
Theoretical and Computational Studies of Peptides and Receptors of the Insulin Family


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Brief Historical Account
1.2. Structural Biology of the Insulin Family
- (1)
- Intracellular kinase domains: The first crystal structures of the human IR kinase domain (IRKD) in inactive and active forms were determined in 1994 [64] and 1997 [65], respectively. Similar inactive and active structures of the IGF1R kinase domain (IGF1RKD) were later determined in 2001 [66] and 2002 [67], respectively.
- (2)
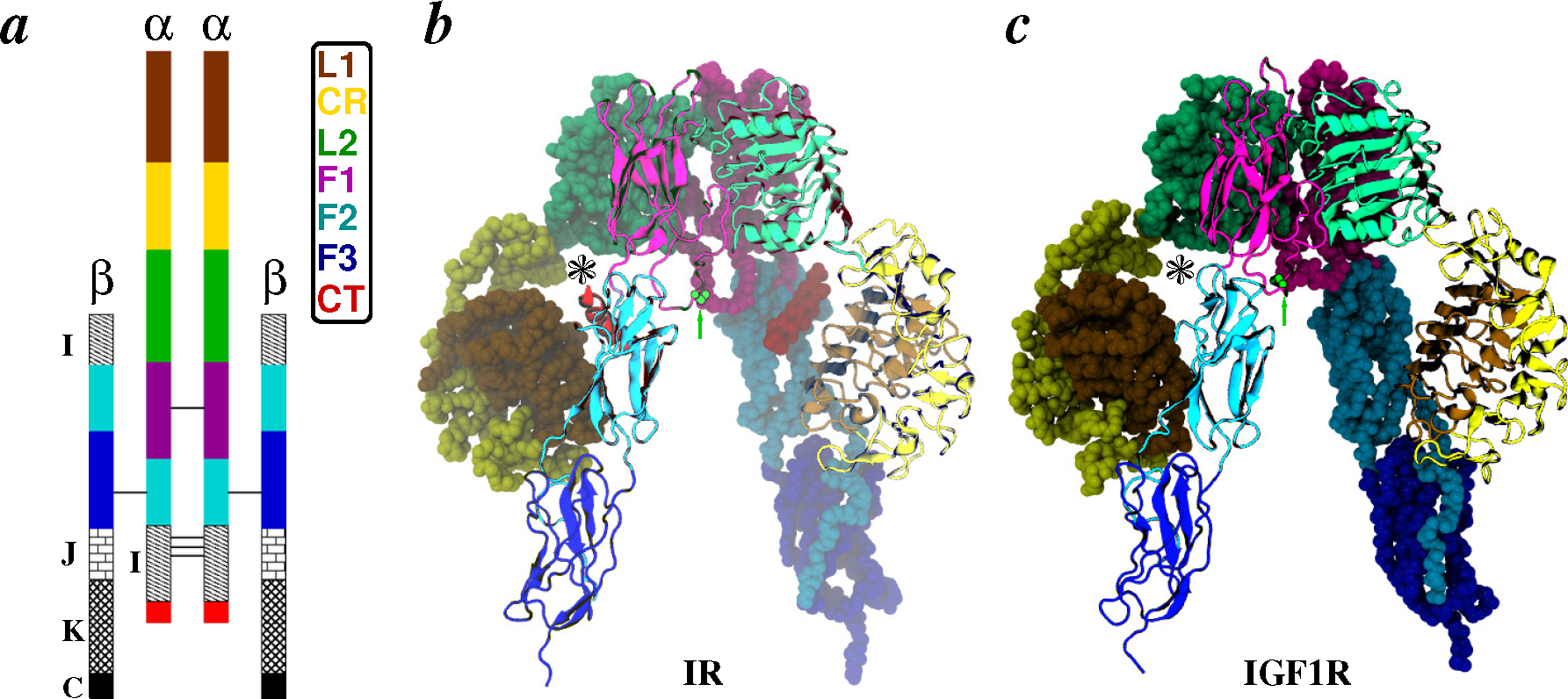
- Apo-ectodomains: In 1998 came the first breakthrough when Colin Ward and colleagues reported the atomic crystal structure of the first three domains of IGF1R [68]. The next year, they further reported the first electron microscopy (EM) images of the human insulin receptor ectodomain and its complexes with antibody fragments [69]. After a gap of seven years, the same group reported ground-breaking discoveries on the crystal structures of the first three domains of IR [70], as well as of the IR ectodomain (IRΔβ) [71]. The original IR ectodomain structure (PDB Code 2DTG) was later improved (PDB Code 3LOH) to include the previously unresolved C-terminal region of the IR α-chain (also known as the αCT peptide) [72]. Consistent with small-angle X-ray scattering (SAXS) data, a homology model of the IGF1R ectodomain (IGF1RΔβ) based on IR crystal structures was constructed in 2009 by Whitten et al. [73]. During the aforementioned seven-year gap, Luo et al. [74] reported the quaternary structure of the insulin-IR complex based on EM images. We note that this quaternary structure has been under debate due to inconsistency with the crystal structure of the IR ectodomain [2,49,71,75].
- (3)
- (4)
- Transmembrane domain: Li et al. [78] have recently reported a solution structure of the transmembrane domain of human IR using NMR spectroscopy.
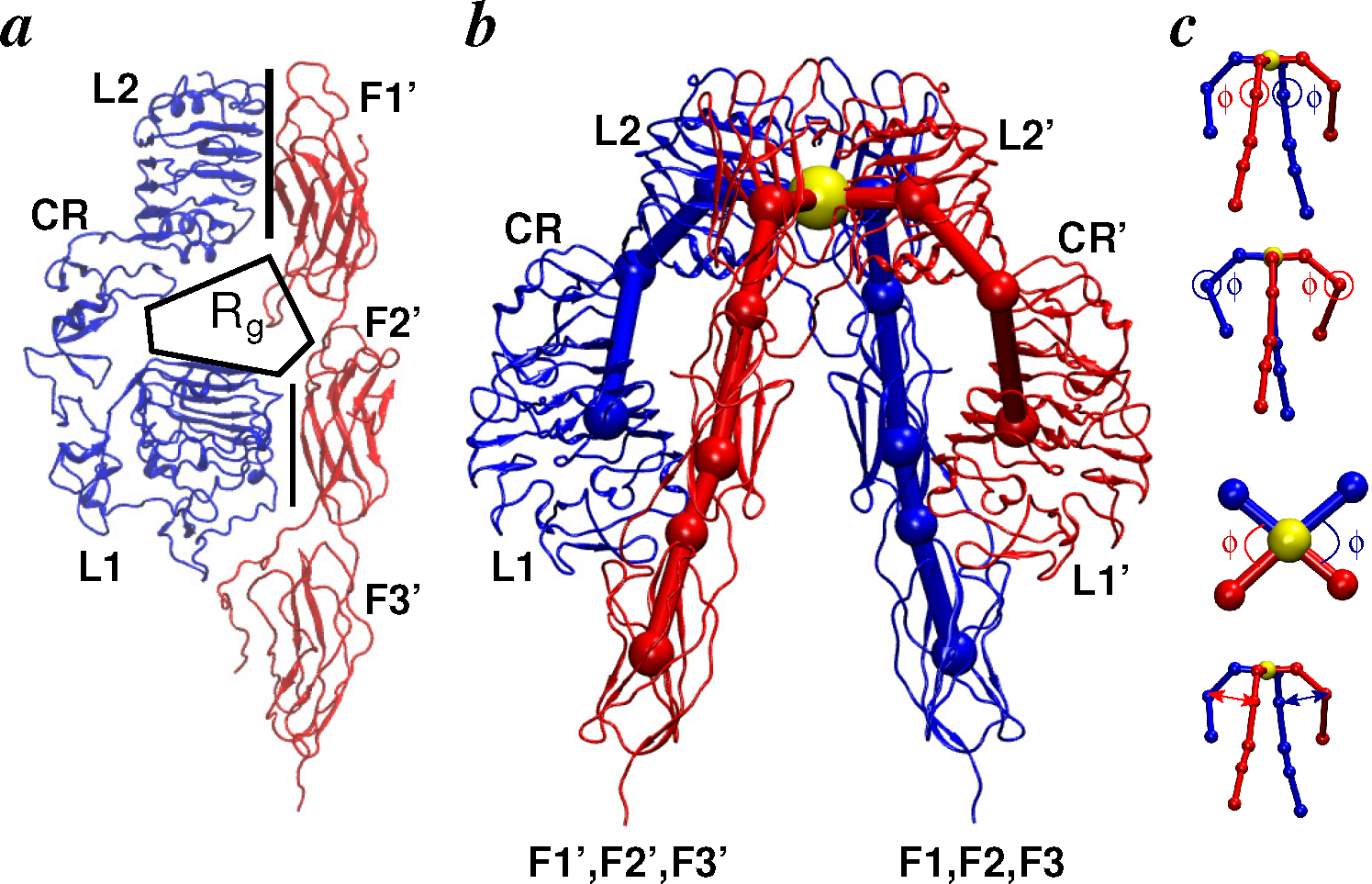
2. Structural Details: Architecture and Nomenclature
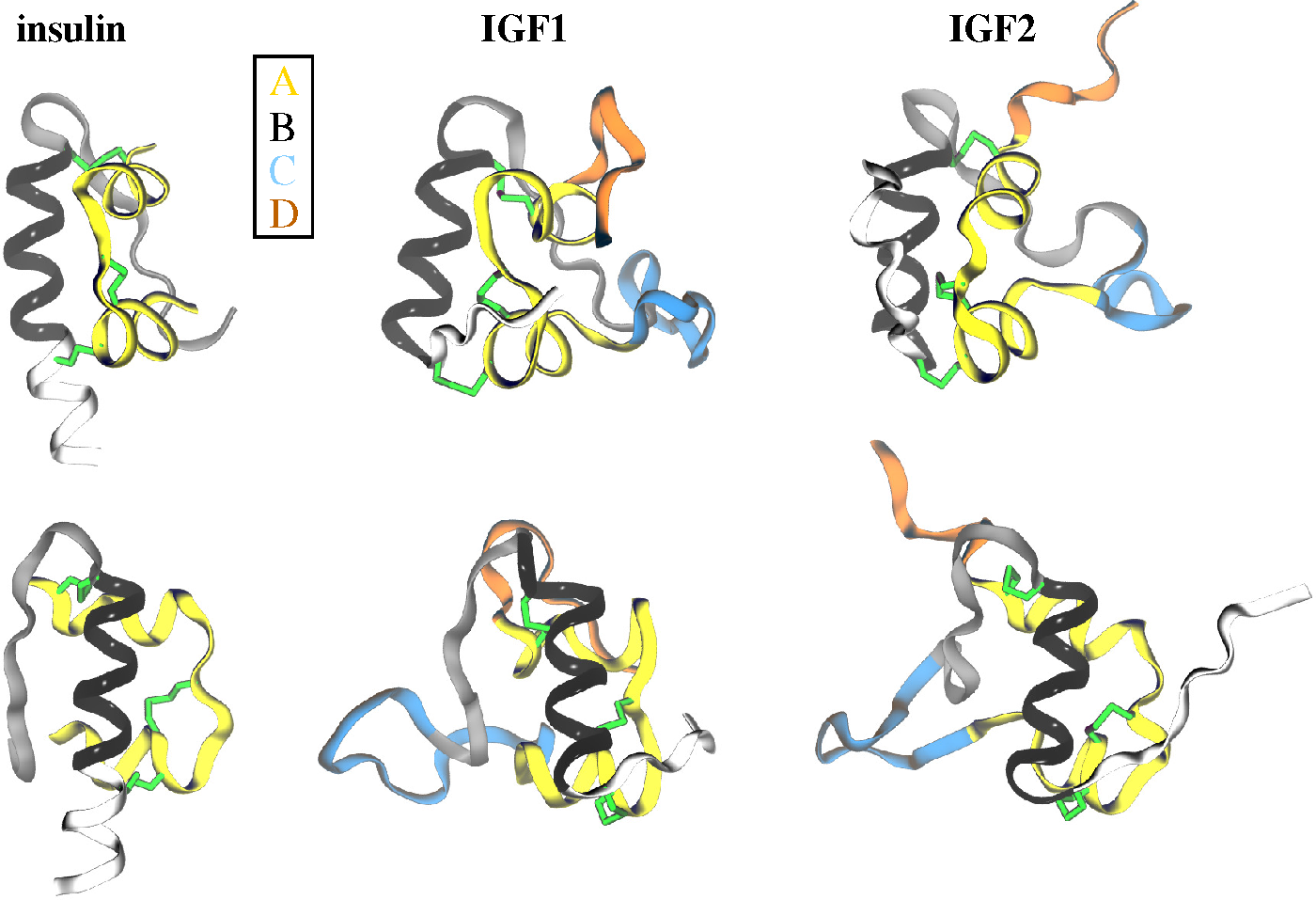
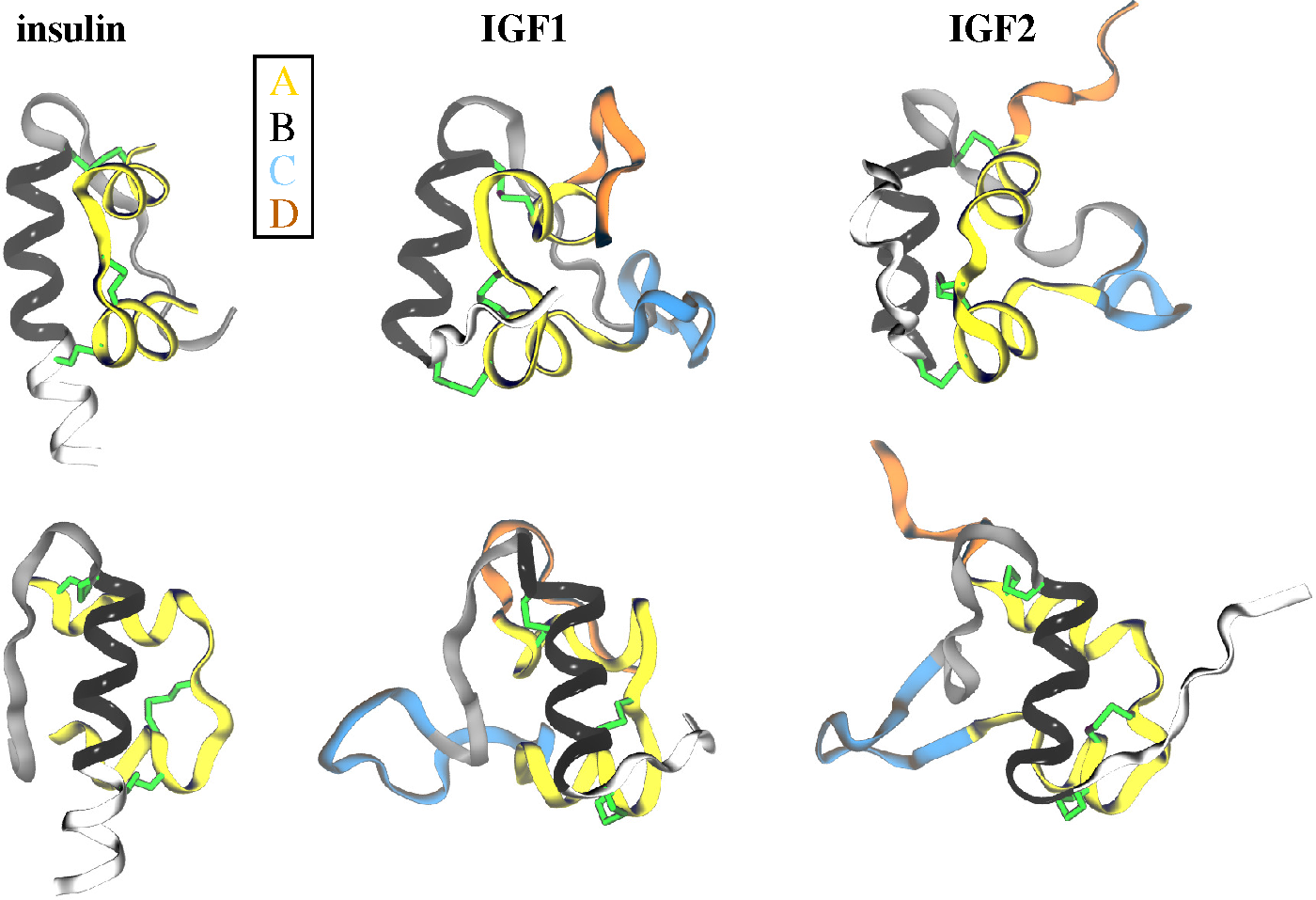
2.1. Ligands
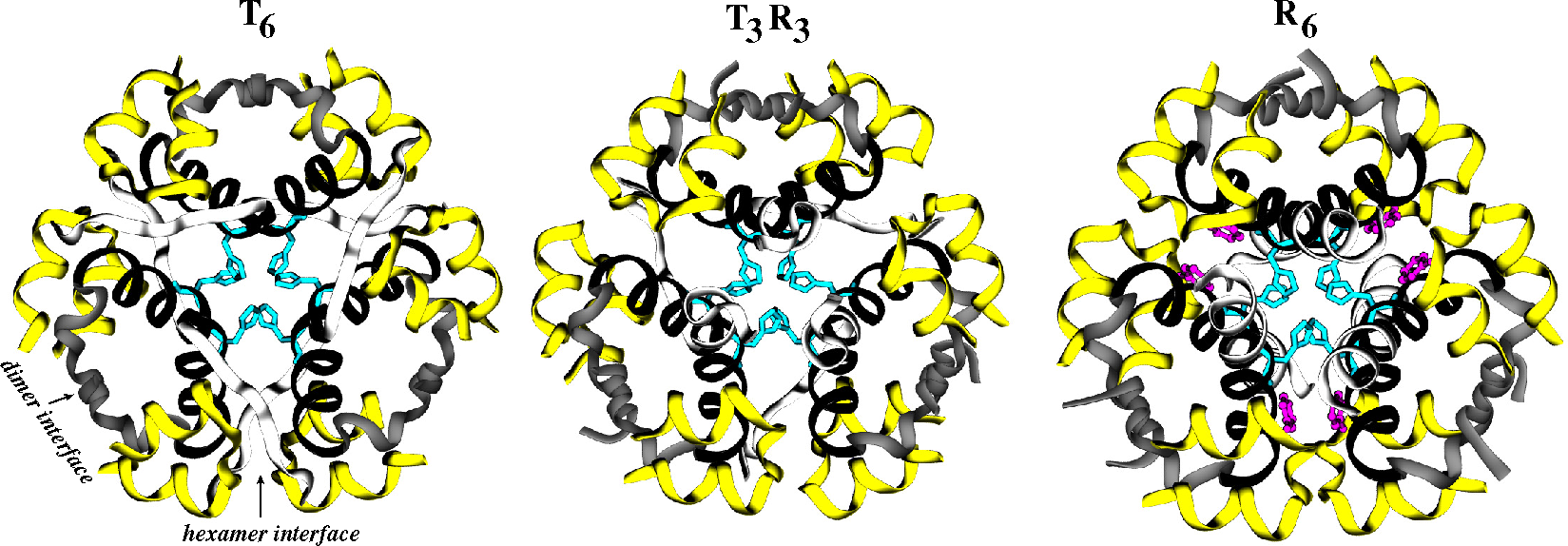
2.2. Receptors
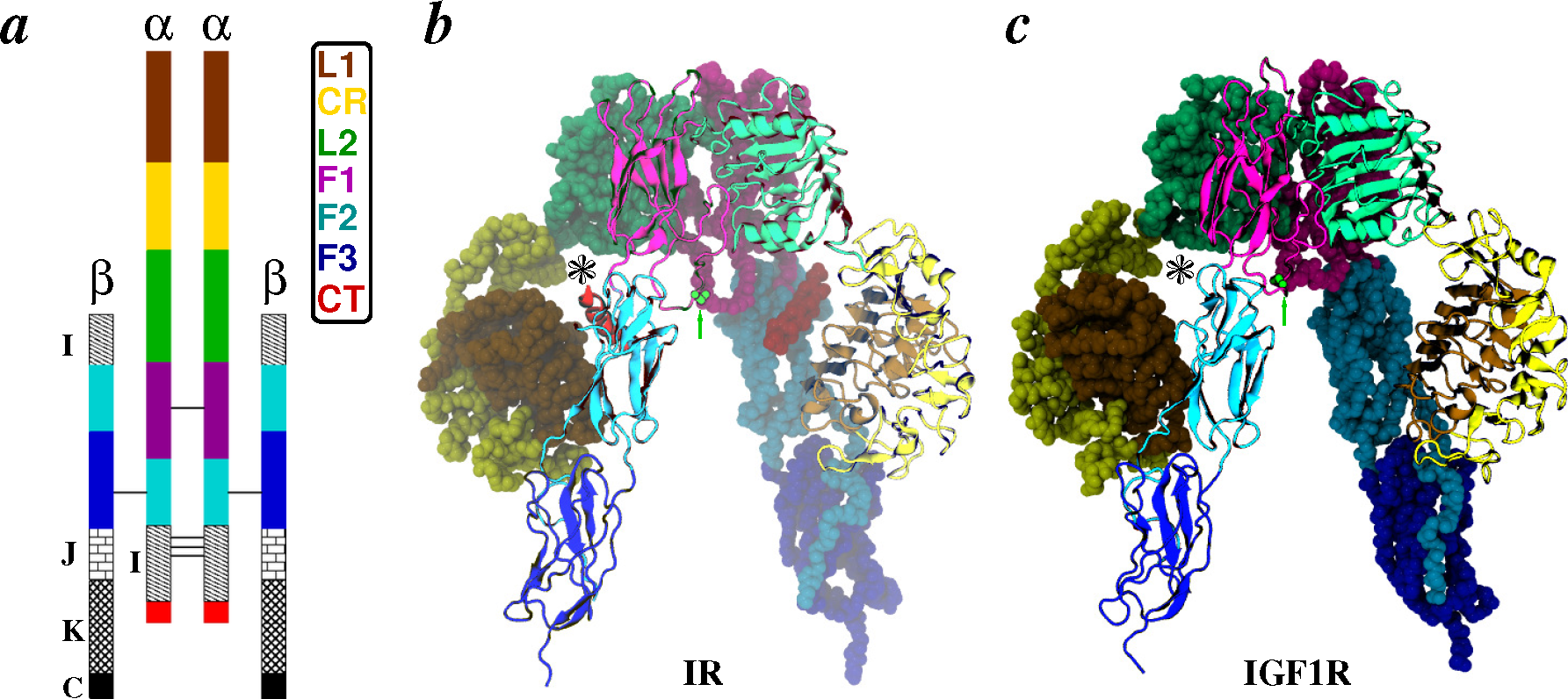
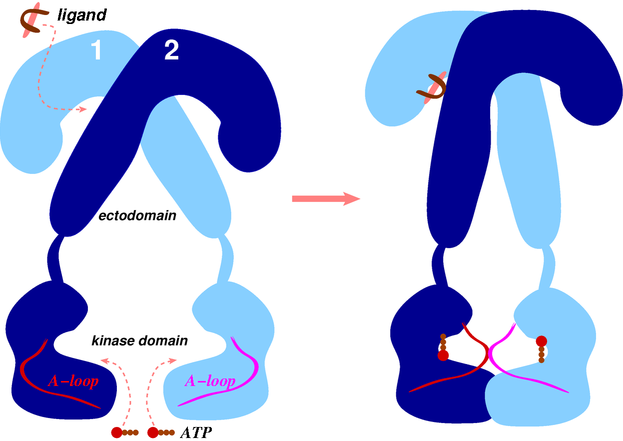
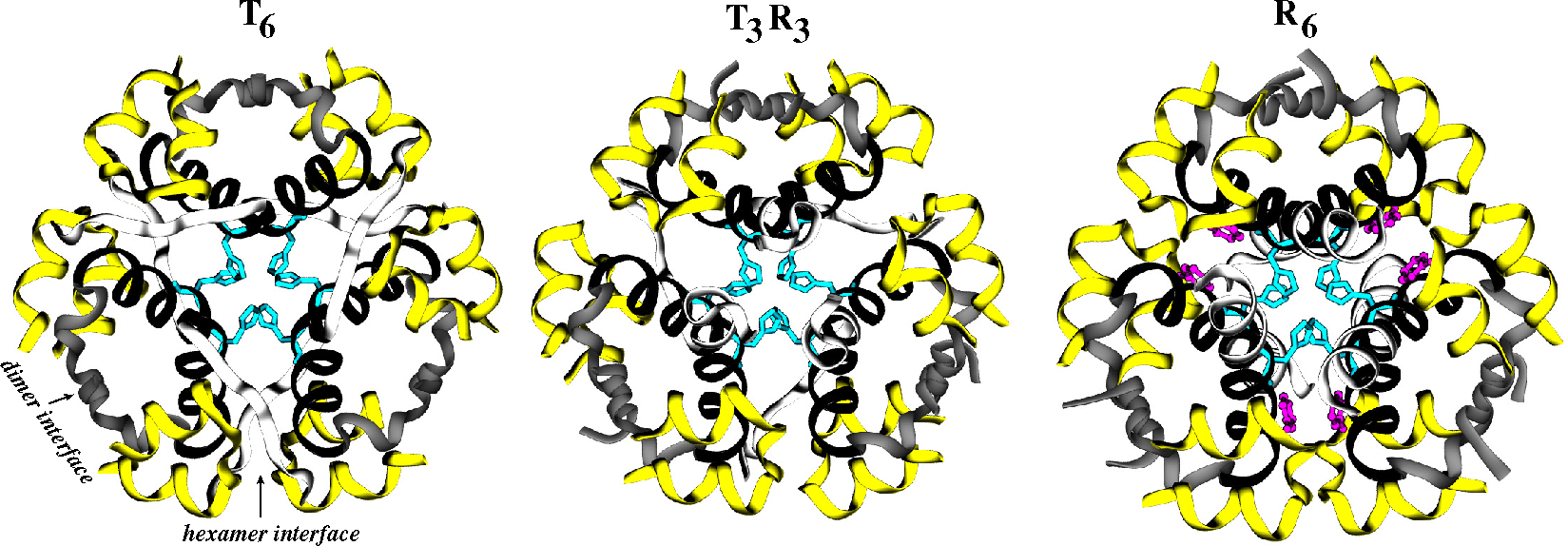
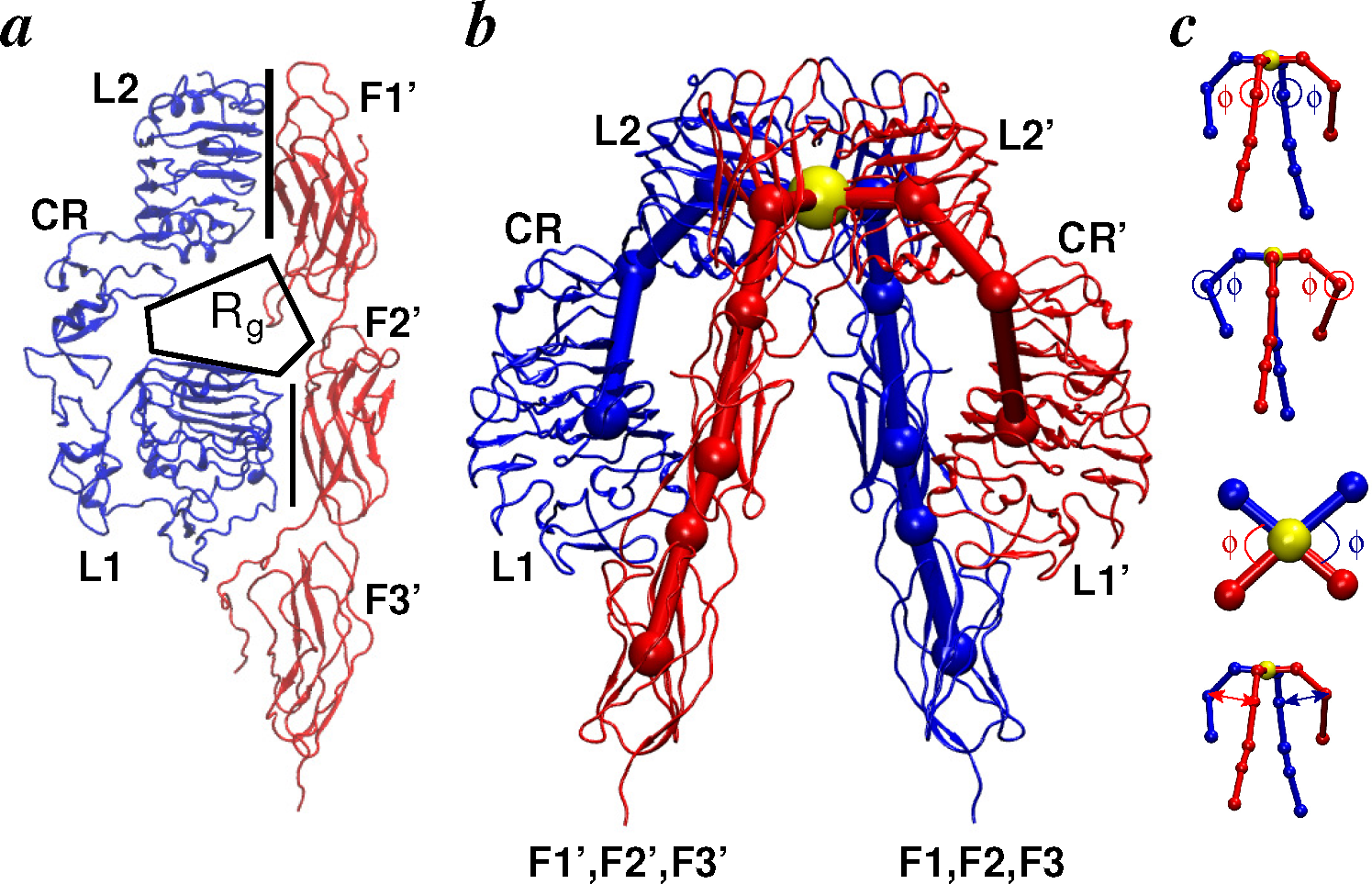
2.2.1. Conformational Metrics of Receptors
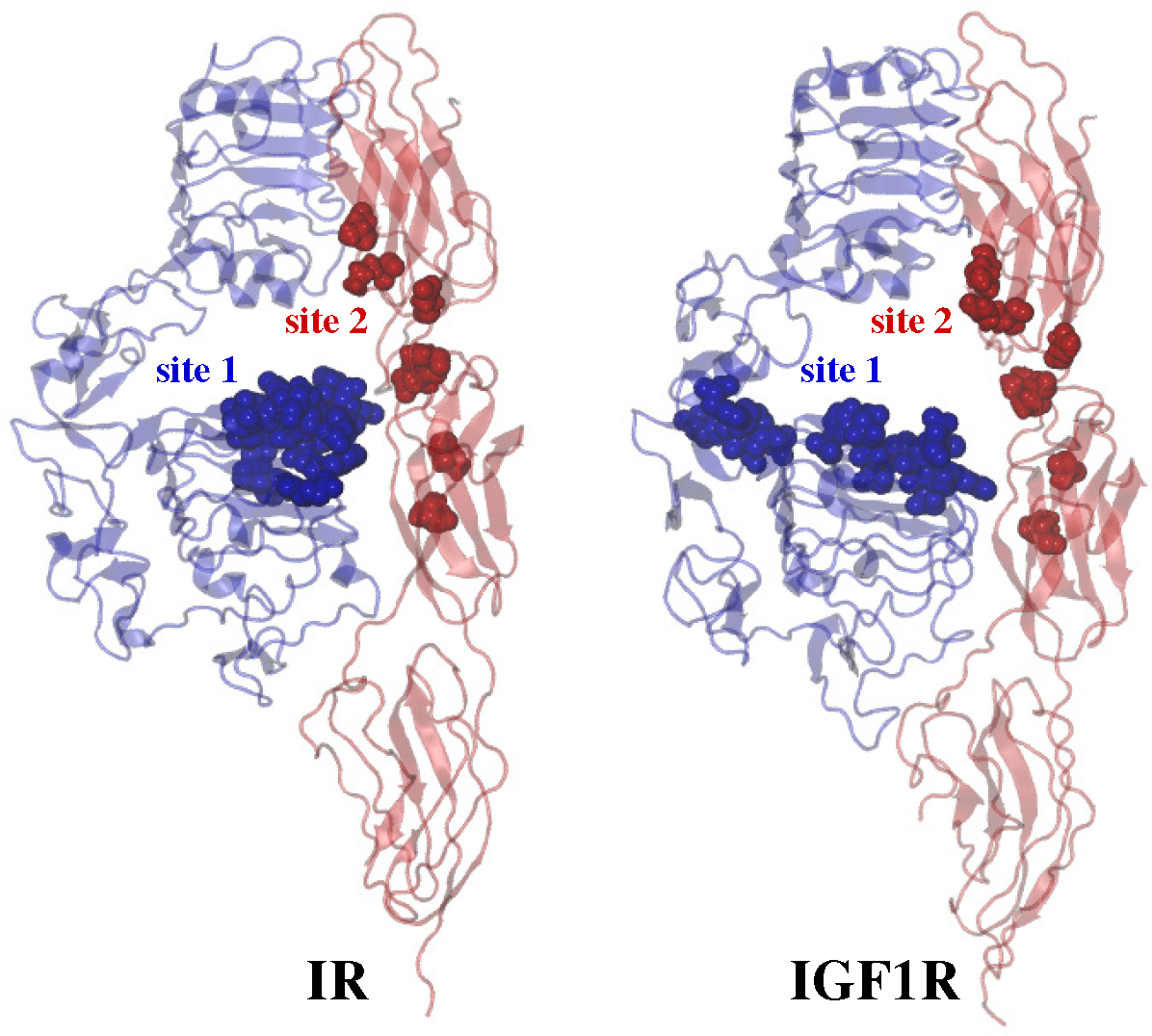
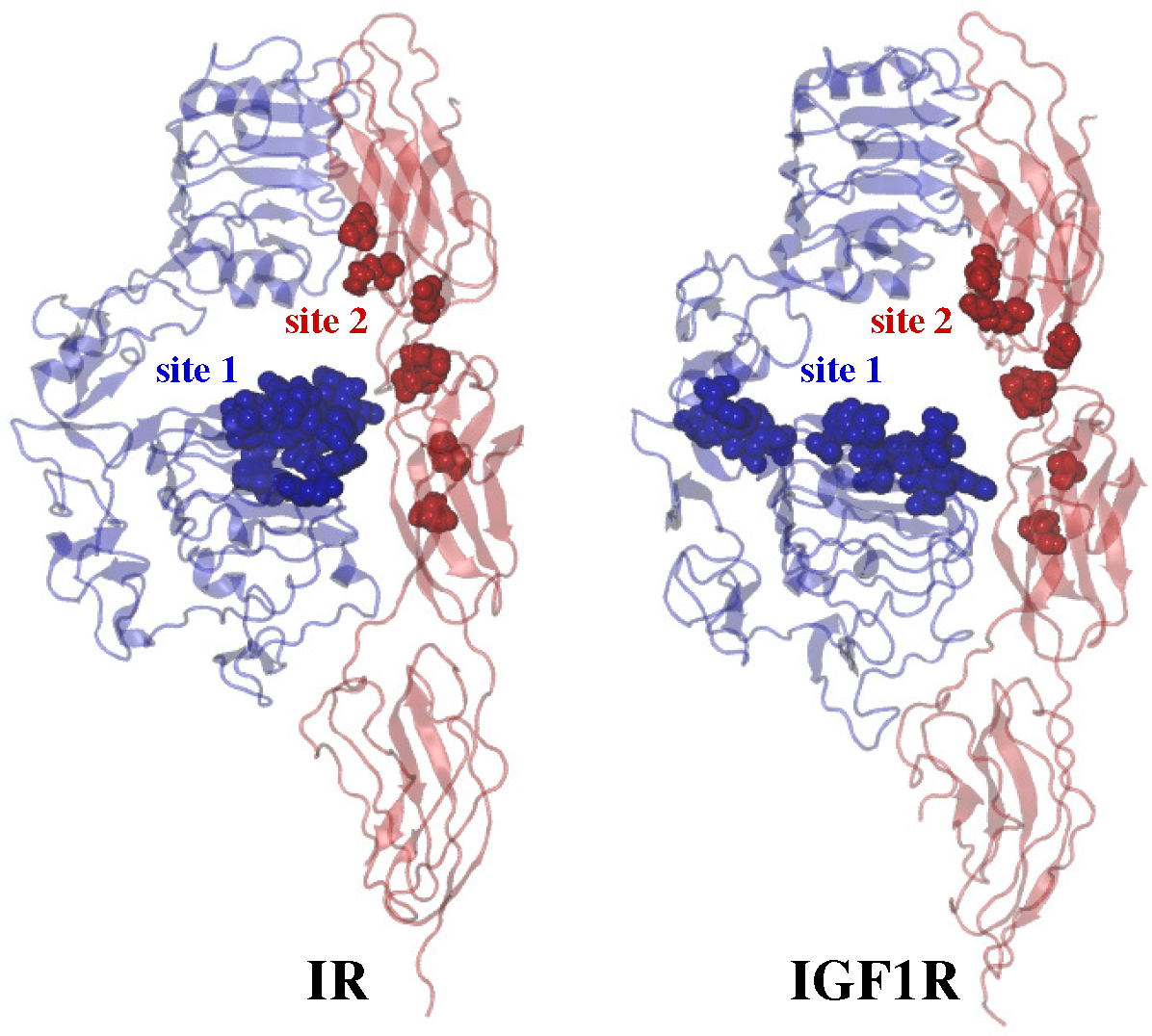
2.3. Ligand/Receptor Interactions
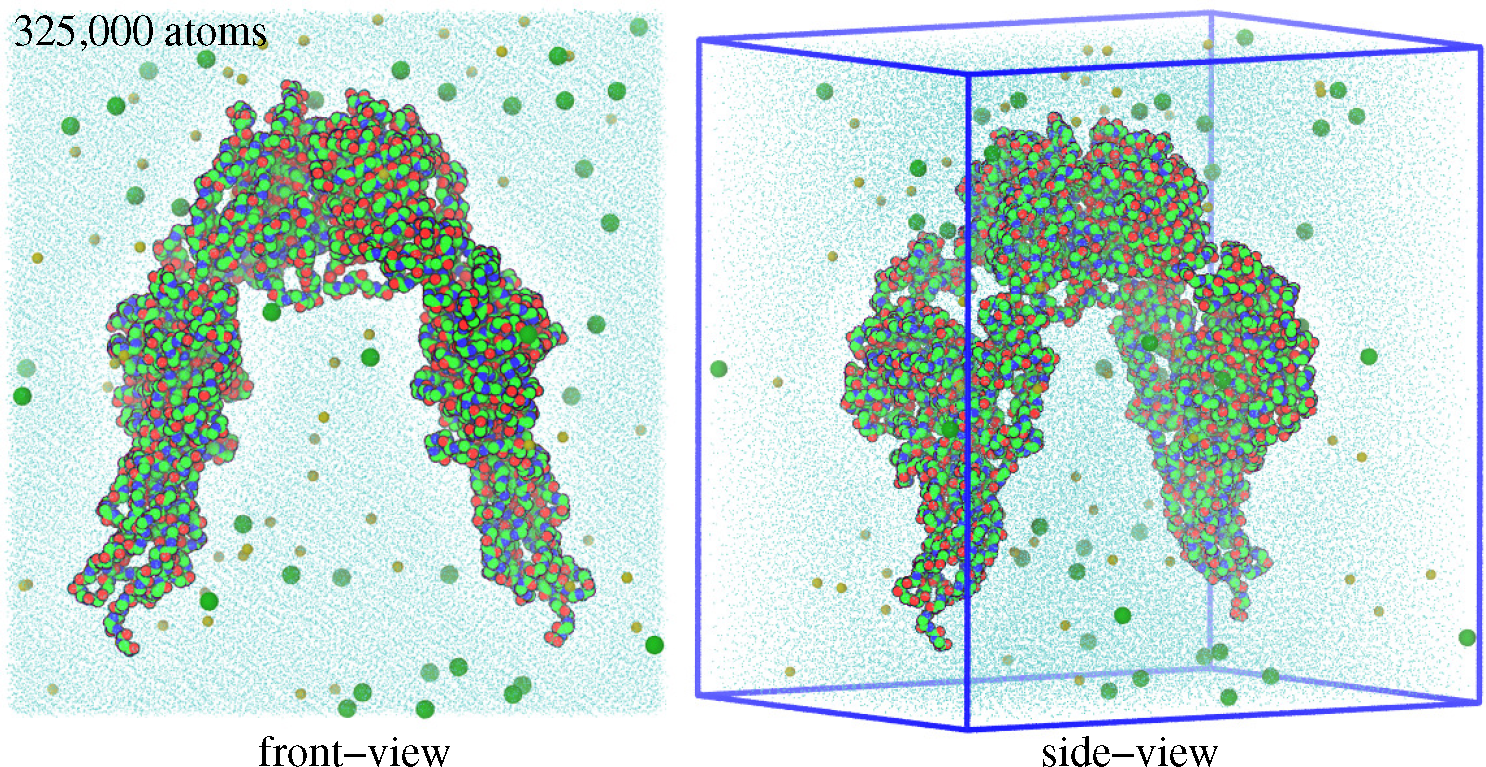
3. Modeling and Simulation Techniques
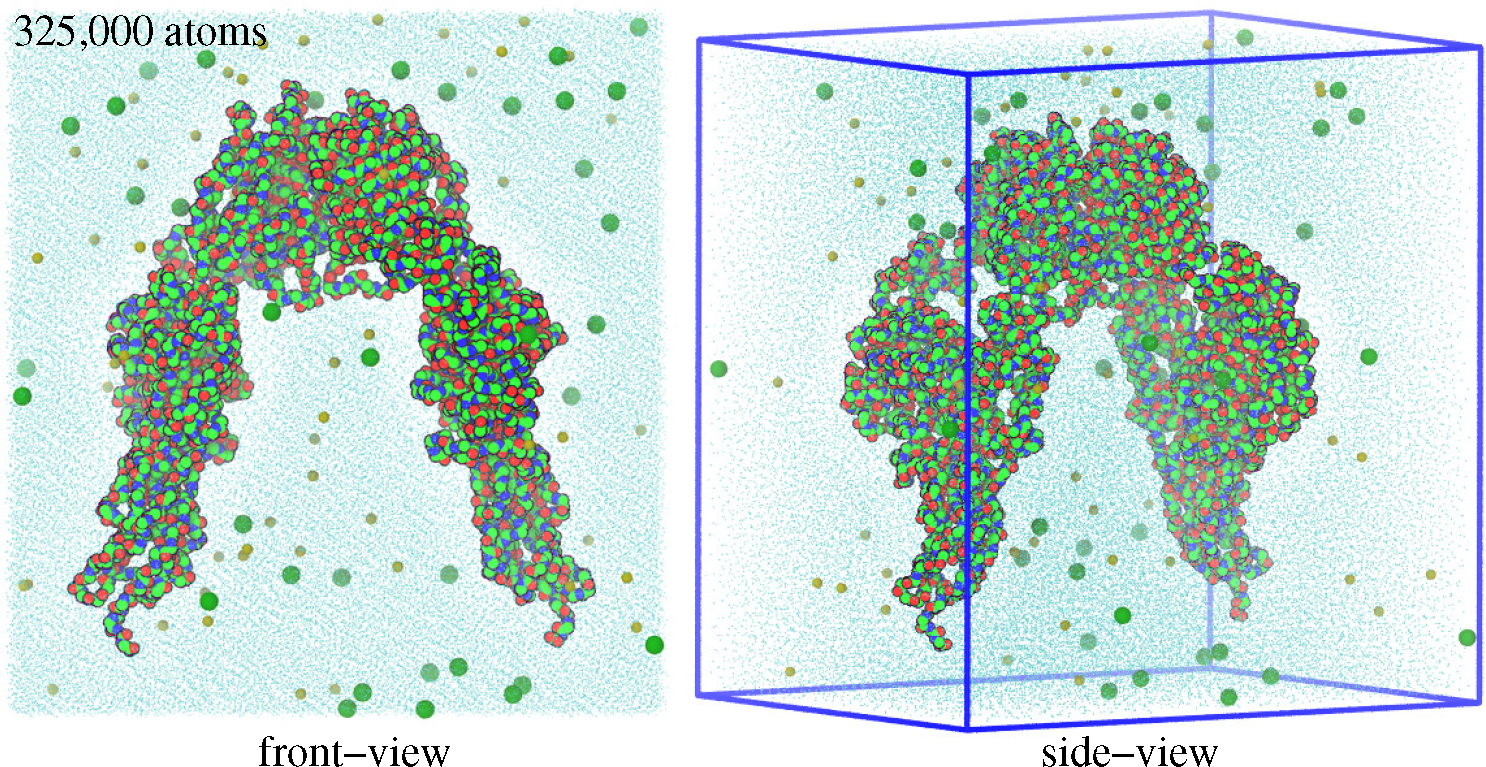
3.1. Molecular Dynamics Simulations
3.2. Monte Carlo Simulations
3.3. Enhanced Sampling and Free Energy Methods
3.3.1. Temperature-Accelerated Molecular Dynamics
3.3.2. String Method in Collective Variables
4. Applications
4.1. Ligands
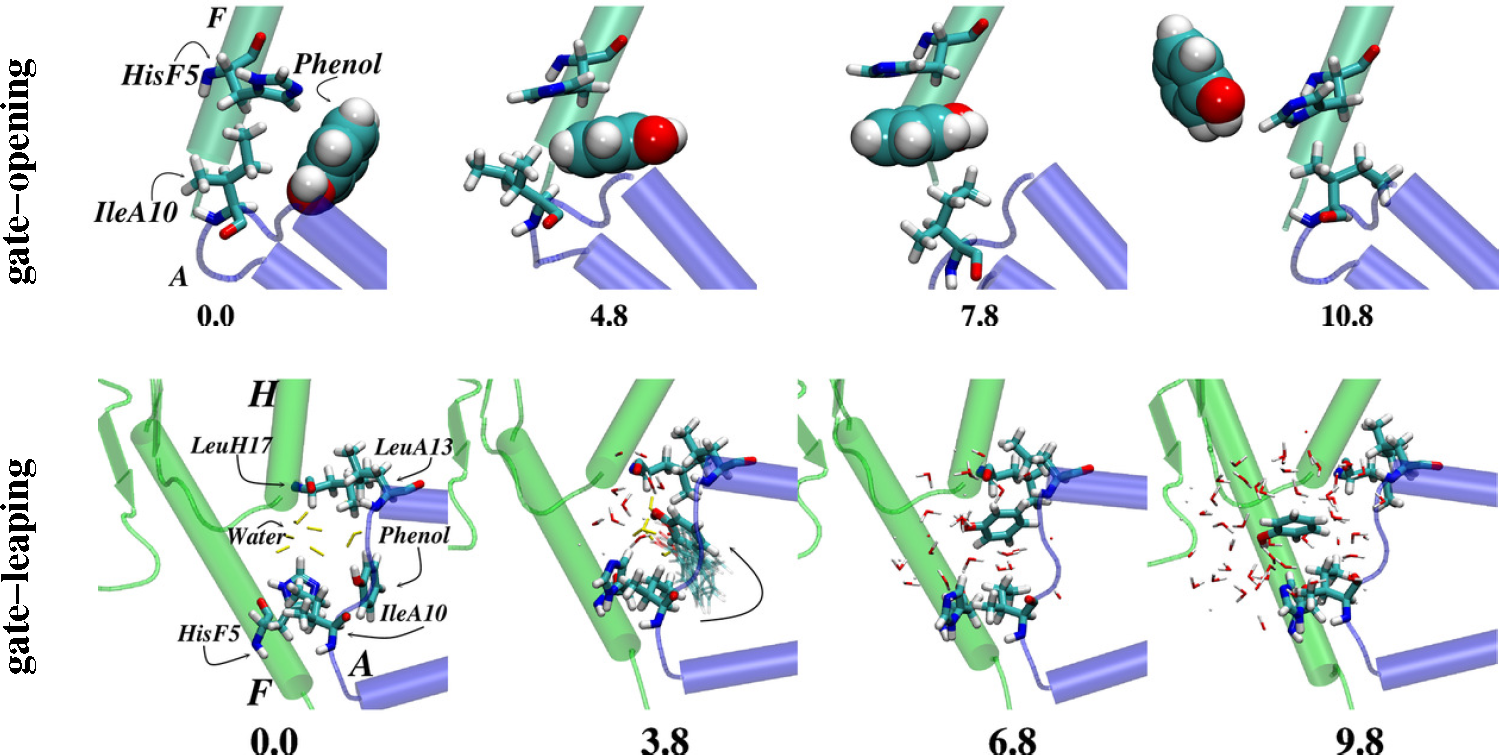
4.2. Receptors
4.2.1. Apo Ectodomains
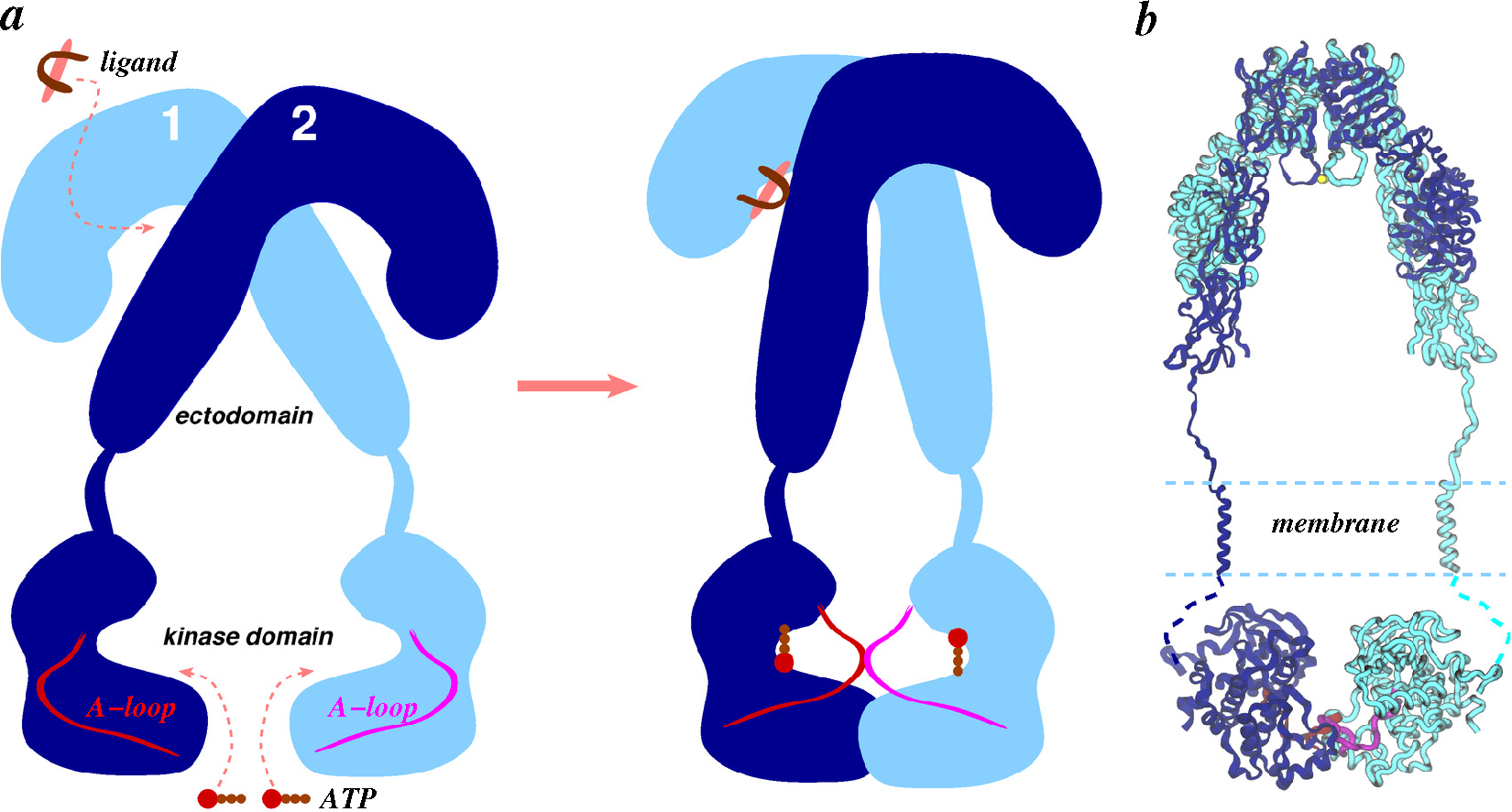
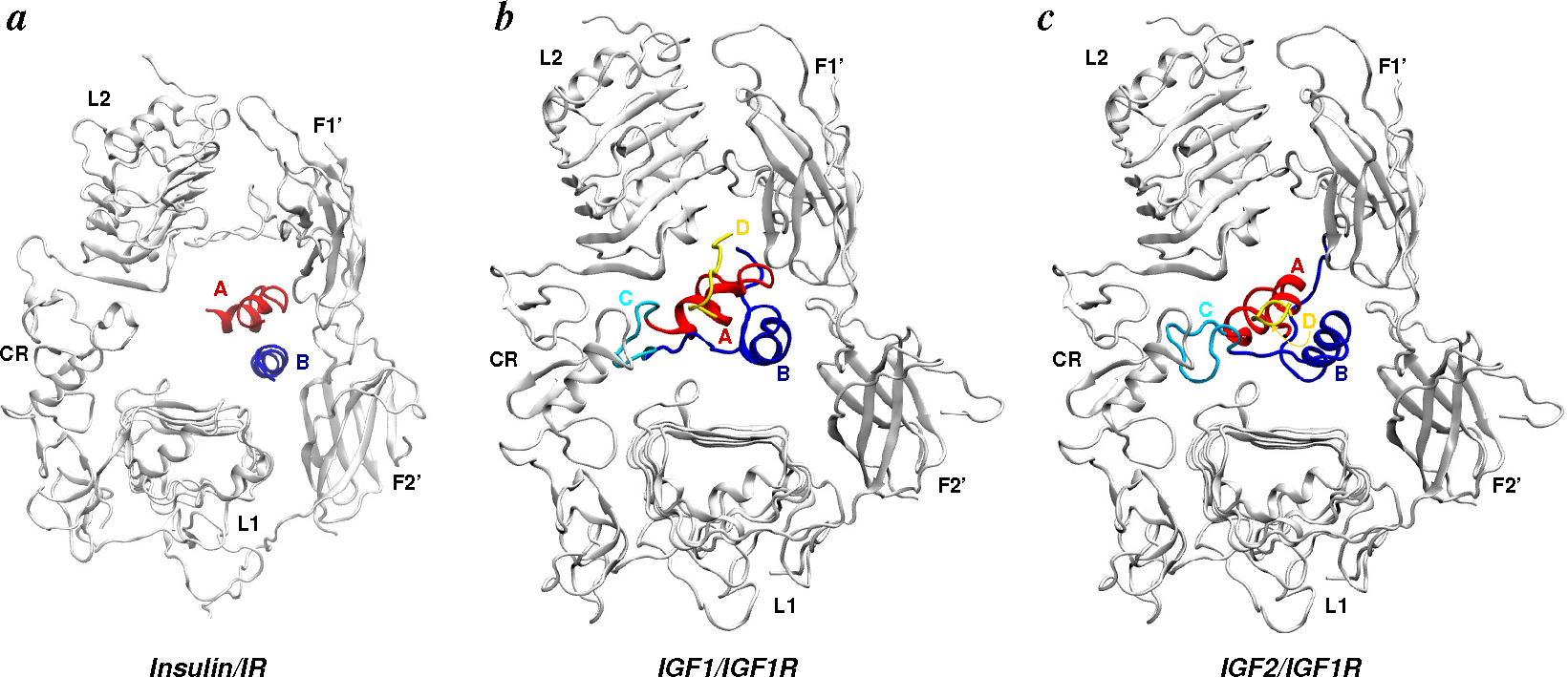
4.2.2. Ligand/Receptor Complexes
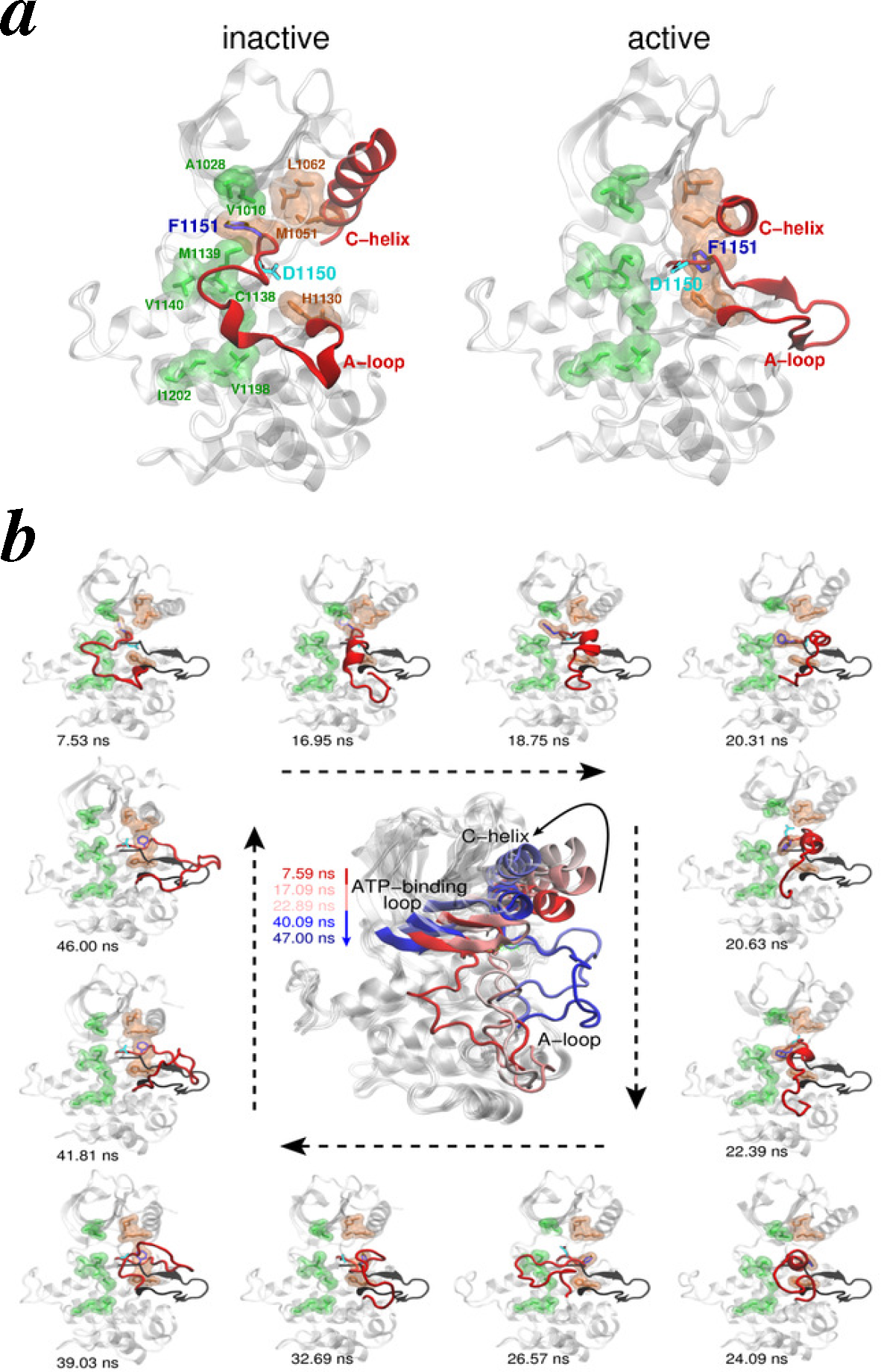
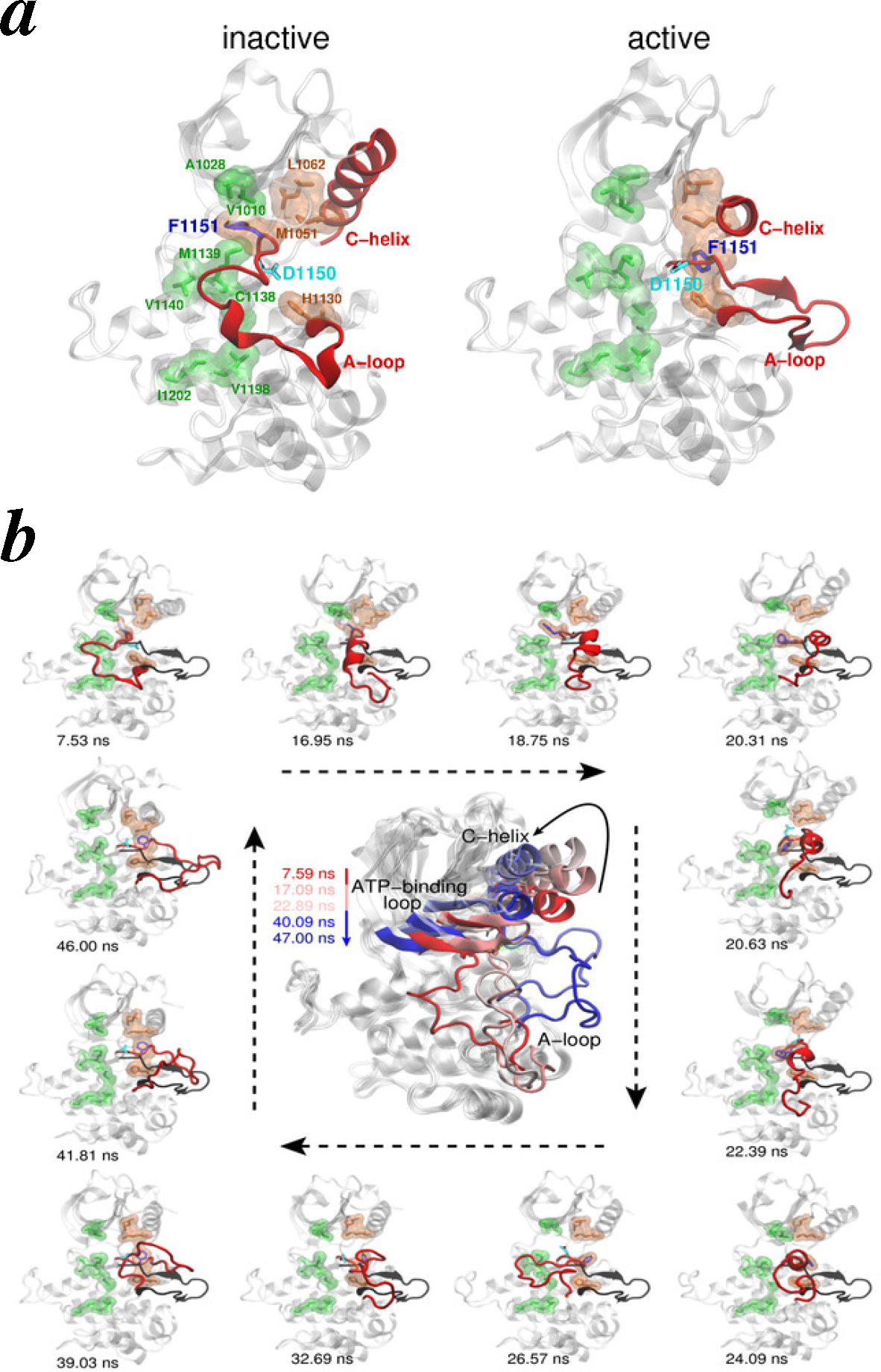
4.2.3. Intracellular Kinase Domains
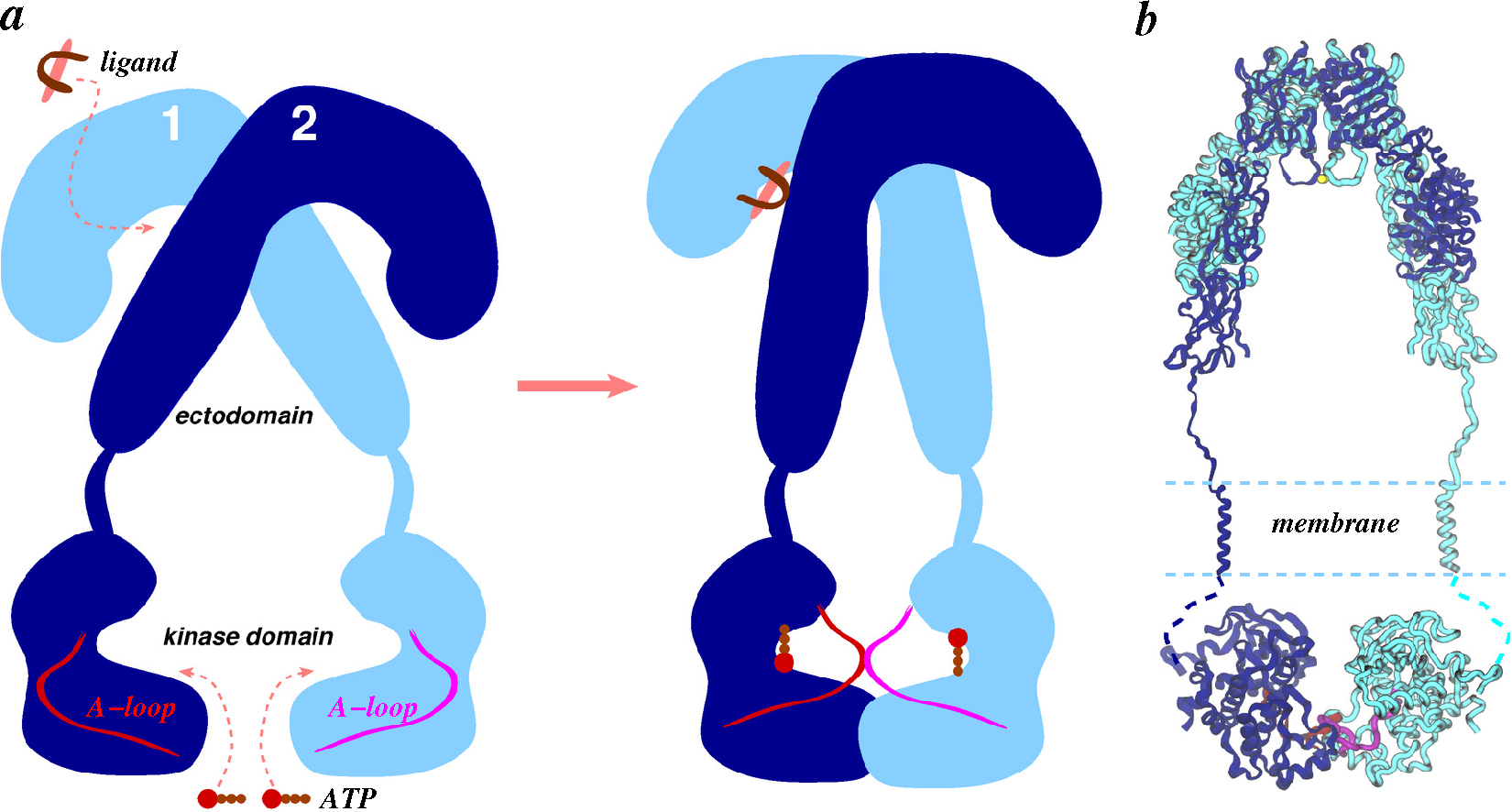
4.3. Limitations of Modeling Studies
5. Outlook
Acknowledgments
Conflicts of Interest
References
- Heller, S.; Kozlovski, P.; Kurtzhals, P. Insulin’s 85th anniversary-An enduring medical miracle. Diabetes Res. Clin. Pract. 2007, 78, 149–158. [Google Scholar]
- De Meyts, P. Insulin and its receptor: Structure, function and evolution. BioEssays 2004, 26, 1351–1362. [Google Scholar]
- Rosenfeld, L. Insulin: Discovery and controversy. Clin. Chem. 2002, 48, 2270–2288. [Google Scholar]
