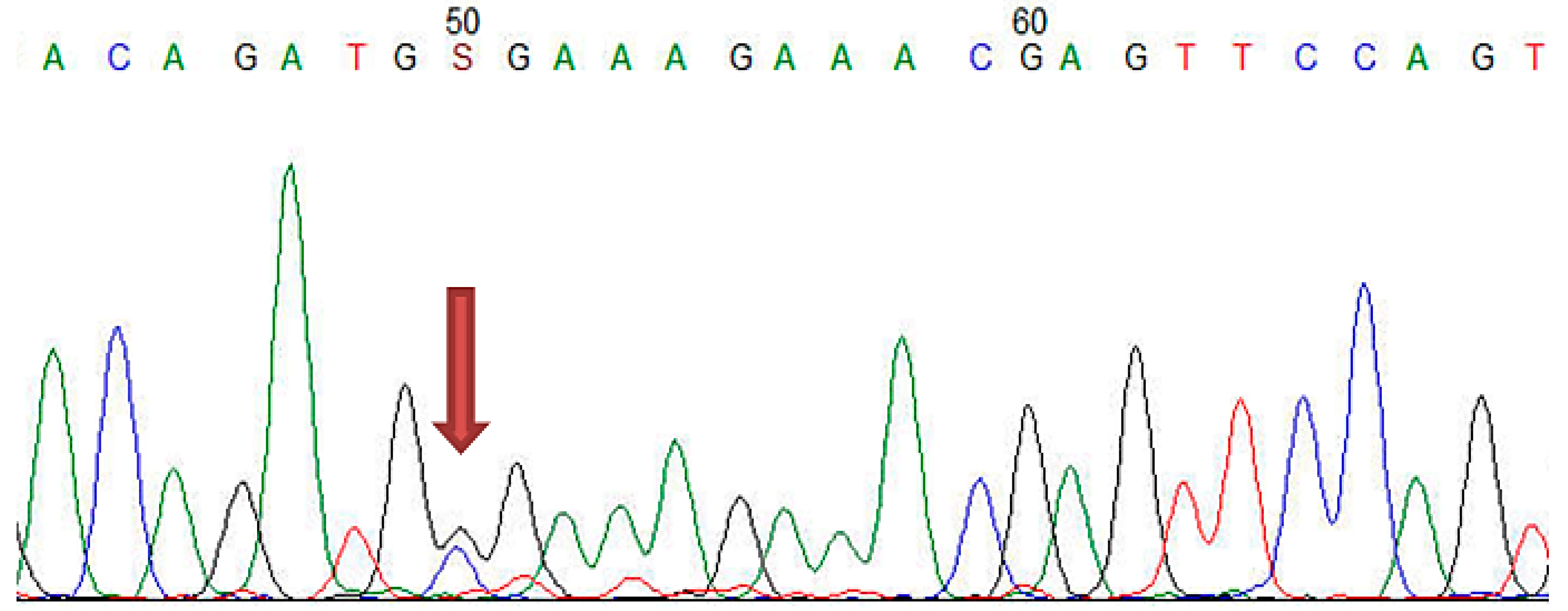
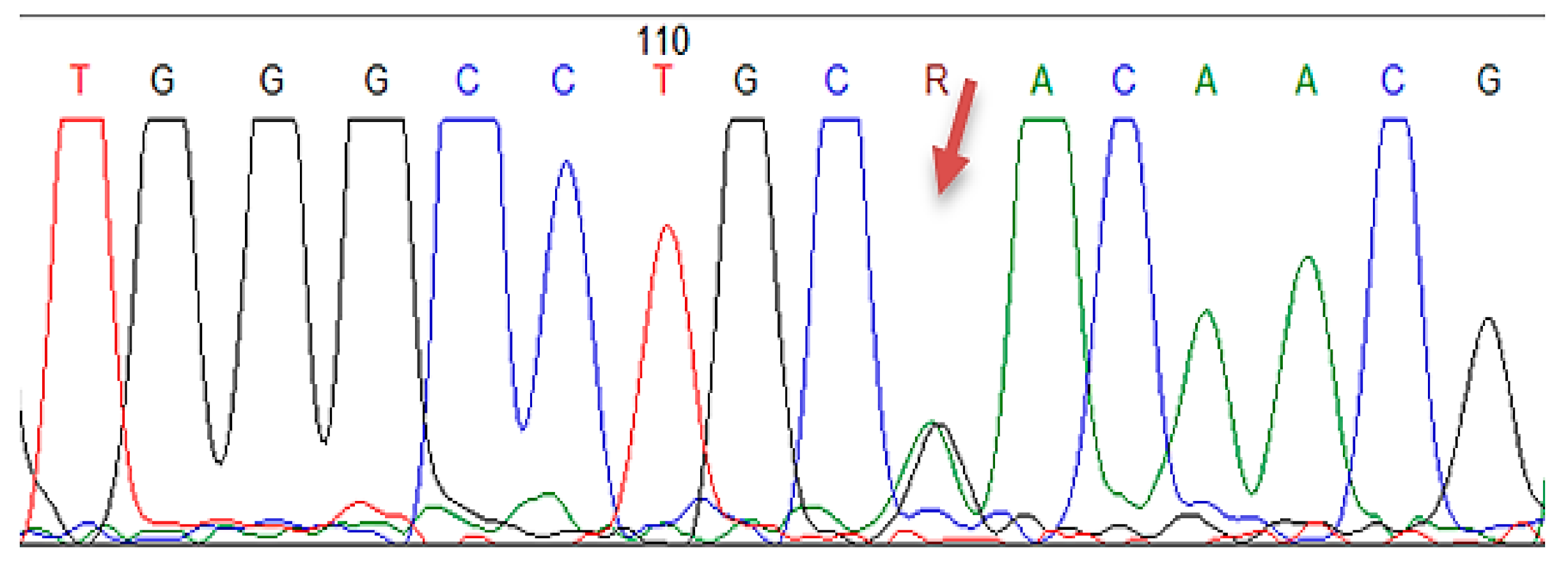
2.4.3. Sanger Sequencing
2.5. Statistical Analysis
3. Results
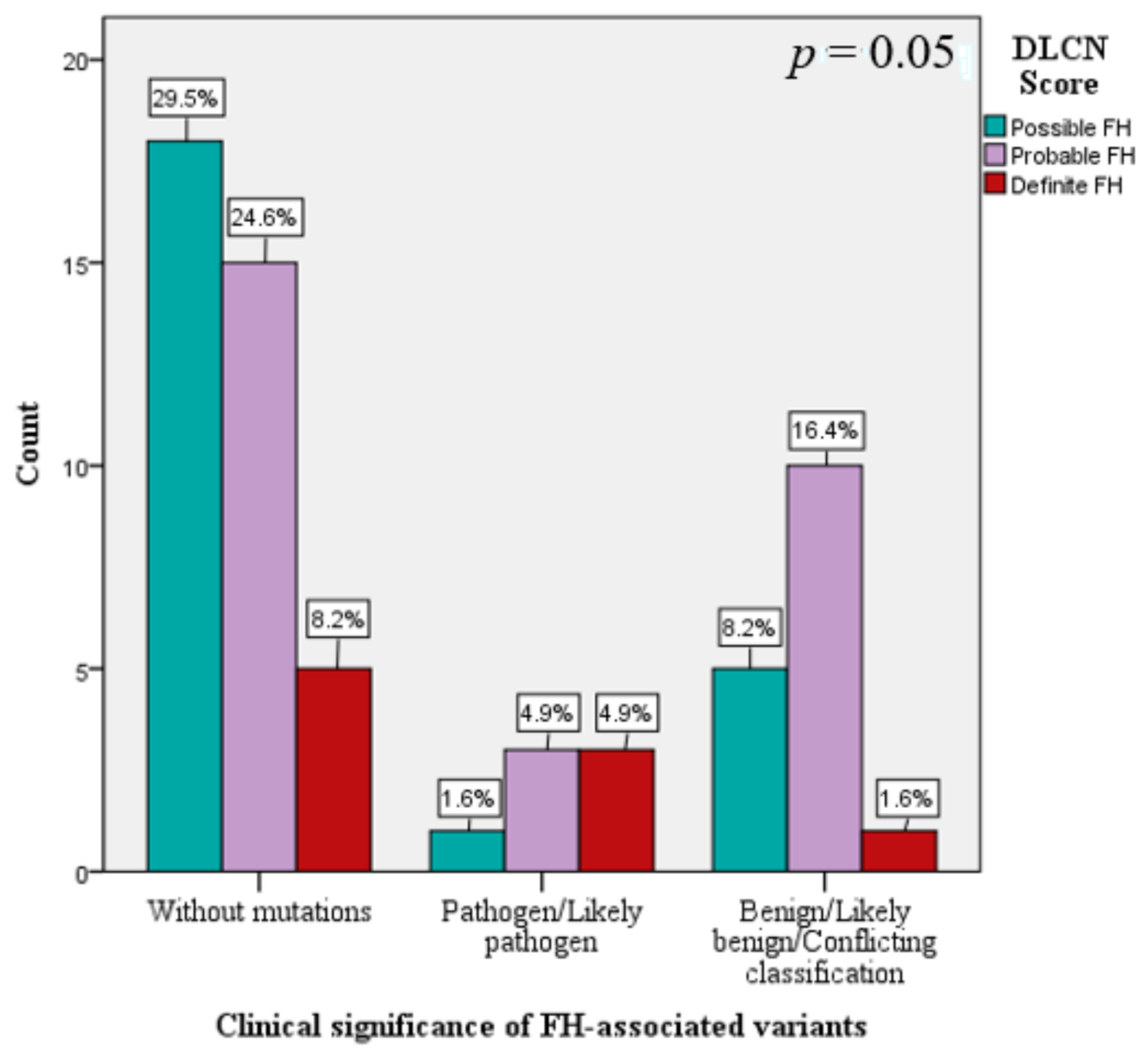
3.1. The Genetic Spectrum of FH in Romania
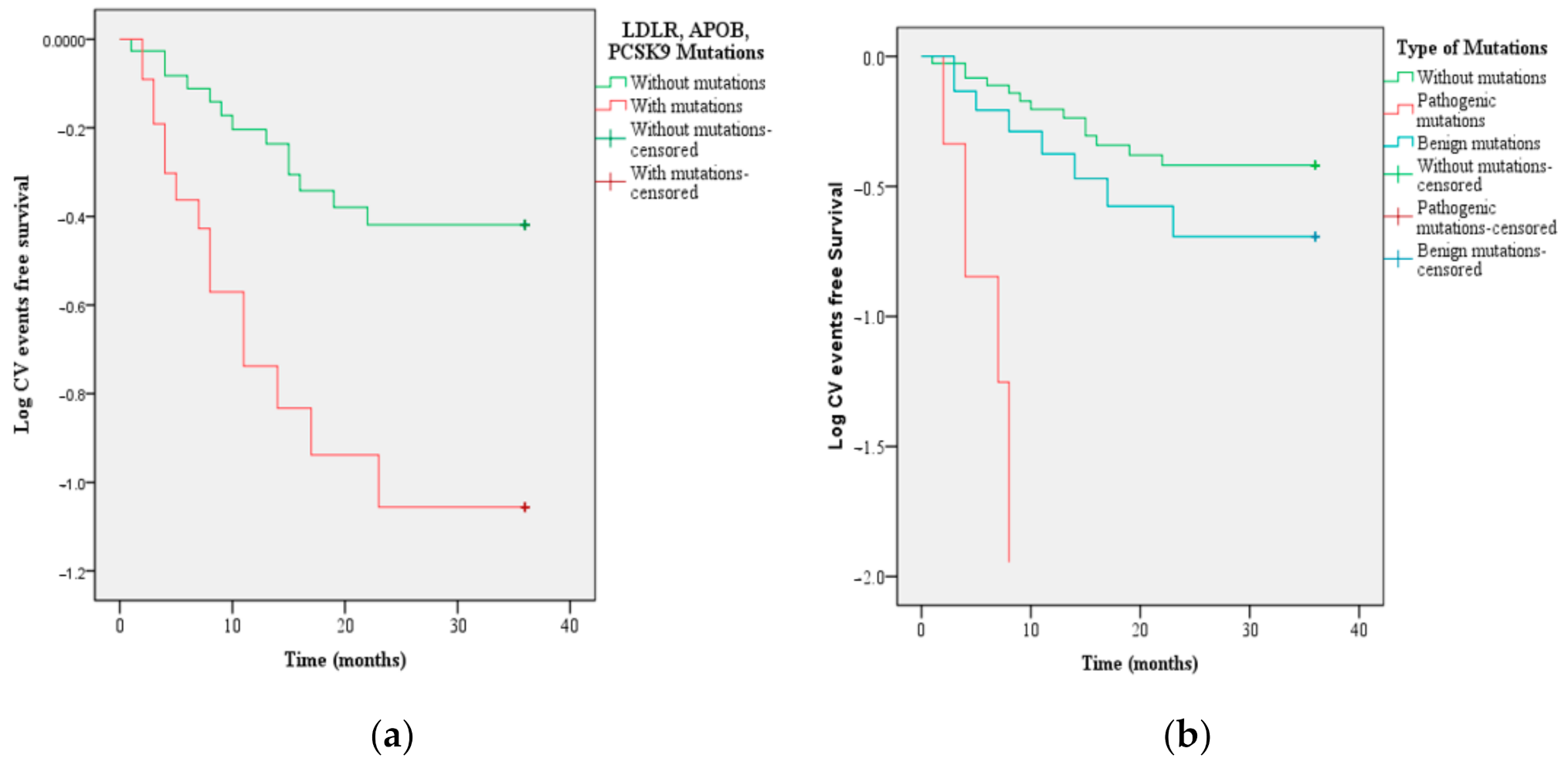
3.2. The New ASCVD in Patients with FH Based on LDLR, APOB, and PCSK9 Mutations
4. Discussion
Study Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trinder, M.; Li, X.; DeCastro, M.L.; Cermakova, L.; Sadananda, S.; Jackson, L.M.; Azizi, H.; Mancini, G.B.J.; Francis, G.A.; Frohlich, J.; et al. Risk of Premature Atherosclerotic Disease in Patients With Monogenic Versus Polygenic Familial Hypercholesterolemia. J. Am. Coll. Cardiol. 2019, 74, 512–522. [Google Scholar] [CrossRef]
- Bertolini, S.; Pisciotta, L.; Rabacchi, C.; Cefalu, A.B.; Noto, D.; Fasano, T.; Signori, A.; Fresa, R.; Averna, M.; Calandra, S. Spectrum of mutations and phenotypic expression in patients with autosomal dominant hypercholesterolemia identified in Italy. Atherosclerosis 2013, 227, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Gabcova, D.; Vohnout, B.; Stanikova, D.; Huckova, M.; Kadurova, M.; Debreova, M.; Kozarova, M.; Fabryova, L.; Stanik, J.; Klimes, I.; et al. The molecular genetic background of familial hypercholesterolemia: Data from the Slovak nation-wide survey. Physiol. Res. 2017, 66, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Jannes, C.E.; Santos, R.D.; de Souza Silva, P.R.; Turolla, L.; Gagliardi, A.C.; Marsiglia, J.D.; Chacra, A.P.; Miname, M.H.; Rocha, V.Z.; Filho, W.S.; et al. Familial hypercholesterolemia in Brazil: Cascade screening program, clinical and genetic aspects. Atherosclerosis 2015, 238, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Cymbron, T.; Mendes, P.; Ramos, A.; Raposo, M.; Kazachkova, N.; Medeiros, A.M.; Bruges-Armas, J.; Bourbon, M.; Lima, M. Familial hypercholesterolemia: Molecular characterization of possible cases from the Azores Islands (Portugal). Meta Gene 2014, 2, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Validation of LDLr Activity as a Tool to Improve Genetic Diagnosis of Familial Hypercholesterolemia: A Retrospective on Functional Characterization of LDLr Variants. Int. J. Mol. Sci. 2018, 19, 1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paththinige, C.S.; Rajapakse, J.; Constantine, G.R.; Sem, K.P.; Singaraja, R.R.; Jayasekara, R.W.; Dissanayake, V.H.W. Spectrum of low-density lipoprotein receptor (LDLR) mutations in a cohort of Sri Lankan patients with familial hypercholesterolemia-A preliminary report. Lipids Health Dis. 2018, 17, 100. [Google Scholar] [CrossRef] [Green Version]
- Ekrami, M.; Torabi, M.; Ghafouri-Fard, S.; Mowla, J.; Mohammad Soltani, B.; Hashemi-Gorji, F.; Mohebbi, Z.; Miryounesi, M. Genetic Analysis of Iranian Patients with Familial Hypercholesterolemia. Iran. Biomed. J. 2018, 22, 117–122. [Google Scholar]
- Sharifi, M.; Walus-Miarka, M.; Idzior-Walus, B.; Malecki, M.T.; Sanak, M.; Whittall, R.; Li, K.W.; Futema, M.; Humphries, S.E. The genetic spectrum of familial hypercholesterolemia in south-eastern Poland. Metabolism 2016, 65, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Al-Khateeb, A.; Zahri, M.K.; Mohamed, M.S.; Sasongko, T.H.; Ibrahim, S.; Yusof, Z.; Zilfalil, B.A. Analysis of sequence variations in low-density lipoprotein receptor gene among Malaysian patients with familial hypercholesterolemia. BMC Med. Genet. 2011, 12, 40. [Google Scholar] [CrossRef] [Green Version]