- King, K.M. A history of insulin: From discovery to modern alternatives. Br. J. Nurs. 2003, 12, 1137–1141. [Google Scholar]
- Ward, C.W.; Lawrence, M.C. Landmarks in insulin research. Front. Endocrin. 2011, 2, 76. [Google Scholar]
- Vashisth, H. Molecular simulation studies of the insulin receptor family. Ph.D. Thesis, Drexel University, Philadelphia, PA, USA, 2010. [Google Scholar]
- Steiner, D. Adventures with insulin in the Islets of Langerhans. J. Biol. Chem. 2011, 286, 17399–17421. [Google Scholar]
- Banting, F.; Best, C.; Collip, J.; Campbell, W.; Fletcher, A. Prancreatic extracts in the treatment of diabetes mellitus. Can. Med. Assoc. J 1922, 12, 141–146. [Google Scholar]
- Banting, F.; Best, C. The internal secretion of the pancreas. J. Lab. Clin. Med. 1922, 7, 251–266. [Google Scholar]
- Poulsen, J.E. The impact of August Krogh on the insulin treatment of diabetes and our present status. Acta. Med. Scand. 1975, 578 Suppl, 7–14. [Google Scholar]
- Scott, D.; Fisher, A. The effect of zinc salts on the action of insulin. J. Pharmacol. Exp. Ther. 1935, 55, 206. [Google Scholar]
- Hagedorn, H.; Jensen, B.; Krarup, N.; Wodstrup, I. Portamine Insulinate. J. Am. Med. Assoc. 1936, 106, 177–180. [Google Scholar]
- Steiner, D.F.; James, D.E. Cellular and molecular biology of the beta cell. Diabetologia 1992, 35, S41–S48. [Google Scholar]
- Yalow, R.S. Radioimmunoassay. Annu. Rev. Biophys. Bioeng. 1980, 9, 327–345. [Google Scholar]
- Yalow, R.S. Radioimmunoassay: A probe for fine structure of biologic systems. Scand. J. Immunol. 1992, 35, 4–20. [Google Scholar]
- Felig, P. Protamine insulin-Hagedorn’s pioneering contribution to drug delivery in the management of diabetes. J. Am. Med. Assoc. 1984, 251, 393–396. [Google Scholar]
- Brange, J.; Ribel, U.; Hansen, J.F.; Dodson, G.; Hansen, M.T.; Havelund, S.; Melberg, S.G.; Norris, F.; Norris, K.; Snel, L.; et al. Monomeric insulins obtained by protein engineering and their medical implications. Nature 1988, 333, 679–682. [Google Scholar]
- Pandyarajan, V.; Weiss, M.A. Design of non-standard insulin analogs for the treatment of diabetes mellitus. Curr. Diab. Rep. 2012, 12, 697–704. [Google Scholar]
- Bakaysa, D.L.; Radziuk, J.; Havel, H.A.; Brader, M.L.; Li, S.; Dodd, S.W.; Beals, J.M.; Pekar, A.H.; Brems, D.N. Physicochemical basis for the rapid time-action of Lys(B28)Pro(B29)-insulin: Dissociation of a protein-ligand complex. Prot. Sci. 1996, 5, 2521–2531. [Google Scholar]
- Birnbaum, D.T.; Kilcomons, M.A.; DeFelippis, M.R.; Beals, J.M. Assembly and dissociation of human insulin and Lys(B28)Pro(B29)-insulin hexamers: A comparison study. Pharm. Res. 1997, 14, 25–36. [Google Scholar]
- Lowman, A.M.; Morishita, M.; Kajita, M.; Nagai, T.; Peppas, N.A. Oral delivery of insulin using pH-responsive complexation gels. J. Pharm. Sci. 1999, 88, 933–937. [Google Scholar]
- Langer, R.; Peppas, N.A. Advances in biomaterials, drug delivery, and bionanotechnology. AIChE J 2003, 49, 2990–3006. [Google Scholar]
- Foss, A.C.; Goto, T.; Morishita, M.; Peppas, N.A. Development of acrylic-based copolymers for oral insulin delivery. Euro. J. Pharm. Biopharm. 2004, 57, 163–169. [Google Scholar]
- Morishita, M.; Lowman, A.M.; Takayama, K.; Nagai, T.; Peppas, N.A. Elucidation of the mechanism of incorporation of insulin in controlled release systems based on complexation polymers. J. Control. Release. 2002, 81, 25–32. [Google Scholar]
- Mo, R.; Jiang, T.; Di, J.; Tai, W.; Gu, Z. Emerging micro-and nanotechnology based synthetic approaches for insulin delivery. Chem. Soc. Rev. 2014, 43, 3595–3629. [Google Scholar]