- Faiz, F.; Allcock, R.J.; Hooper, A.J.; van Bockxmeer, F.M. Detection of variations and identifying genomic breakpoints for large deletions in the LDLR by Ion Torrent semiconductor sequencing. Atherosclerosis 2013, 230, 249–255. [Google Scholar] [CrossRef]
- Rahman, T.; Hamzan, N.S.; Mokhsin, A.; Rahmat, R.; Ibrahim, Z.O.; Razali, R.; Thevarajah, M.; Nawawi, H. Enhanced status of inflammation and endothelial activation in subjects with familial hypercholesterolaemia and their related unaffected family members: A case control study. Lipids Health Dis. 2017, 16, 81. [Google Scholar] [CrossRef] [Green Version]
- Ruscica, M.; Corsini, A.; Ferri, N.; Banach, M.; Sirtori, C.R. Clinical approach to the inflammatory etiology of cardiovascular diseases. Pharmacol. Res. 2020, 159, 104916. [Google Scholar] [CrossRef]
- Jeenduang, N.; Promptmas, C.; Pongrapeeporn, K.S.; Porntadavity, S. Molecular modeling of D151Y and M391T mutations in the LDL receptor. Biochem. Biophys. Res. Commun. 2008, 377, 355–360. [Google Scholar] [CrossRef]
- Maurer, F.; Pradervand, S.; Guilleret, I.; Nanchen, D.; Maghraoui, A.; Chapatte, L.; Bojkowska, K.; Bhuiyan, Z.A.; Jacquemont, N.; Harshman, K.; et al. Identification and molecular characterisation of Lausanne Institutional Biobank participants with familial hypercholesterolaemia-A proof-of-concept study. Swiss Med. Wkly. 2016, 146, w14326. [Google Scholar] [CrossRef]
- Mozas, P.; Castillo, S.; Tejedor, D.; Reyes, G.; Alonso, R.; Franco, M.; Saenz, P.; Fuentes, F.; Almagro, F.; Mata, P.; et al. Molecular characterization of familial hypercholesterolemia in Spain: Identification of 39 novel and 77 recurrent mutations in LDLR. Hum. Mutat. 2004, 24, 187. [Google Scholar] [CrossRef]
- Blanchard, V.; Khantalin, I.; Ramin-Mangata, S.; Chemello, K.; Nativel, B.; Lambert, G. PCSK9: From biology to clinical applications. Pathology 2019, 51, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Seidah, N.G.; Benjannet, S.; Wickham, L.; Marcinkiewicz, J.; Jasmin, S.B.; Stifani, S.; Basak, A.; Prat, A.; Chretien, M. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): Liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 928–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macchi, C.; Banach, M.; Corsini, A.; Sirtori, C.R.; Ferri, N.; Ruscica, M. Changes in circulating pro-protein convertase subtilisin/kexin type 9 levels-Experimental and clinical approaches with lipid-lowering agents. Eur. J. Prev. Cardiol. 2019, 26, 930–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlad, C.E.; Foia, L.; Popescu, R.; Ivanov, I.; Luca, M.C.; Delianu, C.; Toma, V.; Statescu, C.; Rezus, C.; Florea, L. Apolipoproteins A and B and PCSK9: Nontraditional Cardiovascular Risk Factors in Chronic Kidney Disease and in End-Stage Renal Disease. J. Diabetes Res. 2019, 2019, 6906278. [Google Scholar] [CrossRef] [PubMed]
- Rogacev, K.S.; Heine, G.H.; Silbernagel, G.; Kleber, M.E.; Seiler, S.; Emrich, I.; Lennartz, S.; Werner, C.; Zawada, A.M.; Fliser, D.; et al. PCSK9 Plasma Concentrations Are Independent of GFR and Do Not Predict Cardiovascular Events in Patients with Decreased GFR. PLoS ONE 2016, 11, e0146920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Primers 2017, 3, 17093. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019, 290, 140–205. [Google Scholar] [CrossRef] [Green Version]
- Perez de Isla, L.; Alonso, R.; Mata, N.; Fernandez-Perez, C.; Muniz, O.; Diaz-Diaz, J.L.; Saltijeral, A.; Fuentes-Jimenez, F.; de Andres, R.; Zambon, D.; et al. Predicting Cardiovascular Events in Familial Hypercholesterolemia: The SAFEHEART Registry (Spanish Familial Hypercholesterolemia Cohort Study). Circulation 2017, 135, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.P.; Ahmed, K.Z.; Yaqub, Z.; Ghani, R. Carotid intima-media thickness correlation with lipid profile in patients with familial hypercholesterolemia versus controls. J. Coll. Physicians Surg. Pak. 2011, 21, 30–33. [Google Scholar] [PubMed]
- Sivapalaratnam, S.; van Loendersloot, L.L.; Hutten, B.A.; Kastelein, J.J.; Trip, M.D.; de Groot, E. Long-term LDL-c lowering in heterozygous familial hypercholesterolemia normalizes carotid intima-media thickness. Atherosclerosis 2010, 212, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Resmerita, I.; Cozma, R.S.; Popescu, R.; Radulescu, L.M.; Panzaru, M.C.; Butnariu, L.I.; Caba, L.; Ilie, O.D.; Gavril, E.C.; Gorduza, E.V.; et al. Genetics of Hearing Impairment in North-Eastern Romania-A Cost-Effective Improved Diagnosis and Literature Review. Genes 2020, 11, 1506. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Mollaki, V.; Progias, P.; Drogari, E. Familial Hypercholesterolemia in Greek children and their families: Genotype-to-phenotype correlations and a reconsideration of LDLR mutation spectrum. Atherosclerosis 2014, 237, 798–804. [Google Scholar] [CrossRef]
- Iacocca, M.A.; Chora, J.R.; Carrie, A.; Freiberger, T.; Leigh, S.E.; Defesche, J.C.; Kurtz, C.L.; DiStefano, M.T.; Santos, R.D.; Humphries, S.E.; et al. ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum. Mutat. 2018, 39, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information ClinVar. 2020. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 19 February 2021).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Yamamura, T.; Sakai, N.; Miyata, T.; Kokubo, Y.; Yamamoto, A. Update of Japanese common LDLR gene mutations and their phenotypes: Mild type mutation L547V might predominate in the Japanese population. Atherosclerosis 2009, 203, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Tosi, I.; Toledo-Leiva, P.; Neuwirth, C.; Naoumova, R.P.; Soutar, A.K. Genetic defects causing familial hypercholesterolaemia: Identification of deletions and duplications in the LDL-receptor gene and summary of all mutations found in patients attending the Hammersmith Hospital Lipid Clinic. Atherosclerosis 2007, 194, 102–111. [Google Scholar] [CrossRef]
- Futema, M.; Plagnol, V.; Whittall, R.A.; Neil, H.A.; Simon Broome Register, G.; Humphries, S.E. Uk10K Use of targeted exome sequencing as a diagnostic tool for Familial Hypercholesterolaemia. J. Med. Genet. 2012, 49, 644–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, N.; Binder, G.; Keller, C. Mutations in the low-density-lipoprotein receptor gene in German patients with familial hypercholesterolaemia. J. Inherit. Metab. Dis. 2000, 23, 778–790. [Google Scholar] [CrossRef]
- Dedoussis, G.V.; Skoumas, J.; Pitsavos, C.; Choumerianou, D.M.; Genschel, J.; Schmidt, H.; Stefanadis, C. FH clinical phenotype in Greek patients with LDL-R defective vs. negative mutations. Eur. J. Clin. Investig. 2004, 34, 402–409. [Google Scholar] [CrossRef]