- Stretton, A.O.W. The first sequence: Fred Sanger and insulin. Genetics 2002, 162, 527–532. [Google Scholar]
- Adams, M.J.; Blundell, T.; Dodson, E.; Dodson, G.; Vijayan, M.; Baker, E.; Harding, M.; Hodgkin, D.; Rimmer, B.; Sheat, S. Structure of rhombohedral 2 zinc insulin crystals. Nature 1969, 224, 491–495. [Google Scholar]
- Blundell, T.L.; Dodson, G.G.; Hodgkin, D.C.; Mercola, D. Insulin: The structure in the crystal and its reflection in chemistry and biology. Adv. Protein Chem. 1972, 26, 279–402. [Google Scholar]
- Bentley, G.A.; Dodson, E.J.; Dodson, G.G.; Hodgkin, D.C.; Mercola, D. Structure of insulin in 4-zinc insulin. Nature 1976, 261, 166–168. [Google Scholar]
- Baker, E.N.; Blundell, T.L.; Cutfield, J.F.; Cutfield, S.M.; Dodson, G.G.; Hodgkin, D.M.C.; Hubbard, R.E.; Issacs, M.W.; Reynolds, C.D.; Sakabe, K.; et al. The structure of 2Zn pig insulin crystals at 1.5 Å resolution. Phil. Tran. Roy. Soc. Ser. B 1988, 319, 369–456. [Google Scholar]
- Derewenda, U.; Derewenda, Z.; Dodson, E.J.; Dodson, G.G.; Reynolds, C.D.; Smith, G.D.; Sparks, C.; Sweson, D. Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer. Nature 1989, 338, 594–596. [Google Scholar]
- Smith, G.D.; Dodson, G.G. Structure of a rhombohedral R6 insulin/phenol complex. Proteins Struct. Funct. Genet. 1992, 14, 401–408. [Google Scholar]
- Smith, G.D.; Ciszak, E.; Pangborn, W. A novel complex of a phenolic derivative with insulin: Structural features related to the T↔R transition. Protein Sci. 1996, 5, 1502–1511. [Google Scholar]
- Smith, G.D.; Blessing, R.H. Lessons from an aged, dried crystal of T6 human insulin. Acta Crystallogr. D Biol. Crystallogr. 2003, D59, 1384–1394. [Google Scholar]
- Williamson, K.L.; Williams, R.J.P. Conformational analysis by nuclear magnetic resonance: Insulin. Biochemistry 1979, 18, 5966–5972. [Google Scholar]
- Chang, X.; Jorgensen, A.M.M.; Bardrum, P.; Led, J.J. Solution structures of the R6 human insulin hexamer. Biochemistry 1997, 36, 9409–9422. [Google Scholar]
- O’Donoghue, S.I.; Chang, X.; Abseher, R.; Nilges, M.; Led, J.J. Unraveling the symmetry ambiguity in a hexamer: Calculation of the R6 human insulin structure. J. Biomol. NMR. 2000, 16, 93–108. [Google Scholar]
- Cooke, R.M.; Harvey, T.S.; Campbell, I.D. Solution structure of human insulin-like growth factor 1: A nuclear magnetic resonance and restrained molecular dynamics study. Biochemistry 1991, 30, 5484–5491. [Google Scholar]
- Torres, A.M.; Forbes, B.E.; Aplin, S.E.; Wallace, J.C.; Francise, G.L.; Norton, R.S. Solution structure of human insulin-like growthfactor II. Relationship to receptor and binding protein interactions. J. Mol. Biol. 1995, 248, 385–401. [Google Scholar]
- Vajdos, F.F.; Ultsch, M.; Schaffer, M.L.; Deshayes, K.D.; Liu, J.; Skelton, N.J.; de Vos, A.M. Crystal structure of human insulin-like growth factor-1: Detergent binding inhibits binding protein interactions. Biochemistry 2001, 40, 11022–11029. [Google Scholar]
- Sato, A.; Nishimura, S.; Ohkubo, T.; Kyogoku, Y.; Koyama, S.; Kobayashi, M.; Yasuda, T.; Kobayashi, Y. Three-dimensional structure of human insulin-like growth factor-I (IGF-I) determined by 1H-NMR and distance geometry. Int. J. Pept. Protein Res. 1993, 41, 433–440. [Google Scholar]
- Brzozowski, A.M.; Dodson, E.J.; Dodson, G.G.; Murshudov, G.N.; Verma, C.; Turkenburg, J.P.; de Bree, F.M.; Dauter, Z. Structural origins of the functional divergence of human insulin-like growth factor-I and insulin. Biochemistry 2002, 41, 9389–9397. [Google Scholar]
- Cuatrecasas, P. Insulin-receptor interactions in adipose tissue cells: Direct measurement and properties. Proc. Natl. Acad. Sci. USA. 1971, 68, 1264–1268. [Google Scholar]