- Komarova, T.Y.; Korneva, V.A.; Kuznetsova, T.Y.; Golovina, A.S.; Vasilyev, V.B.; Mandelshtam, M.Y. Familial hypercholesterolemia mutations in Petrozavodsk: No similarity to St. Petersburg mutation spectrum. BMC Med. Genet. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Zakharova, F.M.; Damgaard, D.; Mandelshtam, M.Y.; Golubkov, V.I.; Nissen, P.H.; Nilsen, G.G.; Stenderup, A.; Lipovetsky, B.M.; Konstantinov, V.O.; Denisenko, A.D.; et al. Familial hypercholesterolemia in St-Petersburg: The known and novel mutations found in the low density lipoprotein receptor gene in Russia. BMC Med. Genet. 2005, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Gorski, B.; Kubalska, J.; Naruszewicz, M.; Lubinski, J. LDL-R and Apo-B-100 gene mutations in Polish familial hypercholesterolemias. Hum. Genet. 1998, 102, 562–565. [Google Scholar] [CrossRef]
- Lopez, G.; Bernal, L.M.; Gelvez, N.; Gomez, L.F.; Nova, A.; Sanchez, A.I.; Tamayo, M.L. Mutational analysis of the LDLR gene in a cohort of Colombian families with familial hypercholesterolemia. Atherosclerosis 2018, 277, 434–439. [Google Scholar] [CrossRef]
- Al-Allaf, F.A.; Athar, M.; Abduljaleel, Z.; Bouazzaoui, A.; Taher, M.M.; Own, R.; Al-Allaf, A.F.; AbuMansour, I.; Azhar, Z.; Ba-Hammam, F.A.; et al. Identification of a novel nonsense variant c.1332dup, p.(D445*) in the LDLR gene that causes familial hypercholesterolemia. Hum. Genome Var. 2014, 1, 14021. [Google Scholar] [CrossRef] [Green Version]
- Chiou, K.R.; Charng, M.J.; Chang, H.M. Array-based resequencing for mutations causing familial hypercholesterolemia. Atherosclerosis 2011, 216, 383–389. [Google Scholar] [CrossRef]
- Day, I.N.; Whittall, R.A.; O’Dell, S.D.; Haddad, L.; Bolla, M.K.; Gudnason, V.; Humphries, S.E. Spectrum of LDL receptor gene mutations in heterozygous familial hypercholesterolemia. Hum. Mutat. 1997, 10, 116–127. [Google Scholar] [CrossRef]
- Etxebarria, A.; Benito-Vicente, A.; Stef, M.; Ostolaza, H.; Palacios, L.; Martin, C. Activity-associated effect of LDL receptor missense variants located in the cysteine-rich repeats. Atherosclerosis 2015, 238, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Tichy, L.; Fajkusova, L.; Zapletalova, P.; Schwarzova, L.; Vrablik, M.; Freiberger, T. Molecular genetic background of an autosomal dominant hypercholesterolemia in the Czech Republic. Physiol. Res. 2017, 66, S47–S54. [Google Scholar] [CrossRef] [PubMed]
- Lelli, N.; Ghisellini, M.; Calandra, S.; Gaddi, A.; Ciarrocchi, A.; Coviello, D.A.; Bertolini, S. Duplication of exons 13, 14 and 15 of the LDL-receptor gene in a patient with heterozygous familial hypercholesterolemia. Hum. Genet. 1991, 86, 359–362. [Google Scholar] [CrossRef]
- Medeiros, A.M.; Alves, A.C.; Bourbon, M. Mutational analysis of a cohort with clinical diagnosis of familial hypercholesterolemia: Considerations for genetic diagnosis improvement. Genet. Med. 2016, 18, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Sampietro, T.; Pieroni, S.; Molinaro, S.; Sbrana, F.; Dal Pino, B.; Bigazzi, F.; Ruscica, M.; Sirtori, C.R.; Franchini, M. Inherited atherogenic dyslipidemias: Are they correctly reported? Eur. J. Prev. Cardiol. 2020, 12, 2047487320930308. [Google Scholar] [CrossRef]
- Greco, M.F.; Sirtori, C.R.; Corsini, A.; Ezhov, M.; Sampietro, T.; Ruscica, M. Lipoprotein(a) Lowering-From Lipoprotein Apheresis to Antisense Oligonucleotide Approach. J. Clin. Med. 2020, 9, 2103. [Google Scholar] [CrossRef]