- Kasuga, M.; Karlsson, F.A.; Kahn, C.R. Insulin stimulates the phosphorylation of the 95,000-Dalton subunit of its own receptor. Science 1982, 215, 185–187. [Google Scholar]
- Roth, R.A.; Cassell, D.J. Insulin Receptor-Evidence that it is a protein kinase. Science 1983, 219, 299–301. [Google Scholar]
- Ebina, Y.; Ellis, L.; Jarnagin, K.; Edery, M.; Graf, L.; Clauser, E.; Ou, J.h.; Masiarz, F.; Kan, Y.; Goldfine, I.; et al. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signalling. Cell 1985, 40, 747–758. [Google Scholar]
- Ullrich, A.; Bell, J.R.; Chen, E.Y.; Herrera, R.; Petruzzelli, L.M.; Dull, T.J.; Gray, A.; Coussens, L.; Liao, Y.C.; Tsubokawa, M.; et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985, 313, 756–761. [Google Scholar]
- Ullrich, A.; Gray, A.; Tam, A.W.; Yangfeng, T.; Tsubokawa, M.; Collins, C.; Henzel, W.; Lebon, T.; Kathuria, S.; Chen, E.; et al. Insulin-like growth factor-I receptor primary structure-Comparison with insulin-receptor suggests structural determinants that define functional specificity. EMBO J 1986, 5, 2503–2512. [Google Scholar]
- De Meyts, P.; Whittaker, J. Structural biology of insulin and IGF1 receptors: Implications for drug design. Nat. Rev. Drug. Discov. 2002, 1, 769–783. [Google Scholar]
- Adams, T.E.; Epa, V.C.; Garrett, T.P.J.; Ward, C.W. Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci. 2000, 57, 1050–1093. [Google Scholar]
- Hubbard, R.D.; Wilsbacher, J.L. Advances towards the development of ATP-competitive small-molecule inhibitors of the insulin-like growth factor receptor (IGF-IR). ChemMedChem 2007, 2, 41–46. [Google Scholar]
- Ward, C.W.; Lawrence, M.C. Ligand-induced activation of the insulin receptor: A multi-step process involving structural changes in both the ligand and the receptor. BioEssays 2009, 31, 422–434. [Google Scholar]
- Humbel, R.E. Insulin-like growth factors I and II. Eur. J. Biochem. 1990, 190, 445–462. [Google Scholar]
- Cohick, W.; Clemmons, D. The insulin-like growth factors. Annu. Rev. Physiol. 1993, 55, 131–153. [Google Scholar]
- Khandwala, H.M.; McCutcheon, I.E.; Flyvbjerg, A.; Friend, K.E. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr. Rev. 2000, 21, 215–244. [Google Scholar]
- Zhang, X.; Yee, D. Tyrosine kinase signalling in breast cancer insulin-like growth factors and their receptors in breast cancer. Breast Cancer Res. 2000, 2, 170–175. [Google Scholar]
- Yu, H.; Rohan, T. Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst. 2000, 92, 1472–1489. [Google Scholar]
- Leroith, D.; Roberts, C.T., Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003, 195, 127–137. [Google Scholar]
- Denley, A.; Wang, C.X.C.; McNeil, K.A.; Walenkamp, M.J.E.; Duyvenvoorde, H.V.; Wit, J.M.; Wallace, J.C.; Norton, R.S.; Karperien, M.; Forbes, B.E. Structural and functional characteristics of the Val(44)Met insulin-like growth factor I missense mutation: Correlation with effects on growth and development. Mol. Endocrinol. 2005, 19, 711–721. [Google Scholar]
- Belfiore, A. The role of insulin receptor isoforms and hybrid Insulin/IGF-I receptors in human cancer. Curr. Pharm. Des. 2007, 13, 671–686. [Google Scholar]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets for cancer therapy. Nat. Rev. Cancer. 2004, 4, 361–370. [Google Scholar]
- Deyev, I.E.; Sohet, F.; Vassilenko, K.P.; Serova, O.V.; Popova, N.V.; Zozulya, S.A.; Burova, E.B.; Houillier, P.; Rzhevsky, D.I.; Berchatova, A.A.; et al. Insulin receptor-related receptor as an extracellular alkali sensor. Cell Metab. 2011, 13, 679–689. [Google Scholar]
- Bhattacharya, A. Protein structures: Structures of desire. Nature 2009, 459, 24–27. [Google Scholar]