- Vlad, C.E.; Foia, L.; Florea, L.; Costache, I.I.; Covic, A.; Popescu, R.; Reurean-Pintilei, D.; Covic, A. Evaluation of cardiovascular risk factors in patients with familial hypercholesterolemia from the North-Eastern area of Romania. Lipids Health Dis. 2021, 20, 4. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Patients with FH | ||||
|---|---|---|---|---|---|
| Overall | Without Mutation | Benign/Likely Benign Mutation | Pathogenic/Likely Pathogenic Mutation | p | |
| n | 61 | 38 | 16 | 7 | |
| Age—yo (mean ± SD) | 48.4 ± 12.5 | 50.3 ± 11.6 | 43.4 ± 13.6 | 50.1 ± 13.4 | 0.18 |
| Gender (male) n (%) | 22 (36.1%) | 14 (36.8%) | 5 (31.3%) | 3 (42.9%) | 0.86 |
| Smoker n (%) | 18 (29.5 %) | 12 (31.6%) | 5 (31.3%) | 1(14.3%) | 0.64 |
| High blood pressure n (%) | 31 (50.8 %) | 22 (57.9%) | 6 (37.5%) | 3(42.9%) | 0.35 |
| CHD history n (%) | 13 (21.3%) | 10 (26.3%) | 3 (18.8%) | 0 | 0.06 |
| PAD history n (%) | 9 (14.8%) | 7 (18.4%) | 2 (12.5%) | 0 | |
| CHD + PAD history n (%) | 14 (23%) | 6 (15.8%) | 3 (18.8%) | 5 (71.4%) | |
| Obesity n (%) | 22 (36.1%) | 14(36.8%) | 4 (25%) | 4 (57.4%) | 0.33 |
| Type 2 diabetes n (%) | 8 (13.1%) | 5 (13.2%) | 2 (12.5%) | 1 (14.3%) | 0.99 |
| Physical inactivity n (%) | 30 (49.2%) | 21 (55.3%) | 5 (31.3%) | 4 (57.1%) | 0.25 |
| TC mg/dL (median ± IQR) | 315 ± 56 | 307.5 ± 44 | 320 ± 41 | 353 ± 206 | 0.02 * |
| LDL–C mg/dL (mean ± SD) | 254.2 ± 53 | 246.2 ± 46.2 | 255.4 ± 46.5 | 294.7 ± 85.1 | 0.31 |
| HDL–C mg/dL (median ± IQR) | 45.8 ± 18 | 45 ± 12.3 | 48.5 ± 16.1 | 39 ± 16.2 | 0.28 |
| TG mg/dL (mean ± SD) | 174.4 ± 92 | 179.5 ± 92.2 | 160.6 ± 102.3 | 178.9 ± 74.2 | 0.61 |
| hsCRP mg/L (mean ± SD) | 5.85 ± 2.29 | 5.8 ± 2.2 | 6.3 ± 2.2 | 7.4 ± 2.4 | 0.26 |
| ECG changes n (%) | 25 (41%) | 13 (34.2%) | 7 (43.8%) | 5 (71.4%) | 0.18 |
| EF % (mean ± SD) | 53.2 ± 9.8 | 53.8 ± 9.4 | 54.1 ± 9.6 | 47.6 ± 11.8 | 0.35 |
| ABI (mean ± SD) | 0.96 ± 0.93 | 0.85 ± 0.07 | 0.85 ± 0.08 | 0.77 ± 0.11 | 0.15 |
| cIMT mm (mean ± SD) | 0.95 ± 0.33 | 0.91 ± 0.32 | 0.93 ± 0.36 | 1.21 ± 0.31 | 0.09 |
| Lipid-Lowering Agents | |||||
| Statin n (%) | 22 (36.1%) | 14 (36.8%) | 6 (37.5%) | 2 (28.6%) | 0.41 |
| Statin + ezetimibe n (%) | 18 (29.5%) | 9 (23.7%) | 7 (43.8%) | 2 (28.6%) | |
| Statin + fenofibrate n (%) | 8 (13.1 %) | 7 (18.4%) | 1 (6.3%) | 0 | |
| Statin + ezetimibe + fenofibrate n (%) | 13 (21.3%) | 8 (21.1%) | 2 (12.5%) | 3 (42.9%) | |
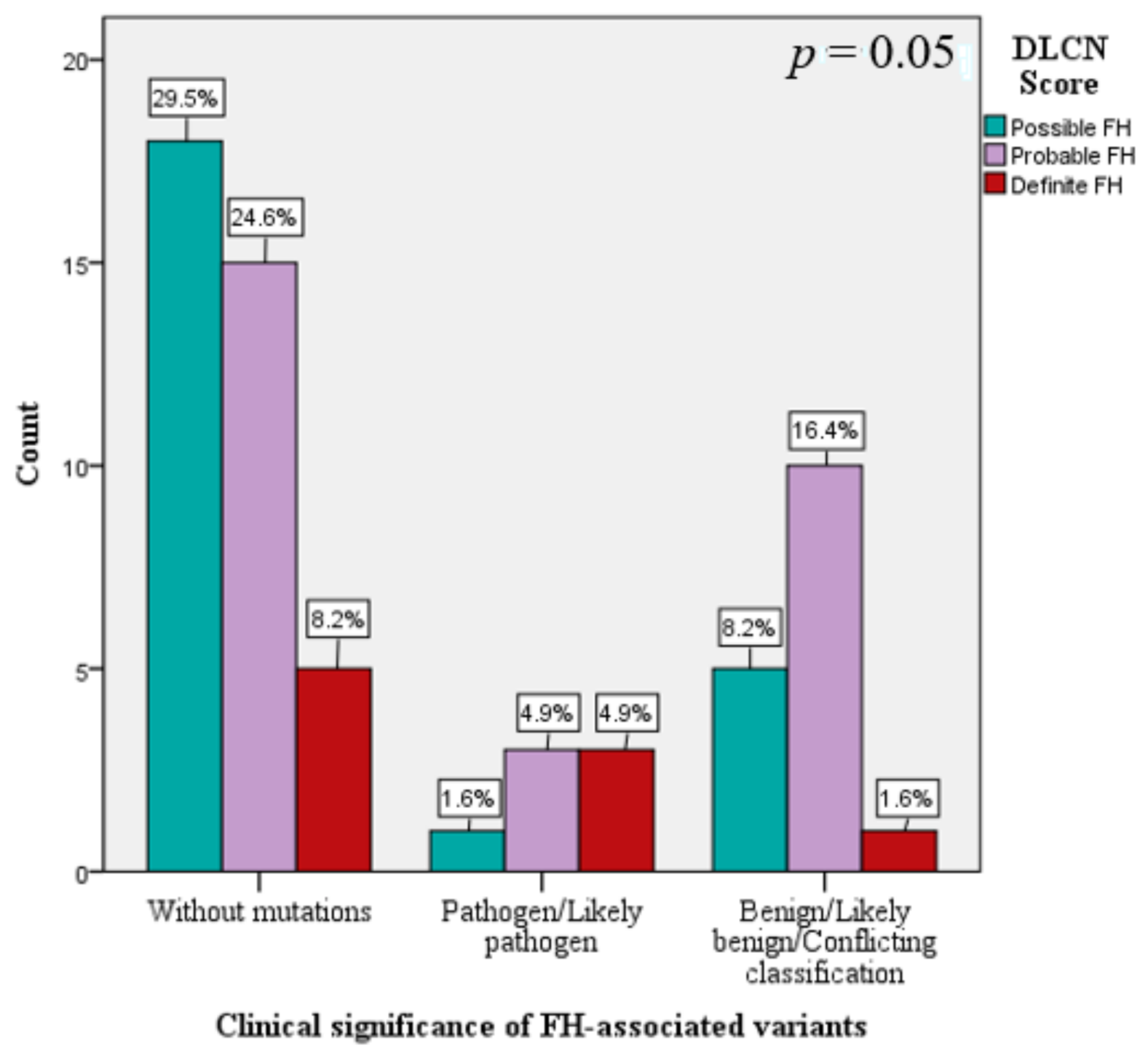
| DLCN Score (mean ± SD) | 6.4 ± 2.9 | 5.9 ± 2.5 | 6.2 ± 1.9 | 9.6 ± 4.9 | 0.02 * |
| Gene | Location | Nucleotide Change | Protein Change | Number of Carriers |
|---|---|---|---|---|
| Pathogenic Variants | ||||
| LDLR | Exon 2 | c.81C > G | p.(Cys27Trp) | 2 |
| LDLR | Exon 4 | c.502G > A | p.(Asp168Asn) | 1 |
| LDLR | Exon 11 | c.1618G > A | p.(Ala540Thr) | 3 |
| LDLR | Exon 13–15 | c.(1845+1_1846-1)_(2311+1_2312-1)dup | p (?) | 1 |
| Benign Variants | ||||
| LDLR | Exon 2 | c.81C > T | p.(Cys27=) | 3 |
| LDLR | Exon 10 | c.1413A > G | p.(Arg471=) | 15 |
| LDLR | Exon 11 | c.1617C > T | p.(Pro539=) | 3 |
| LDLR | Exon 12 | c.1773C > T | p.(Asn591=) | 12 |
| LDLR | Exon 13 | c.1959T > C | p.(Val653=) | 12 |
| LDLR | Exon 15 | c.2232A > G | p.(Arg744=) | 14 |
| APOB | Exon 26 | c.10740C > T | p.(Asn3580=) | 3 |
| Conflicting interpretations | ||||
| LDLR | Exon 3 | c.211G > A | p.(Gly71Arg) | 1 |
| LDLR | Exon 7 | c.1060+7= | p (?) | 18 |
| LDLR | Intron 7 | c.1060+10G > A | p (?) | 8 |
| PCSK9 | Exon 7 | c.1026A > G | p.(Gln342=) | 6 |
| Characteristics | Patients with FH | |||||
|---|---|---|---|---|---|---|
| ASCVD- Mutation- | ASCVD+ Mutation- | ASCVD- Benign/Likely Benign Mutation+ | ASCVD+ Benign/Likely Benign Mutation+ | ASCVD+ Pathogenic/Likely PATHOGENIC Mutation+ | p | |
| n (%) | 25 (41%) | 13 (21.31%) | 8 (13.1%) | 8 (13.1%) | 7 (11.5%) | |
| TC baseline mg/dL (median ± IQR) | 312.7 ± 20.4 | 352.8 ± 63.2 | 323.9 ± 26.9 | 352.6 ± 42.2 | 445.6 ± 203.9 | 0.01 * |
| TC 12 mo mg/dL (median ± IQR) | 249.1 ± 17.9 | 284.5 ± 51.8 | 261 ± 13.9 | 278.5 ± 44.2 | 310.3 ± 63.5 | 0.009 * |
| TC 24 mo mg/dL (median ± IQR) | 227.7 ± 19.3 | 254.8 ± 47.4 | 236.1 ± 12.9 | 256.6 ± 29.8 | 278.7 ± 57.9 | 0.01 * |
| TC 36 mo mg/dL (median ± IQR) | 210.8 ± 17.1 | 230.9 ± 14.3 | 229.3 ± 12.2 | 245.6 ± 29.6 | 271.9 ± 53.3 | 0.001 * |
| LDL–C baseline mg/dL (mean ± SD) | 233.4 ± 29.1 | 270.8 ± 62.5 | 240.1 ± 29.2 | 257.8 ± 54.7 | 309.7 ± 78.7 | 0.15 |
| LDL–C 12 mo mg/dL (mean ± SD) | 163.6 ± 21.4 | 204.4 ± 50.9 | 166.6 ± 13.3 | 195.4 ± 52.9 | 234.1 ± 66.7 | 0.002 * |
| LDL–C 24 mo mg/dL (mean ± SD) | 137.4 ± 17.8 | 167.5 ± 47.6 | 139.6 ± 14.2 | 168.5 ± 39.7 | 202.4 ± 59.7 | 0.003 * |
| LDL–C 36 mo mg/dL (mean ± SD) | 113.1 ± 17.3 | 146.5 ± 11.6 | 131.1 ± 14.9 | 159.1 ± 35.1 | 189.3 ± 56.5 | 0.001 * |
| HDL–C baseline mg/dL (median ± IQR) | 51.3 ± 12.5 | 42.4 ± 9.8 | 54.5 ± 16.4 | 55.5 ± 19.9 | 43.1 ± 7.9 | 0.14 |
| HDL–C 12 mo mg/dL (median ± IQR) | 61.7 ± 8.9 | 53.9 ± 9.8 | 64.3 ± 12.1 | 64.4 ± 12.4 | 54.9 ± 10.1 | 0.19 |
| HDL–C 24 mo mg/dL (median ± IQR) | 67.4 ± 7.9 | 62.4 ± 7.1 | 71.1 ± 6.8 | 68.4 ± 9.4 | 59.4 ± 10.8 | 0.05 * |
| HDL–C 36 mo mg/dL (median ± IQR) | 75.1 ± 8.2 | 61.6 ± 7.7 | 73.1 ± 7.9 | 65.1 ± 4.8 | 58.3 ± 5.8 | 0.001 * |
| TG baseline mg/dL (mean ± SD) | 166.8 ± 90.9 | 203.7 ± 93.3 | 204.1 ± 128.1 | 140.1 ± 80.2 | 152.4 ± 47.9 | 0.19 |
| TG 12 mo mg/dL (mean ± SD) | 124.2 ± 48.8 | 142.4 ± 51.3 | 151.6 ± 69.4 | 109.1 ± 39.7 | 124.7 ± 29.7 | 0.26 |
| TG 24 mo mg/dL (mean ± SD) | 116.5 ± 31.2 | 124.9 ± 30.4 | 125.1 ± 36.3 | 106.8 ± 34.3 | 122.4 ± 30.5 | 0.41 |
| TG 36 mo mg/dL (mean ± SD) | 120.6 ± 22.3 | 123.1 ± 22.3 | 126.1 ± 28.5 | 107.1 ± 33.1 | 121.7 ± 25.4 | 0.25 |
| hsCRP baseline mg/L (mean ± SD) | 5.1 ± 1.9 | 7.3 ± 1.8 | 6.1 ± 2.7 | 6.7 ± 1.6 | 7.1 ± 2.5 | 0.02 * |
| hsCRP 12 mo mg/L (mean ± SD) | 4.1 ± 1.8 | 6.1 ± 1.3 | 4.8 ± 1.9 | 5.7 ± 1.6 | 5.8 ± 2.4 | 0.02 * |
| hsCRP 24 mo mg/L (mean ± SD) | 3.5 ± 1.4 | 5.3 ± 1.3 | 4.1 ± 1.6 | 4.4 ± 1.7 | 4.7 ± 2.3 | 0.01 * |
| hsCRP 36 mo mg/L (mean ± SD) | 0.6 ± 0.2 | 7.2 ± 1.2 | 2.4 ± 2.7 | 5.9 ± 1.1 | 6.7 ± 1.8 | 0.001 * |
| EF baseline % (mean ± SD) | 56.4 ± 7.6 | 48.7 ± 10.5 | 53.8 ± 8.6 | 49.4 ± 10.8 | 53.3 ± 12.9 | 0.13 |
| EF 12 mo % (mean ± SD) | 55.4 ± 6.6 | 48.1 ± 9.6 | 52.5 ± 7.6 | 48.8 ± 8.4 | 50.7 ± 14.8 | 0.11 |
| EF 24 mo % (mean ± SD) | 54.8 ± 6.3 | 43.1 ± 10.7 | 52.5 ± 7.6 | 47.5 ± 8.1 | 44.3 ± 16. 2 | 0.006 * |
| EF 36 mo % (mean ± SD) | 53.8 ± 6.7 | 41.2 ± 12.1 | 51.9 ± 7.5 | 45 ± 10.4 | 37.9 ± 17.3 | 0.002 * |
| ABI baseline (mean ± SD) | 0.86 ± 0.06 | 0.81 ± 0.06 | 0.89 ± 0.08 | 0.79 ± 0.06 | 0.78 ± 0.11 | 0.01 * |
| ABI 12 mo (mean ± SD) | 0.89 ± 0.05 | 0.85 ± 0.05 | 0.91 ± 0.06 | 0.81 ± 0.06 | 0.81 ± 0.08 | 0.001 * |
| ABI 24 mo (mean ± SD) | 0.94 ± 0.07 | 0.89 ± 0.71 | 0.94 ± 0.08 | 0.84 ± 0.13 | 0.85 ± 0.07 | 0.004 * |
| ABI 36 mo (mean ± SD) | 0.94 ± 0.04 | 0.82 ± 0.11 | 0.94 ± 0.03 | 0.81 ± 0.09 | 0.83 ± 0.09 | 0.001 * |