- Hubbard, S.R.; Wei, L.; Elis, L.; Hendrickson, W.A. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 1994, 372, 746–754. [Google Scholar]
- Hubbard, S.R. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J 1997, 16, 5572–5581. [Google Scholar]
- Favelyukis, S.; Till, J.H.; Hubbard, S.R.; Miller, W.T. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Mol. Biol. 2001, 8, 1058–1063. [Google Scholar]
- Munshi, S.; Kornienko, M.; Hall, D.L.; Reid, J.C.; Waxman, L.; Stirdivant, S.M.; Darke, P.L.; Kuo, L.C. Crystal structure of the Apo, unactivated insulin-like growth factor-1 receptor kinase implication for inhibitor specificity. J. Biol. Chem. 2002, 277, 38797–38802. [Google Scholar]
- Garrett, T.P.J.; McKern, N.M.; Lou, M.Z.; Frenkel, M.J.; Bentley, J.D.; Lovrecz, G.; Elleman, T.C.; Cosgrove, L.J.; Ward, C.W. Crystal structure of the first three domains of the type-1 insulin-like growth factor receptor. Nature 1998, 394, 395–399. [Google Scholar]
- Tulloch, P.; Lawrence, L.; McKern, N.; Robinson, C.; Bentley, J.; Cosgrove, L.; Ivancic, N.; Lovrecz, G.; Siddle, K.; Ward, C. Single-molecule imaging of human insulin receptor ectodomain and its Fab complexes. J. Struc. Biol. 1999, 125, 11–18. [Google Scholar]
- Lou, M.; Garrett, T.; McKern, N.; Hoyne, P.; Epa, V.; Bentley, J.; Lovrecz, G.; Cosgrove, L.; Frenkel, M.; Ward, C. The first three domains of the insulin receptor differ structurally from the insulin-like growth factor 1 receptor in the regions governing ligand specificity. Proc. Natl. Acad. Sci. USA. 2006, 103, 12429–12434. [Google Scholar]
- McKern, N.; Lawrence, M.; Streltsov, V.; Lou, M.; Adams, T.; Lovrecz, G.; Elleman, T.; Richards, K.; Bentley, J.; Pilling, P.; et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 2006, 443, 218–221. [Google Scholar]
- Smith, B.J.; Huang, K.; Kong, G.; Chan, S.J.; Nakagawa, S.; Menting, J.G.; Hu, S.Q.; Whittaker, J.; Steiner, D.F.; Katsoyannis, P.G.; et al. Structural resolution of a tandem hormone-binding element in the insulin receptor and its implications for design of peptide agonists. Proc. Natl. Acad. Sci. USA. 2010, 107, 6771–6776. [Google Scholar]
- Whitten, A.E.; Smith, B.J.; Menting, J.G.; Margetts, M.B.; McKern, N.M.; Lovrecz, G.O.; Adams, T.E.; Richards, K.; Bentley, J.D.; Trewhella, J.; et al. Solution structure of ectodomains of the insulin receptor family: The ectodomain of the Type 1 insulin-like growth factor receptor displays asymmetry of ligand binding accompanied by limited conformational change. J. Mol. Biol. 2009, 394, 878–892. [Google Scholar]
- Luo, R.Z.T.; Beniac, D.R.; Fernandes, A.; Yip, C.C.; Ottensmeyer, F.P. Quaternary structure of the insulin-insulin receptor complex. Science 1999, 285, 1077–1080. [Google Scholar]
- Ward, C.; Lawrence, M.; Streltsov, V.; Adams, T.; McKern, N. The insulin and EGF receptor structures: New insights into ligand-induced receptor activation. Trends Biochem. Sci. 2007, 32, 129–137. [Google Scholar]
- Menting, J.G.; Whittaker, J.; Margetts, M.B.; Whittaker, L.J.; Kong, G.K.W.; Smith, B.J.; Watson, C.J.; Žáková, L.; Kletvíková, E.; Jiráček, J.; et al. How insulin engages its primary binding site on the insulin receptor. Nature 2013, 493, 241–245. [Google Scholar]
- Menting, J.G.; Yang, Y.; Chan, S.J.; Phillips, N.B.; Smith, B.J.; Whittaker, J.; Wickramasinghe, N.P.; Whittaker, L.J.; Pandyarajan, V.; Wan, Z.L.; et al. Protective hinge in insulin opens to enable its receptor engagement. Proc. Natl. Acad. Sci. USA. 2014, 111, E3395–E3404. [Google Scholar]
- Li, Q.; Wong, Y.L.; Kang, C. Solution structure of the transmembrane domain of the insulin receptor in detergent micelles. Biochim. Biophys. Acta. 1313–1321.