| cIMT baseline mm (mean ± SD) | 0.82 ± 0.28 | 1.08 ± 0.32 | 0.89 ± 0.36 | 1.13 ± 0.31 | 1.04 ± 0.42 | 0.03 * |
| cIMT 12 mo mm (mean ± SD) | 0.75 ± 0.23 | 1.03 ± 0.25 | 0.81 ± 0.31 | 1.05 ± 0.24 | 1.06 ± 0.31 | 0.002 * |
| cIMT 24 mo mm (mean ± SD) | 0.74 ± 0.25 | 0.99 ± 0.31 | 0.75 ± 0.29 | 0.96 ± 0.31 | 0.94 ± 0.36 | 0.01 * |
| cIMT 36 mo mm (mean ± SD) | 0.73 ± 0.15 | 1.07 ± 0.18 | 0.76 ± 0.12 | 1.08 ± 0.21 | 1.13 ± 0.15 | 0.001 * |
| Variable | B | SE | Wald | df | p | OR | 95.0% CI for HR | |
|---|---|---|---|---|---|---|---|---|
| Lower | Upper | |||||||
| Type of mutations Pathogenic Mutations Benign Mutations | 5.41 | 2 | 0.05 * | |||||
| 1.57 | 0.68 | 5.29 | 1 | 0.02 * | 4.81 | 1.26 | 18.32 | |
| 0.37 | 0.48 | 0.61 | 1 | 0.44 | 1.45 | 0.57 | 3.73 | |
| EF baseline | 0.01 | 0.03 | 0.05 | 1 | 0.82 | 1.01 | 0.95 | 1.08 |
| cIMT baseline | 0.33 | 1.12 | 0.08 | 1 | 0.77 | 1.39 | 0.15 | 13.29 |
| ABI baseline | −3.96 | 4.74 | 0.69 | 1 | 0.41 | 0.02 | 0.001 | 20.73 |
| hsCRP baseline | 0.16 | 0.13 | 1.51 | 1 | 0.22 | 1.18 | 0.91 | 1.53 |
| LDL–C baseline | 0.04 | 0.08 | 0.33 | 1 | 0.57 | 1.01 | 0.99 | 1.02 |
| DLCN Score Possible FH Probable FH Definite FH | ||||||||
| ref | ref | 1.69 ref | 2 ref | 0.43 ref | ref | ref | ref | |
| 0.48 | 0.66 | 0.53 | 1 | 0.47 | 1.62 | 0.45 | 5.87 | |
| −0.39 | 1.51 | 0.07 | 1 | 0.79 | 0.67 | 0.04 | 12.94 | |
| Simon Broome score | 0.56 | 0.73 | 0.59 | 1 | 0.44 | 1.75 | 0.42 | 7.37 |
| Lipid-lowering DrugsStatin Statin + Ezetimibe Statin + Fenofibrate Statin + Fenofibrate + Ezetimibe | ||||||||
| ref | ref | 1.35 ref | 3 ref | 0.72 ref | ref | ref | ref | |
| 0.15 | 0.61 | 0.06 | 1 | 0.81 | 1.17 | 0.35 | 3.84 | |
| 0.46 | 0.76 | 0.37 | 1 | 0.54 | 1.59 | 0.36 | 7.09 | |
| −0.44 | 0.67 | 0.44 | 1 | 0.51 | 0.64 | 0.18 | 2.36 | |
| Locality | Country | Diagnostic Criteria | Number of Patients | Number of Patients with Mutations | Technique- Molecular Analysis | Gene | Number of Detected Mutations |
|---|---|---|---|---|---|---|---|
| Western Europe | Italy [2] | DLCN | 1018 | 94 | MLPAN orthern blot analysis and RT-PCR amplification In silico analysis | LDLR APOB PCSK9 | 984 LDLR 22 APOB 2 PCSK9 |
| Switzerland [15] | LDL–C 95th percentile | 94 | NA | NGS (Illumina) Sanger sequencing | LDLR APOB PCSK9 | 43 LDLR 5 APOB3 6 PCSK9 | |
| UK-1 [45] | SB criteria | 791 | 134 | SSCP analysis | LDLR | 51 LDLR | |
| UK-2 [35] | SB criteria | 280 | 171 He FH patients 28 Ho/ compound He | MLPA sequencing of amplified fragments of genomic DNA or mRNA | LDLR LDLRAP1PCSK9 APOB | 98 LDLR 2 PCSK9 5 LDLRAP1 14 APOB | |
| UK-3 [36] | SB FH register SB criteria | 48 | 14 | MLPA NGS (Illumina) Sanger sequencing | LDLR LDLRAP1PCSK9 APOB | 17 LDLR 1 LDLRAP1 2 PCSK9 3 APOB | |
| Spain [16] | Spanish FH Registry | 476 | 329 | SSCP analysis | LDLRAPOB | 116 LDLR 4 APOB | |
| Germany [37] | LDL–C 90th percentile | 162 | 27 | MLPA Direct sequencing on LDLR gene | LDLR | 24 LDLR | |
| Portugal (Azores Island) [5] | SB | 33 | 33 | LIPOchip® Array version 7 (DNA array) Direct sequencing for exons 2–6 | LDLR | 18 LDLR | |
| Central and Eastern Europe | Slovakia [3] | LDL–C 95th percentile + HCH in family | 359 | 16 for APOB 164 for LDLR | TaqMan SNP Genotyping Assay ID Bidirectional sequencing on LDLR gene MLPA | LDLR APOB | 54 LDLR 1 APOB |
| Greece-1 [38] | LDL–C 95th percentile CVD history CVD family history tendon xanthomas | 183 | 78 | DGGE analysis | LDLR APOB | 17 LDLR 0 APOB | |