- Ward, C.W.; Garrett, T.P. The relationship between the L1 and L2 domains of the insulin and epidermal growth factor receptors and leucine-rich repeat modules. BMC Bioinform. 2001, 2, 4. [Google Scholar] [Green Version]
- Lawrence, M.; McKern, N.; Ward, C. Insulin receptor structure and its implications for the IGF-1 receptor. Curr. Opin. Struct. Biol. 2007, 17, 699–705. [Google Scholar]
- Ward, C.; Lawrence, M.; Streltsov, V.; Garrett, T.; McKern, N.; Lou, M.; Lovrecz, G.; Adams, T. Structural insights into ligand-induced activation of the insulin receptor. Acta Physiol. 2008, 192, 3–9. [Google Scholar]
- Ward, C.; Lawrence, M. Similar but different: Ligand-induced activation of the insulin and epidermal growth factor receptor families. Curr. Opin. Struct. Biol. 2012, 22, 1–7. [Google Scholar]
- Ward, C.W.; Menting, J.G.; Lawrence, M.C. The insulin receptor changes conformation in unforeseen ways on ligand binding: Sharpening the picture of insulin receptor activation. Bioessays 2013, 35, 945–954. [Google Scholar]
- De Meyts, P. The structural basis of insulin and insulin-like growth factor-I receptor binding and negative co-operativity, and its relevance to mitogenic versus metabolic signalling. Diabetologia 1994, 37, S135–S148. [Google Scholar]
- De Meyts, P. The insulin receptor: A prototype for dimeric, allosteric membrane receptors? Trends Biochem. Sci. 2008, 33, 376–384. [Google Scholar]
- Jensen, M.; de Meyts, P. Molecular mechanisms of differential intracellular signaling from the insulin receptor. Vitam. Horm. 2009, 80, 51–75. [Google Scholar]
- Hubbard, S.R.; Mohammadi, M.; Schlessinger, J. Autoregulatory mechanisms in protein-tyrosine kinases. J. Biol. Chem. 1998, 273, 11987–11990. [Google Scholar]
- Hubbard, S.R.; Till, J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar]
- Hubbard, S.R. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2004, 5, 464–471. [Google Scholar]
- Hubbard, S.R.; Miller, W.T. Receptor tyrosine kinases: Mechanisms of activation and signaling. Curr. Opin. Cell Biol. 2007, 19, 117–123. [Google Scholar]
- Hubbard, S.R. Structural biology: Insulin meets its receptor. Nature 2013, 493, 171–172. [Google Scholar]
- Kahn, C.R.; White, M. The insulin receptor and the molecular mechanism of insulin action. J. Clin. Invest. 1988, 82, 1151–1156. [Google Scholar]
- Tavaré, J.M.; Siddle, K. Mutational analysis of insulin receptor function: Consensus and controversy. Biochim. Biophys. Acta. 1993, 1178, 21–39. [Google Scholar]
- White, M.F.; The, IRS-signalling. system: A network of docking proteins that mediate insulin action. Mol. Cellul. Biochem. 1998, 182, 3–11. [Google Scholar]
- Di Guglielmo, G.M.; Baass, P.C.; Authier, F.; Posner, B.I.; Bergeron, J.J. Insulin receptor internalization and signalling. Mol. Cellul. Biochem. 1998, 182, 59–63. [Google Scholar]
- Marino-Buslje, C.; Martin-Martinez, M.; Mizuguchi, K.; Siddle, K.; Blundell, T. The insulin receptor: From protein sequence to structure. Biochem. Soc. Trans. 1999, 27, 715–726. [Google Scholar]
- Kitamura, T.; Kahn, C.; Accili, D. Insulin receptor knockout mice. Annu. Rev. Physiol. 2003, 65, 313–332. [Google Scholar]
- Ussar, S.; Vienberg, S.G.; Kahn, C.R. Receptor antibodies as novel therapeutics for diabetes. Sci. Transl. Med. 2011, 3, 113ps47. [Google Scholar]
- Maruyama, I.N. Mechanisms of activation of receptor tyrosine kinases: Monomers or dimers. Cells 2014, 3, 304–330. [Google Scholar]
- Weiss, M.A. Proinsulin and the genetics of diabetes mellitus. J. Biol. Chem. 2009, 284, 19159–19163. [Google Scholar]
- Kamrava, M.; Gius, D.; Casagrande, G.; Kohn, E. Will targeting insulin growth factor help us or hurt us?: An oncologist’s perspective. Ageing Res. Rev. 2011, 10, 62–70. [Google Scholar]
- Kaarsholm, N.C.; Ko, H.C.; Dunn, M.F. Comparison of solution structural flexibility and zinc binding domains for insulin, proinsulin, and miniproinsulin. Biochemistry 1989, 28, 4427–4435. [Google Scholar]
- Roy, M.; Brader, M.L.; Lee, R.W.K.; Kaarsholm, N.C.; Hansen, J.F.; Dunn, M.F. Spectroscopic signatures of the T to R conformational transition in the insulin hexamer. J. Mol. Biol. 1989, 264, 19081–19085. [Google Scholar]
- Berchtold, H.; Hilgenfeld, R. Binding of phenol to R6 insulin hexamers. Biopolymers 1999, 51, 165–172. [Google Scholar]
- Sohma, Y.; Pentelute, B.L.; Whittaker, J.; Hua, Q.; Whittaker, L.J.; Weiss, M.A.; Kent, S.B.H. Comparative properties of insulin-like growth factor 1 (IGF-1) and [Gly7D-Ala]IGF-1 prepared by total chemical synthesis. Angew. Chem. Int. Ed. 2008, 47, 1102–1106. [Google Scholar]
- Cosgrove, L.; Lovrecz, G.O.; Verkuylen, A.; Cavaleri, L.; Black, L.A.; Bentley, J.D.; Howlett, G.J.; Gray, P.P.; Ward, C.W.; McKern, N.M. Purification and properties of insulin receptor ectodomain from large-scale mammalian cell culture. Protein Expr. Purif. 1995, 6, 789–798. [Google Scholar]
- Sparrow, L.G.; Gorman, J.J.; Strike, P.M.; Robinson, C.P.; McKern, N.M.; Epa, V.C.; Ward, C.W. The location and characterisation of the O-linked glycans of the human insulin receptor. Proteins 2007, 66, 261–265. [Google Scholar]
- Sparrow, L.G.; Lawrence, M.C.; Gorman, J.J.; Strike, P.M.; Robinson, C.P.; McKern, N.M.; Ward, C.W. N-linked glycans of the human insulin receptor and their distribution over the crystal structure. Proteins 2008, 71, 426–439. [Google Scholar]
- Massague, J.; Pilch, P.F.; Czech, M.P. Electrophoretic resolution of three major insulin receptor structures with unique subunit stoichiometries. Proc. Natl. Acad. Sci. USA. 1980, 77, 7137–7141. [Google Scholar]
- Sparrow, L.G.; McKern, N.M.; Gorman, J.J.; Strike, P.M.; Robinson, C.P.; Bentley, J.D.; Ward, C.W. The disulfide bonds in the C-terminal domains of the human insulin receptor ectodomain. J. Biol. Chem. 1997, 272, 29460–29467. [Google Scholar]
- Vashisth, H.; Abrams, C. Docking of insulin to a structurally equilibrated insulin receptor ectodomain. Proteins 2010, 78, 1531–1543. [Google Scholar]
- Vashisth, H.; Abrams, C. All-atom structural models for complexes of insulin-like growth factors IGF1 and IGF2 with their cognate receptor. J. Mol. Biol. 2010, 400, 645–658. [Google Scholar]
- Pullen, R.; Lindsay, D.; Wood, S.; Tickle, I.; Blundell, T.; Wollmer, A.; Krail, G.; Brandenburg, D.; Zahn, H.; Gliemann, J.; Gammeltoft, S. Receptor-binding region of insulin. Nature 1976, 259, 369–373. [Google Scholar]
- De Meyts, P.; Obberghen, E.; Roth, J.; Wollmer, A.; Brandenburg, D. Mapping of the residues responsible for the negative cooperativity of the receptor-binding region of insulin. Nature 1978, 273, 504–509. [Google Scholar]
- Kristensen, C.; Kjeldsen, T.; Wiberg, F.; Schäffer, L.; Hach, M.; Havelund, S.; Bass, J.; Steiner, D.; Andersen, A. Alanine scanning mutagenesis of insulin. J. Biol. Chem. 1997, 272, 12978–12983. [Google Scholar]
- Chen, H.; Shi, M.; Guo, Z.; Tang, Y.; Qiao, Z.; Liang, Z.; Feng, Y. Four new monomeric insulins obtained by alanine scanning the dimer-forming surface of the insulin molecule. Protein Eng. 2000, 13, 779–782. [Google Scholar]
- Nakamura, T.; Takahashi, H.; Takahashi, M.; Shimba, N.; Suzuki, E.I.; Shimada, I. Direct determination of the insulin-insulin receptor interface using transferred cross-saturation experiments. J. Med. Chem. 2010, 53, 1917–1922. [Google Scholar]
- Williams, P.; Mynarcik, D.; Yu, G.; Whittaker, J. Mapping of an NH2-terminal ligand binding site of the insulin receptor by alanine scanning mutagenesis. J. Biol. Chem. 1995, 270, 3012–3016. [Google Scholar]
- Whittaker, J.; Whittaker, L. Characterization of the functional insulin binding epitopes of the full-length insulin receptor. J. Biol. Chem. 2005, 280, 20932–20936. [Google Scholar]
- Mynarcik, D.; Williams, P.; Schaffer, L.; Yu, G.; Whittaker, J. Analog binding properties of insulin receptor mutants. J. Biol. Chem. 1997, 272, 2077–2081. [Google Scholar]
- Schaefer, E.; Siddle, K.; Ellis, L. Deletion analysis of the human insulin receptor ectodomain reveals independently folded soluble subdomains and insulin binding by a monomeric α-subunit. J. Biol. Chem. 1990, 265, 13248–13253. [Google Scholar]
- Kristensen, C.; Andersen, A.; Hach, M.; Wiberg, F.; Schäffer, L.; Kjeldsen, T. A single-chain insulin-like growth factor I/insulin hybrid binds with high affinity to the insulin receptor. Biochem. J 1995, 305, 981–986. [Google Scholar]
- Schlein, M.; Havelund, S.; Kristensen, C.; Dunn, M.; Kaarsholm, N. Ligand-induced conformational change in the minimized insulin receptor. J. Mol. Biol. 2000, 303, 161–169. [Google Scholar]
- Brandt, J.; Andersen, A.; Kristensen, C. Dimeric fragment of the insulin receptor α-subunit binds insulin with full holoreceptor affinity. J. Biol. Chem. 2001, 276, 12378–12384. [Google Scholar]
- Surinya, K.; Molina, L.; Soos, M.; Brandt, J.; Kristensen, C.; Siddle, K. Role of insulin receptor dimerization domains in ligand binding, cooperativity, and modulation by anti-receptor antibodies. J. Biol. Chem. 2002, 277, 16718–16725. [Google Scholar]
- Kristensen, C.; Andersen, A.; Østergaard, S.; Hansen, P.; Brandt, J. Functional reconstitution of insulin receptor binding site from non-binding receptor fragments. J. Biol. Chem. 2002, 277, 18340–18345. [Google Scholar]
- Kurose, T.; Pashmforoush, M.; Yoshimasa, Y.; Carroll, R.; Schwartz, G.; Burke, G.; Katsoyannis, P.; Steiner, D. Cross-linking of a B25 Azidophenylalanine insulin derivative to the carboxyl-terminal regions of the α-subunit of the insulin receptor. J. Biol. Chem. 1994, 269, 29190–29197. [Google Scholar]
- Mynarcik, D.; Yu, G.; Whittaker, J. Alanine-scanning mutagenesis of a C-terminal ligand binding domain in the insulin receptor α subunit. J. Biol. Chem. 1996, 271, 2439–2442. [Google Scholar]
- Kristensen, C.; Wiberg, F.; Andersen, A. Specificity of insulin and insulin-like growth factor I receptors investigated using chimeric mini-receptors. J. Biol. Chem. 1999, 274, 37351–37356. [Google Scholar]
- Molina, L.; Marino-Buslje, C.; Quinn, D.; Siddle, K. Structural domains of the insulin receptor and IGF receptor required for dimerization and ligand binding. FEBS Lett. 2000, 467, 226–230. [Google Scholar]
- Whittaker, L.; Hao, C.; Fu, W.; Whittaker, J. High-affinity insulin binding: Insulin interacts with two receptor ligand binding sites. Biochemistry 2008, 47, 12900–12909. [Google Scholar]
- Zhang, B.; Roth, R. A region of the insulin receptor important for ligand binding (residues 450-601) is recognized by patients’ autoimmune antibodies and inhibitory monoclonal antibodies. Proc. Natl. Acad. Sci. USA. 1991, 88, 9858–9862. [Google Scholar]
- Fabry, M.; Schaefer, E.; Ellis, L.; Kojro, E.; Fahrenholz, F.; Brandenburg, D. Detection of a new hormone contact site within the insulin receptor ectodomain by the use of a novel photoreactive insulin. J. Biol. Chem. 1992, 267, 8950–8956. [Google Scholar]
- Schumacher, R.; Soos, M.; Schlessinger, J.; Brandenburg, D.; Siddle, K.; Ullrich, A. Signaling-competent receptor chimeras allow mapping of major insulin receptor binding domain determinants. J. Biol. Chem. 1993, 268, 1087–1094. [Google Scholar]
- Hao, C.; Whittaker, L.; Whittaker, J. Characterization of a second ligand binding site of the insulin receptor. Biochem. Biophys. Res. Commun. 2006, 347, 334–339. [Google Scholar]
- Benyoucef, S.; Surinya, K.; Hadaschik, D.; Siddle, K. Characterization of insulin/IGF hybrid receptors: Contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. Biochem. J 2007, 403, 603–613. [Google Scholar]
- Renteria, M.; Gandhi, H.; Vinuesa, P.; Helmerhorst, E.; Mancera, R. A comparative structural bioinformatics analysis of the insulin receptor family ectodomain based on phylogenetic information. PLoS One. 2008, 3, e3667. [Google Scholar]
- Bayne, M.L.; Applebaum, J.; Chicchi, G.G.; Hayes, N.S.; Green, B.G.; Cascieri, M.A. Structural analogs of human insulin-like growth factor-I with reduced affinity for serum binding-proteins and the type-2 insulin-like growth-factor receptor. J. Biol. Chem. 1988, 263, 6233–6239. [Google Scholar]
- Cascieri, M.A.; Chicchi, G.G.; Applebaum, J.; Hayes, N.S.; Green, B.G.; Bayne, M.L. Identification of the domains of IGF-I responsible for high-affinity binding to the type-1 and type-2 IGF receptors (IGF-R1, IGF-R2), insulin-receptor (IR) and binding-proteins (BP). FASEB J 1988, 2, A1773. [Google Scholar]
- Cascieri, M.A.; Chicchi, G.G.; Applebaum, J.; Green, B.G.; Hayes, N.S.; Bayne, M.L. Structural analogs of human insulin-like growth-factor (IGF)-I with altered affinity for type-2 IGF receptors. J. Biol. Chem. 1989, 264, 2199–2202. [Google Scholar]
- Bayne, M.L.; Applebaum, J.; Chicchi, G.G.; Miller, R.E.; Cascieri, M.A. The roles of Tyrosine-24, Tyrosine-31, and Tyrosine-60 in the high-affinity binding of insulin-like growth factor-I to the type-I insulin-like growth-factor receptor. J. Biol. Chem. 1990, 265, 15648–15652. [Google Scholar]