| Greece-2 [30] | HeFH | 561 | 140 | DNA sequencing of the LDLR gene | LDLR | 26 LDLR | |
| Russia 1 [39] | DLCN | 80 | 80 | Sanger sequencing In silico analysis | LDLR APOB | 12 LDLR 0 APOB | |
| Russia 2 [40] | LDL–C 95th percentile CVD history CVD family history tendon xanthomas | 45 | 24 | Automated DNA sequencing | LDLR APOB | 21 LDLR 0 APOB | |
| Poland-1 [41] | LDL–C 90th percentile | 30 families | 17 families | SSCP analysis sequencing of polymerase chain reaction restriction enzyme patterns on Southern blots and long-PCR | LDLR APOB | 11 LDLR 1 APOB | |
| Poland-2 [9] | SB | 161 | 40 | High resolution melt Direct sequencing MLPA | LDLR APOB | 39 LDLR 1 APOB | |
| Czech Republic [47] | personal history and/or family history of premature CHD elevated TC, LDL95th percentile | 3914 | 1296 | denaturing high-performance liquid chromatography (dHPLC) PCR-RFLP Sanger sequencing MLPA | LDLR APOB | 864 LDLR 32 APOB | |
| Worldwide | Canada [1] | DLCN The British Columbia FH Registry | 626 | 275 | NGS (Illumina) | LDLR APOB PCSK9LDLRAP1 | 131 unique FH-causing SNVs 38 CNV LDLR 0 CNV PCSK9, APOB |
| Brazil [4] | DLCN SB | 248 | 125 | MLPA | LDLR APOB PCSK9 | 71 LDLR 2 APOB 0 PCSK9 | |
| Colombia [42] | MedPed | 24 families | NA | Sanger sequencing | LDLR | 18 LDLR 3 pathogenic LDLR | |
| Australia [11] | mutations previously determined | 30 | NA | Ion Torrent Personal Genome Machine (PGM) sequencing Sanger sequencing MLPA | LDLR | 2179 LDLR | |
| Malaysia [10] | SB | 164 | 117 | Denaturing High-Performance Liquid Chromatography MLPA In silico analyses of variant effects | LDLR | 8 mutation LDLR21 variants | |
| Sri Lanka [7] | Modified SB DCLN | 27 | 5 | Sanger sequencing | LDLR | 4 variants He 1 mutation He compound | |
| Saudi Arabia [43] | DLCN | 2 | 2 | Sanger sequencing | LDLR APOB PCSK9 | 2 LDLR mutations 0 APOB 0 PCSK9 | |
| Iran [8] | SB | 80 | NA | ARMS-PCR PCR- RFLP assay | LDLR APOB PCSK9 | 2 LDLR mutations 6 LDLR polymorphism 0 APOB 0 PCSK9 | |
| Taiwan [44] | SB | 125 | 76 | Microarray resequencing Sanger sequencing | LDLR APOB | 66 LDLR mutations 10 APOB mutations | |
| Japan [34] | criteria suggested by the Japan Atherosclerosis Society | 205 | 118 | SSCP assay MLPA | LDLR | 53 LDLR mutations 21 large rearrangements |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlad, C.-E.; Foia, L.G.; Popescu, R.; Popa, I.; Aanicai, R.; Reurean-Pintilei, D.; Toma, V.; Florea, L.; Kanbay, M.; Covic, A. Molecular Genetic Approach and Evaluation of Cardiovascular Events in Patients with Clinical Familial Hypercholesterolemia Phenotype from Romania. J. Clin. Med. 2021, 10, 1399. https://doi.org/10.3390/jcm10071399
Vlad C-E, Foia LG, Popescu R, Popa I, Aanicai R, Reurean-Pintilei D, Toma V, Florea L, Kanbay M, Covic A. Molecular Genetic Approach and Evaluation of Cardiovascular Events in Patients with Clinical Familial Hypercholesterolemia Phenotype from Romania. Journal of Clinical Medicine. 2021; 10(7):1399. https://doi.org/10.3390/jcm10071399
Chicago/Turabian StyleVlad, Cristiana-Elena, Liliana Georgeta Foia, Roxana Popescu, Ioana Popa, Ruxandra Aanicai, Delia Reurean-Pintilei, Vasilica Toma, Laura Florea, Mehmet Kanbay, and Adrian Covic. 2021. "Molecular Genetic Approach and Evaluation of Cardiovascular Events in Patients with Clinical Familial Hypercholesterolemia Phenotype from Romania" Journal of Clinical Medicine 10, no. 7: 1399. https://doi.org/10.3390/jcm10071399