- Sakano, K.I.; Enjoh, T.; Numata, F.; Fujiwara, H.; Marumoto, Y.; Higashihashi, N.; Sato, Y.; Perdue, J.F.; Fujitayamaguchi, Y. The design, expression, and characterization of human insulin-like growth factor-II (IGF-II) mutants specific for either the IGF-II cation-independent Mannose 6-Phosphate receptor or IGF-1 receptor. J. Biol. Chem. 1991, 266, 20626–20635. [Google Scholar]
- Roth, B.V.; Burgisser, D.M.; Luthi, C.; Humbel, R.E. Mutants of human insulin-like growth factor-II-Expression and characterization of analogs with a substitution of Tyr27 and/or a deletion of residues 62–67. Biochem. Biophys. Res. Commun. 1991, 181, 907–914. [Google Scholar]
- Burgisser, D.M.; Roth, B.V.; Giger, R.; Luthi, C.; Weigl, S.; Zarn, J.; Humbel, R.E. Mutants of human insulin-like growth factor-II with altered affinities for the type-1 and type-2 insulin-like growth-factor receptor. J. Biol. Chem. 1991, 266, 1029–1033. [Google Scholar]
- King, R.; Wells, J.R.E.; Krieg, P.; Snoswell, M.; Brazier, J.; Bagley, C.J.; Wallace, J.C.; Ballard, F.J.; Ross, M.; Francis, G.L. Production and characterization of recombinant insulin-like growth-factor-I (IGF-I) and potent analogs of IGF-I, with Gly or Arg substituted for Glu3, following their expression in Escherichia-Coli as fusion proteins. J. Mol. Endocrinol. 1992, 8, 29–41. [Google Scholar]
- Francis, G.L.; Ross, M.; Ballard, F.J.; Milner, S.J.; Senn, C.; McNeil, K.A.; Wallace, J.C.; King, R.; Wells, J.R.E. Novel recombinant fusion protein analogs of insulin-like growth-factor (IGF)-1 indicate the relative importance of IGF-binding protein and receptor-binding for enhanced biological potency. J. Mol. Endocrinol. 1992, 8, 213–223. [Google Scholar]
- Luthi, C.; Roth, B.V.; Humbel, R.E. Mutants of human insulin-like growth factor-II (IGF-II)-Expression and characterization of truncated IGF-II and of 2 naturally-occurring variants. Eur. J. Biochem. 1992, 205, 483–490. [Google Scholar]
- Zhang, W.G.; Gustafson, T.A.; Rutter, W.J.; Johnson, J.D. Positively charged side-chains in the insulin-like growth-factor-I C-regions and D-regions determine receptor-binding specificity. J. Biol. Chem. 1994, 269, 10609–10613. [Google Scholar]
- Hodgson, D.R.; May, F.; Westley, B.R. Mutations at position-11 and position-60 of insulin-like growth factor-1 reveal differences between its interactions with the type-I insulin-like-growth-factor receptor and the insulin receptor. Eur. J. Biochem. 1995, 233, 299–309. [Google Scholar]
- Shooter, G.K.; Magee, B.; Soos, M.A.; Francis, G.L.; Siddle, K.; Wallace, J.C. Insulin-like growth factor (IGF)-I A- and B-domain analogues with altered type 1 IGF and insulin receptor binding specificities. J. Mol. Endocrinol. 1996, 17, 237–246. [Google Scholar]
- Hodgson, D.R.; May, F.; Westley, B.R. Involvement of phenylalanine 23 in the binding of IGF-1 to the insulin and type I IGF receptor. Regul. Peptides. 1996, 66, 191–196. [Google Scholar]
- Jansson, M.; Uhlen, M.; Nilsson, B. Structural changes in insulin-like growth factor (IGF) I mutant proteins affecting binding kinetic rates to IGF binding protein 1 and IGF-I receptor. Biochemistry 1997, 36, 4108–4117. [Google Scholar]
- Jansson, M.; Andersson, G.; Uhlen, M.; Nilsson, B.; Kordel, J. The insulin-like growth factor (IGF)binding protein 1 binding epitope on IGF-I probed by heteronuclear NMR spectroscopy and mutational analysis. J. Biol. Chem. 1998, 273, 24701–24707. [Google Scholar]
- Yandell, C.A.; Francis, G.L.; Wheldrake, J.F.; Upton, Z. Kangaroo IGF-II is structurally and functionally similar to the human [Ser(29)]-IGF-II variant. J. Endocrinol. 1999, 161, 445–453. [Google Scholar]
- Magee, B.A.; Shooter, G.K.; Wallace, J.C.; Francis, G.L. Insulin-like growth factor I and its binding proteins: A study of the binding interface using B-domain analogues. Biochemistry 1999, 38, 15863–15870. [Google Scholar]
- Dubaquie, Y.; Mortensen, D.L.; Intintoli, A.; Hogue, D.A.; Nakamura, G.; Rancatore, P.; Lester, P.; Sadick, M.D.; Filvaroff, E.; Fielder, P.J.; et al. Binding protein-3-selective insulin-like growth factor I variants: Engineering, biodistributions, and clearance. Endocrinology 2001, 142, 165–173. [Google Scholar]
- Forbes, B.E.; McNeil, K.A.; Scott, C.D.; Surinya, K.H.; Cosgrove, L.J.; Wallace, J.C. Contribution of residues A54 and L55 of the human insulin-like growth factor-II (IGF-II) A domain to type 2 IGF receptor binding specificity. Growth Factors. 2001, 19, 163–173. [Google Scholar]
- Sørensen, H.; Whittaker, L.; Hinrichsen, J.; Groth, A.; Whittaker, J. Mapping of the insulin-like growth factor II binding site of the type I insulin-like growth factor receptor by alanine scanning mutagenesis. FEBS Lett. 2004, 565, 19–22. [Google Scholar]
- Gauguin, L.; Delaine, C.; Alvino, C.L.; McNeil, K.A.; Wallace, J.C.; Forbes, B.E.; de Meyts, P. Alanine scanning of a putative receptor binding surface of insulin-like growth factor-I. J. Biol. Chem. 2008, 283, 20821–20829. [Google Scholar]
- Alvino, C.L.; McNeil, K.A.; Ong, S.C.; Delaine, C.; Booker, G.W.; Wallace, J.C.; Whittaker, J.; Forbes, B.E. A novel approach to identify two distinct receptor binding surfaces of insulin-like growth factor II. J. Biol. Chem. 2009, 284, 7656–7664. [Google Scholar]
- Gauguin, L.; Klaproth, B.; Sajid, W.; Andersen, A.S.; McNeil, K.A.; Forbes, B.E.; de Meyts, P. Structural basis for the lower affinity of the insulin-like growth factors for the insulin receptor. J. Biol. Chem. 2008, 282, 2604–2613. [Google Scholar]
- Andersen, A.S.; Kjeldsen, T.; Wiberg, F.C.; Christensen, P.M.; Ramussen, J.S.; Norris, K.; Møller, K.B.; Møller, N.P.H. Changing the insulin-receptor to possess insulin-like growth factor-I ligand specificity. Biochemistry 1990, 29, 7363–7366. [Google Scholar]
- Kjeldsen, T.; Andersen, A.S.; Wiberg, F.C.; Ramussen, J.S.; Schaffer, L.; Balschmidt, P.; Møller, K.B.; Møller, N.P.H. The ligand specificities of the insulin-receptor and the insulin-like growth factor-I receptor reside in different regions of a common binding-site. Proc. Natl. Acad. Sci. USA. 1991, 88, 4404–4408. [Google Scholar]
- Schumacher, R.; Mosthaf, L.; Schlessinger, J.; Brandenburg, D.; Ullrich, A. Insulin and insulin-like growth factor-I binding-specificity is determined by distinct regions of their cognate receptors. J. Biol. Chem. 1991, 266, 19288–19295. [Google Scholar]
- Zhang, B.; Roth, R.A. Binding-properties of chimeric insulin-receptors containing the cysteine-rich domain of either the insulin-like growth factor-I receptor or the insulin-receptor related receptor. Biochemistry 1991, 30, 5113–5117. [Google Scholar]
- Hoyne, P.A.; Cosgrove, L.J.; Mckern, N.M.; Bentley, J.D.; Ivancic, N.; Elleman, T.C.; Ward, C.W. High affinity insulin binding by soluble insulin receptor extracellular domain fused to a leucine zipper. FEBS Lett. 2000, 479, 15–18. [Google Scholar]
- Gill, R.; Wallach, B.; Verma, C.; Urso, B.; de Wolf, E.; Grotzinger, J.; Murray-Rust, J.; Pitts, J.; Wollmer, A.; de Meyts, P.; et al. Engineering the C-region of human insulin-like growth factor-1: Implications for receptor binding. Prot. Eng. 1996, 9, 1011–1019. [Google Scholar]
- Keyhanfar, M.; Booker, G.W.; Whittaker, J.; Wallace, J.C.; Forbes, B.E. Precise mapping of an IGF-I-binding site on the IGF-1R. Biochem. J 2007, 401, 269–277. [Google Scholar]
- Kiselyov, V.; Versteyhe, S.; Gauguin, L.; de Meyts, P. Harmonic oscillator model of the insulin and IGF1 receptors’ allosteric binding and activation. Mol. Syst. Biol. 2009, 5, 1–12. [Google Scholar]
- De Meyts, P. Insulin interactions with its receptors: Experimental evidence for negative coooperativity. Biochem. Biophys. Res. Comm. 1973, 55, 154–161. [Google Scholar]
- De Meyts, P.; Bianco, A.R.; Roth, J. Site-site interactions among insulin receptors: Characterization of the negative cooperativity. J. Biol. Chem. 1976, 251, 1877–1888. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulations: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Leach, A.R. Molecular Modelling: Principles and Applications, 2nd ed.; Pearson Education Limited: Harlow, UK, 2001. [Google Scholar]
- Schlick, T. Molecular Modeling and Simulation: An Interdisciplinary Guide, 1st ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Rapaport, D.C. The Art of Molecular Dynamics Simulation, 2nd ed.; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Karplus, M.; Petsko, G.A. Molecular dynamics simulations in biology. Nature 1990, 347, 631–639. [Google Scholar]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar]
- Karplus, M.; Kuriyan, J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA. 2005, 102, 6679–6685. [Google Scholar]
- MacKerell, A.D., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R.L., Jr.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar]
- MacKerell, A., Jr.; Feig, M.; Brooks, C, III. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.V.; Dinola, A.; Haa, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar]
- Hoover, W.G.; Ladd, A.J.C.; Moran, C. High-strain-rate plastic-flow studied via non-equilibrium molecular dynamics. Phys. Rev. Lett. 1982, 48, 1818–1820. [Google Scholar]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar]
- Hoover, W.G. Canonical dynamics-Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar]
- Kalé, L.; Skeel, R.; Bhandarkar, M.; Brunner, R.; Gursoy, A.; Krawetz, N.; Phillips, J.; Shinozaki, A.; Varadarajan, K.; Schulten, K. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar]
- Phillips, J.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar]
- Van Gunsteren, W.F.; Mark, A.E. Validation of molecular dynamics simulation. J. Chem. Phys. 1998, 108, 6109–6116. [Google Scholar] [Green Version]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.N.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar]
- Vashisth, H.; Abrams, C.F. All-atom structural models of insulin binding to the insulin receptor in the presence of a tandem hormone-binding element. Proteins 2013, 81, 1017–1030. [Google Scholar]
- Vashisth, H.; Maragliano, L.; Abrams, C.F. “DFG-flip” in the insulin receptor kinase is facilitated by a helical intermediate state of the activation loop. Biophys. J 2012, 102, 1979–1987. [Google Scholar]
- Vashisth, H.; Skiniotis, G.; Brooks, C.L., III. Collective variable approaches for single molecule flexible fitting and enhanced sampling. Chem. Rev. 2014, 114, 3353–3365. [Google Scholar]
- Abrams, C.; Bussi, G. Enhanced sampling in molecular dynamics using metadynamics, replica-exchange, and temperature-acceleration. Entropy 2013, 16, 163–199. [Google Scholar]
- Maragliano, L.; Vanden-Eijnden, E. A temperature accelerated method for sampling free energy and determining reaction pathways in rare events simulations. Chem. Phys. Lett. 2006, 426, 168–175. [Google Scholar]
- Maragliano, L.; Vanden-Eijnden, E. Single-sweep methods for free energy calculations. J. Chem. Phys. 2008, 128, 184110. [Google Scholar]
- Abrams, C.F.; Vanden-Eijnden, E. Large-scale conformational sampling of proteins using temperature-accelerated molecular dynamics. Proc. Natl. Acad. Sci. USA. 2010, 107, 4961–4966. [Google Scholar]
- Vashisth, H.; Skiniotis, G.; Brooks, C.L, III. Using enhanced sampling and structural restraints to refine atomic structures into low-resolution electron microscopy maps. Structure 2012, 20, 1453–1462. [Google Scholar]
- Vashisth, H.; Brooks, C. L., III. Conformational sampling of maltose-transporter components in Cartesian collective variables is governed by the low-frequency normal modes. J. Phys. Chem. Lett. 2012, 3, 3379–3384. [Google Scholar]
- Vashisth, H.; Storaska, A.J.; Neubig, R.R.; Brooks, C. L., III. Conformational dynamics of a regulator of G-protein signaling protein reveals a mechanism of allosteric inhibition by a small molecule. ACS Chem. Biol. 2013, 8, 2778–2784. [Google Scholar]
- Selwa, E.; Huynh, T.; Ciccotti, G.; Maragliano, L.; Malliavin, T.E. Temperature-accelerated molecular dynamics gives insights into globular conformations sampled in the free state of the AC catalytic domain. Proteins 2014, 82, 2483–2496. [Google Scholar]
- Naveh, Z.M.H.; Malliavin, T.E.; Maragliano, L.; Cottone, G.; Ciccotti, G. Conformational changes in acetylcholine binding protein investigated by temperature accelerated molecular dynamics. PLoS One 2014, 9, e88555. [Google Scholar]
- Maragliano, L.; Fischer, A.; Vanden-Eijnden, E.; Ciccotti, G. String method in collective variables: Minimum free energy paths and isocommittor surfaces. J. Chem. Phys. 2006, 125, 024106. [Google Scholar]
- McCammon, J.A.; Gelin, B.R.; Karplus, M. Dynamics of folded proteins. Nature 1977, 267, 585–590. [Google Scholar]
- Wodak, S.J.; Alard, P.; Delhaise, P.; Renneboogsquilbin, C. Simulation of conformational changes in 2 ZN insulin. J. Mol. Biol. 1985, 181, 317–322. [Google Scholar]
- Krüger, P.; Strassburger, W.; Wollmer, A.; Gunsteren, W.F.V.; Dodson, G.G. The simulated dynamics of the insulin monomer and their relationship to the molecules structure. E. Biophys. J 1987, 14, 449–459. [Google Scholar]
- Mark, A.E.; Berendsen, H.J.C.; Gunsteren, W.F.V. Conformational flexibility of aqueous monomeric and dimeric insulin: A molecular dynamics study. Biochemistry 1991, 30, 10866–10872. [Google Scholar]
- Krüger, P.; Hahnen, J.; Wollmer, A. Comparative studies on the dynamics of cross-linked insulin. Euro. Biophys. J 1994, 23, 177–187. [Google Scholar]
- Tidor, B.; Karplus, M. The contribution of vibrational entropy to molecular association: The dimerization of insulin. J. Mol. Biol. 1994, 238, 405–414. [Google Scholar]
- Falconi, M.; Cambria, M.T.; Cambria, A.; Desideri, A. Structure and stability of the insulin dimer investigated by molecular dynamics simulation. J. Bimol. Struc. Dynamics. 2001, 18, 761–772. [Google Scholar]
- Budi, A.; Legge, S.; Treutlein, H.I.; Yarovsky, I. Effect of external stresses on protein conformation: A computer modelling study. Euro. Biophys. J 2004, 33, 121–129. [Google Scholar]
- Zoete, V.; Meuwly, M.; Karplus, M. A comparison of the dynamic behavior of monomeric and dimeric insulin shows structural rearrangements in the active monomer. J. Mol. Biol. 2004, 342, 913–929. [Google Scholar]
- Isralewitz, B.; Gao, M.; Schulten, K. Steered molecular dynamics and mechanical functions of proteins. Curr. Op. Struct. Biol. 2001, 11, 224–230. [Google Scholar]
- Kim, T.; Rhee, A.; Yip, C.M. Force-induced insulin dimer dissociation: A molecular dynamics study. J. Am. Chem. Soc. 2006, 128, 5330–5331. [Google Scholar]
- Zoete, V.; Meuwly, M. Importance of individual side chains for the stability of a protein fold: Computational alanine scanning of the insulin monomer. J. Comp. Chem. 2006, 27, 1843–1857. [Google Scholar]
- Legge, F.S.; Budi, A.; Treutlein, H.; Yarovsky, I. Protein flexibility: Multiple molecular dynamics simulations of insulin chain B. Biophys. Chem. 2006, 119, 146–157. [Google Scholar]
- Todorova, N.; Marinelli, F.; Piana, S.; Yarovsky, I. Exploring the folding free energy landscape of insulin using bias exchange metadynamics. J. Phys. Chem. B 2009, 113, 3556–3564. [Google Scholar]
- Schlitter, J.; Engels, M.; Krüger, P.; Jacoby, E.; Wollmer, A. Targeted molecular dynamics simulation of conformational change - application to the T→R transition in insulin. Mol. Simul. 1993, 10, 291–308. [Google Scholar]
- Jacoby, E.; Krüger, P.; Schlitter, J.; Roper, D.; Wollmer, A. Simulation of a complex protein structural change: The T ⇔ R transition in the insulin hexamer. Prot. Eng. 1996, 9, 113–125. [Google Scholar]
- Badger, J.; Caspar, D. Water structure in cubic insulin crystals. Proc. Natl. Acad. Sci. USA. 1991, 88, 622–626. [Google Scholar]
- Chai, C.C.; Jhon, M.S. Molecular Dynamics Study on Protein and it’s Water Structure at High Pressure. Mol. Sim. 2000, 23, 257–274. [Google Scholar]
- Bagchi, K.; Roy, S. Sensitivity of water dynamics to biologically significant surfaces of monomeric insulin: Role of topology and electrostatic interactions. J. Phys. Chem. B 2014, 118, 3805–3813. [Google Scholar]
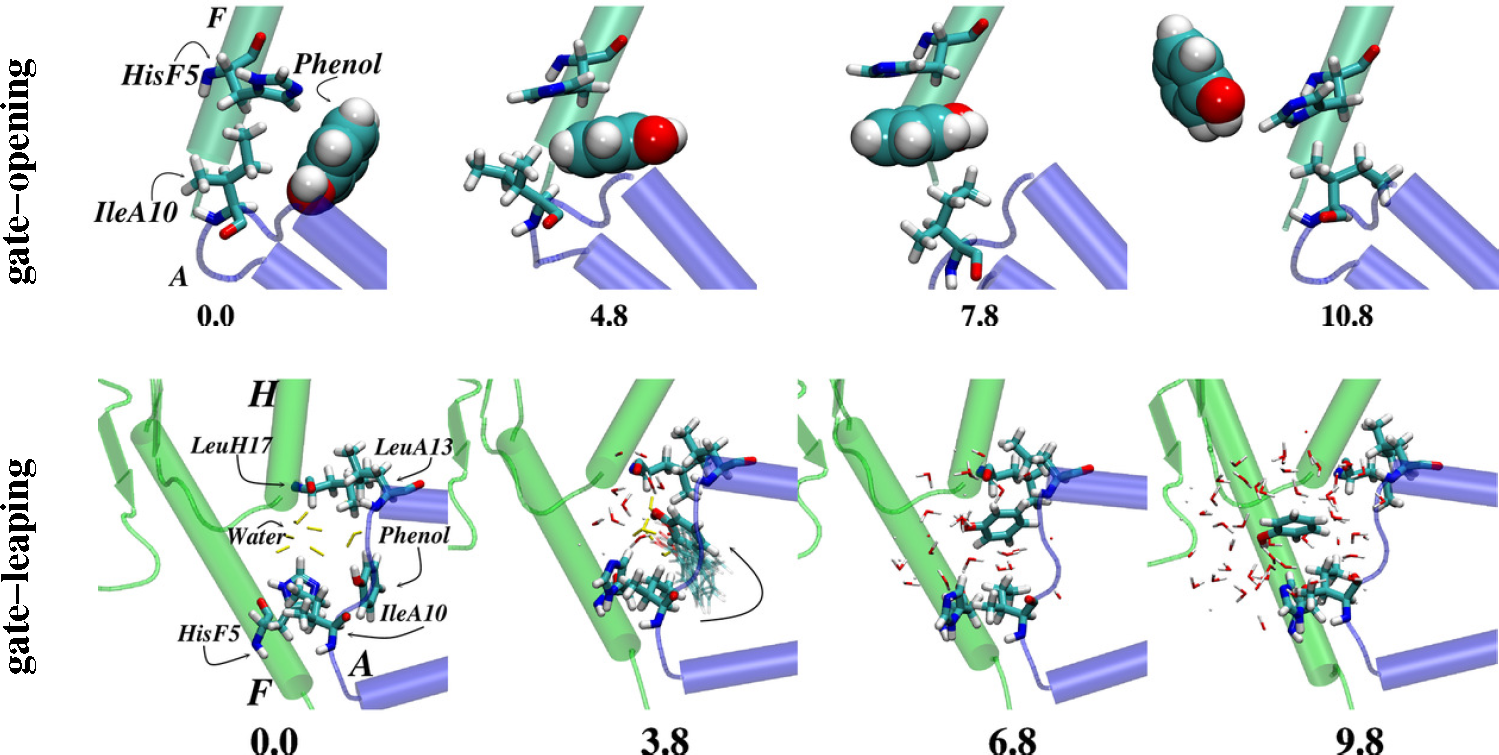
- Vashisth, H.; Abrams, C.F. Ligand escape pathways and (Un)binding free energy calculations for the hexameric insulin-phenol complex. Biophys. J 2008, 95, 4193–4204. [Google Scholar]
- Swegat, W.; Schlitter, J.; Krüger, P.; Wollmer, A. MD simulation of protein-ligand interaction: Formation and dissociation of an insulin-phenol complex. Biophys. J 2003, 84, 1493–1506. [Google Scholar]
- Vashisth, H. Flexibility in the insulin receptor ectodomain enables docking of insulin in crystallographic conformation observed in hormone-bound microreceptor. Membranes 2014, 4, 730–746. [Google Scholar]
- Schäffer, L. A model for insulin binding to the insulin receptor. Eur. J. Biochem. 1994, 221, 1127–1132. [Google Scholar]
- Soos, M.; Field, C.; Lammers, R.; Ullrich, A.; Zhang, B.; Roth, R.; Andersen, A.; Kjeldsen, T.; Siddle, K. A panel of monoclonal antibodies for the type I insulin-like growth factor receptor. Epitope mapping, effects on ligand binding, and biological activity. J. Biol. Chem. 1992, 267, 12955–12963. [Google Scholar]
- Kadowaki, H.; Kadowaki, T.; Cama, A.; Marcus-Samuels, B.; Rovira, A.; Bevins, C.; Taylor, S. Mutagenesis of lysine 460 in the human insulin receptor. Effects upon receptor recycling and cooperative interactions among binding sites. J. Biol. Chem. 1990, 265, 21285–21296. [Google Scholar]
- Christoffersen, C.T.; Bornfeldt, K.E.; Rotella, C.M.; Gonzales, N.; Vissing, H.; Shymko, R.M.; Tenhoeve, J.; Groffen, J.; Heisterkamp, N.; de Meyts, P. Negative cooperativity in the insulin-like growth-factor-I receptor and a chimeric IGF/Insulin receptor. Endocrinology 1994, 135, 472–475. [Google Scholar]
- Chan, S.J.; Nakagawa, S.; Steiner, D.F. Complementation analysis demonstrates that insulin cross-links both α-subunits in a truncated insulin receptor dimer. J. Biol. Chem. 2007, 282, 13754–13758. [Google Scholar]
- Whittaker, J.; Garcia, P.; Yu, G.; Mynarcik, D. Transmembrane domain interactions are necessary for negative cooperativity of the insulin-receptor. Mol. Endocrinol. 1994, 8, 1521–1526. [Google Scholar]
- Bass, J.; Kurose, T.; Pashmforoush, M.; Steiner, D.F. Fusion of insulin receptor ectodomains to immunoglobulin constant domains reproduces high-affinity insulin binding in vitro. J. Biol. Chem. 1996, 271, 19367–19375. [Google Scholar]
- Surinya, K.H.; Forbes, B.E.; Occhiodoro, F.; Booker, G.W.; Francis, G.L.; Siddle, K.; Wallace, J.C.; Cosgrove, L.J. An investigation of the ligand binding properties and negative cooperativity of soluble insulin-like growth factor receptors. J. Biol. Chem. 2008, 283, 5355–5363. [Google Scholar]
- Slaaby, R.; Schaffer, L.; Lautrup-Larsen, I.; Andersen, A.S.; Shaw, A.C.; Mathiasen, I.S.; Brandt, J. Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J. Biol. Chem. 2006, 281, 25869–25874. [Google Scholar]
- Cara, J.F.; Mirmira, R.G.; Nakagawa, S.H.; Tager, H.S. An insulin-like growth factor-I insulin hybrid exhibiting high potency for interaction with the type-I insulin-like growth-factor and insulin-receptors of placental plasma-membranes. J. Biol. Chem. 1990, 265, 17820–17825. [Google Scholar]
- DeWolf, E.; Gill, R.; Geddes, S.; Pitts, J.; Wollmer, A.; Grotzinger, J. Solution structure of a mini IGF-1. Prot. Sci. 1996, 5, 2193–2202. [Google Scholar]
- Denley, A.; Bonython, E.R.; Booker, G.W.; Cosgrove, L.J.; Forbes, B.E.; Ward, C.W.; Wallace, J.C. Structural determinants for high-affinity binding of insulin-like growth factor II to insulin receptor (IR)-A, the exon 11 minus isoform of the IR. Mol. Endocrinol. 2004, 18, 2502–2512. [Google Scholar]
- Bayne, M.L.; Applebaum, J.; Underwood, D.; Chicchi, G.G.; Green, B.G.; Hayes, N.S.; Cascieri, M.A. The C-region of human insulin-like growth factor (IGF) I is required for high-affinity binding to the type-1 IGF receptor. J. Biol. Chem. 1988, 264, 11004–11008. [Google Scholar]
- Epa, V.C.; Ward, C.W. Model for the complex between the insulin-like growth factor I and its receptor: Towards designing antagonists for the IGF-I receptor. Protein Eng. Des. Sel. 2006, 19, 377–384. [Google Scholar]
- Xu, B.; Huang, K.; Chu, Y.C.; Hu, S.Q.; Nakagawa, S.; Wang, S.; Wang, R.Y.; Whittaker, J.; Katsoyannis, P.G.; Weiss, M. Decoding the cryptic active conformation of a protein by synthetic photoscanning: Insulin inserts a detachable arm between receptor domains. J. Biol. Chem. 2009, 284, 14597–14608. [Google Scholar]
- Whittaker, J.; Whittaker, L.; Roberts, C., Jr.; Phillips, N.; Ismail-Beigi, F.; Lawrence, M.; Weiss, M. α-Helical element at the hormone-binding surface of the insulin receptor functions as a signaling element to activate its tyrosine kinase. Proc. Natl. Acad. Sci. USA. 2012, 109, 11166–71. [Google Scholar]
- Huang, K.; Chan, S.; Hua, Q.; Chu, Y.; Wang, R.; Klaproth, B.; Jia, W.; Whittaker, J.; de Meyts, P.; Nakagawa, S.; et al. The A-chain of insulin contacts the insert domain of the insulin receptor. J. Biol. Chem. 2007, 282, 35337–35349. [Google Scholar]
- Menting, J.; Ward, C.; Margetts, M.; Lawrence, M. A thermodynamic study of ligand binding to the first three domains of the human insulin receptor: Relationship between the receptor alpha-chain C-terminal peptide and the site-1 insulin mimetic peptides. Biochemistry 2009, 48, 5492–5500. [Google Scholar]
- Pillutla, R.C.; Hsiao, K.C.; Beasley, J.R.; Brandt, J.; Ostergaard, S.; Hansen, P.H.; Spetzler, J.C.; Danielsen, G.M.; Andersen, A.S.; Brissette, R.E.; et al. Peptides identify the critical hotspots involved in the biological activation of the insulin receptor. J. Biol. Chem. 2002, 277, 22590–22594. [Google Scholar]
- Schaffer, L.; Brissette, R.E.; Spetzler, J.C.; Pillutla, R.C.; Ostergaard, S.; Lennick, M.; Brandt, J.; Fletcher, P.W.; Danielsen, G.M.; Hsiao, K.C.; et al. Assembly of high-affinity insulin receptor agonists and antagonists from peptide building blocks. Proc. Natl. Acad. Sci. USA. 2003, 100, 4435–4439. [Google Scholar]
- Derewenda, U.; Derewenda, Z.; Dodson, E.J.; Dodson, G.G.; Bing, X.; Markussen, J. X-ray analysis of the single chain B29-A1 peptide-linked insulin molecule. A completely inactive analogue. J. Mol. Biol. 1991, 220, 425–433. [Google Scholar]
- Mirmira, R.; Nakagawa, S.; Tager, H. Importance of the character and configuration of residues B24, B25, and B26 in insulin-receptor interactions. J. Biol. Chem. 1991, 266, 1428–1436. [Google Scholar]
- Hua, Q.X.; Shoelson, S.E.; Kochoyan, M.; Weiss, M. Receptor-binding redefined by a structural switch in a mutant human insulin. Nature 1991, 354, 238–241. [Google Scholar]
- Hua, Q.X.; Xu, B.; Huang, K.; Hu, S.Q.; Nakagawa, S.; Jia, W.; Wang, S.; Whittaker, J.; Katsoyannis, P.G.; Weiss, M. Enhancing the activity of a protein by stereospecific unfolding: Conformational life cycle of insulin and its evolutionary origins. J. Biol. Chem. 2009, 284, 14586–14596. [Google Scholar]
- Ellis, L.; Clauser, E.; Morgan, D.O.; Edery, M.; Roth, R.A.; Rutter, W.J. Replacement of insulin-receptor tyrosine residues 1162 and 1163 compromises insulin-stimulated kinase-activity and uptake of 2-deoxyglucose. Cell 1986, 45, 721–732. [Google Scholar]
- Wilden, P.A.; Kahn, C.R.; Siddle, K.; White, M.F. Insulin-receptor kinase domain autophosphorylation regulates receptor enzymatic function. J. Biol. Chem. 1992, 267, 16660–16668. [Google Scholar]
- Kornev, A.P.; Haste, N.M.; Taylor, S.S.; Ten Eyck, L.F. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA. 2006, 103, 17783–17788. [Google Scholar]
- Kornev, A.P.; Taylor, S.S.; Ten Eyck, L.F. A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. USA. 2008, 105, 14377–14382. [Google Scholar]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar]
- Banavali, N.K.; Roux, B. Anatomy of a structural pathway for activation of the catalytic domain of Src kinase Hck. Proteins 2007, 67, 1096–1112. [Google Scholar]
- Gan, W.; Yang, S.; Roux, B. Atomistic view of the conformational activation of Src kinase using the string method with swarms-of-trajectories. Biophys. J 2009, 97, L8–L10. [Google Scholar]
- Berteotti, A.; Cavalli, A.; Branduardi, D.; Gervasio, F.L.; Recanatini, M.; Parrinello, M. Protein conformational transitions: The closure mechanism of a kinase explored by atomistic simulations. J. Am. Chem. Soc. 2009, 131, 244–250. [Google Scholar]
- Yang, S.; Banavali, N.K.; Roux, B. Mapping the conformational transition in Src activation by cumulating the information from multiple molecular dynamics trajectories. Proc. Natl. Acad. Sci. USA. 2009, 106, 3776–3781. [Google Scholar]
- Shan, Y.B.; Seeliger, M.A.; Eastwood, M.P.; Frank, F.; Xu, H.F.; Jensen, M.Ø.; Dror, R.O.; Kuriyan, J.; Shaw, D.E. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. USA. 2009, 106, 139–144. [Google Scholar]
- Flörke, R.; Schnaith, K.; Passlack, W.; Wichert, M.; Kuehn, L.; Fabry, M.; Federwisch, M.; Reinauer, H. Hormone-triggered conformational changes within the insulin-receptor ectodomain: Requirement for transmembrane anchors. Biochem. J 2001, 360, 189–198. [Google Scholar]
- Kavran, J.M.; McCabe, J.M.; Byrne, P.O.; Connacher, M.K.; Wang, Z.; Ramek, A.; Sarabipour, S.; Shan, Y.; Shaw, D.E.; Hristova, K.; et al. How IGF-1 activates its receptor. eLife 2014, 3, e03772. [Google Scholar]
- Lee, J.; Miyazaki, M.; Romeo, G.R.; Shoelson, S.E. Insulin receptor activation with transmembrane domain ligands. J. Biol. Chem. 2014, 289, 19769–19777. [Google Scholar]
- Hubbard, S.R.; Miller, W.T. Closing in on a mechanism for activation. eLife 2014, 3, e04909. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vashisth, H. Theoretical and Computational Studies of Peptides and Receptors of the Insulin Family. Membranes 2015, 5, 48-83. https://doi.org/10.3390/membranes5010048
Vashisth H. Theoretical and Computational Studies of Peptides and Receptors of the Insulin Family. Membranes. 2015; 5(1):48-83. https://doi.org/10.3390/membranes5010048
Chicago/Turabian StyleVashisth, Harish. 2015. "Theoretical and Computational Studies of Peptides and Receptors of the Insulin Family" Membranes 5, no. 1: 48-83. https://doi.org/10.3390/membranes5010048