
Understanding Functional Redundancy and Promiscuity of Multidrug Transporters in E. coli under Lipophilic Cation Stress

 , , , , and
, , , , and
Abstract
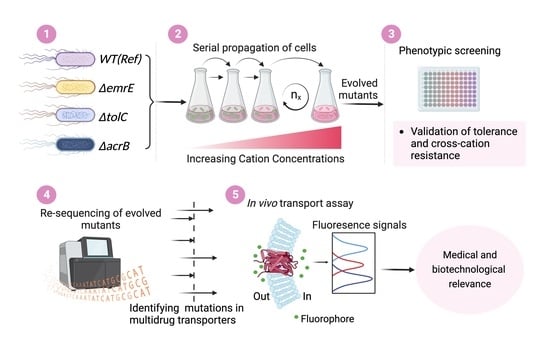
:
1. Introduction
2. Materials and Methods
2.1. Strains, Chemicals, and Culture Media
2.2. Tolerance Adaptive Laboratory Evolution (TALE)
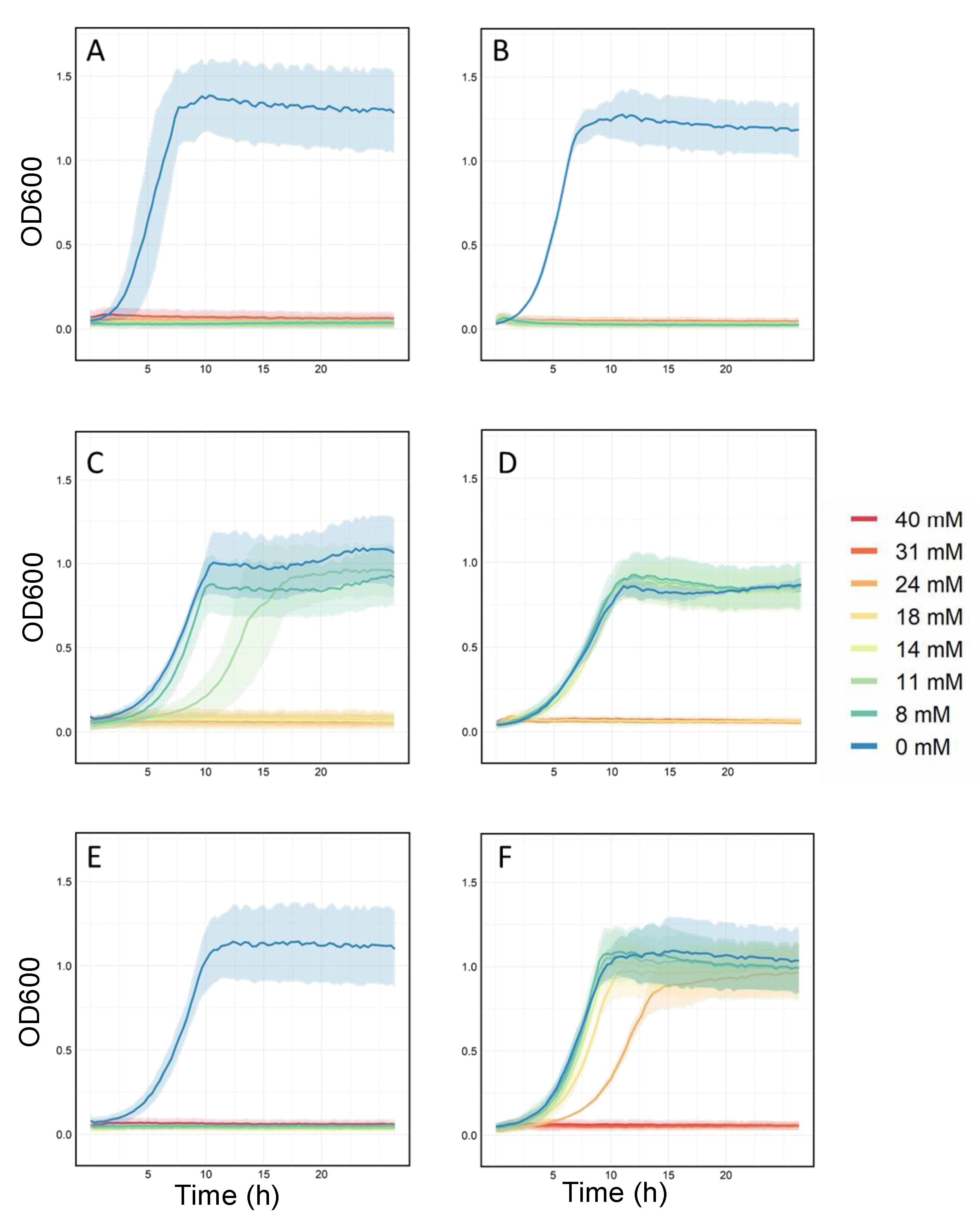
2.3. Validation of Resistant Phenotypes and Cross-Cation Resistance
2.4. Whole Genome Sequencing and Mutational Analysis
2.5. Fluorophore Accumulation Transport Assay
3. Results and Discussion
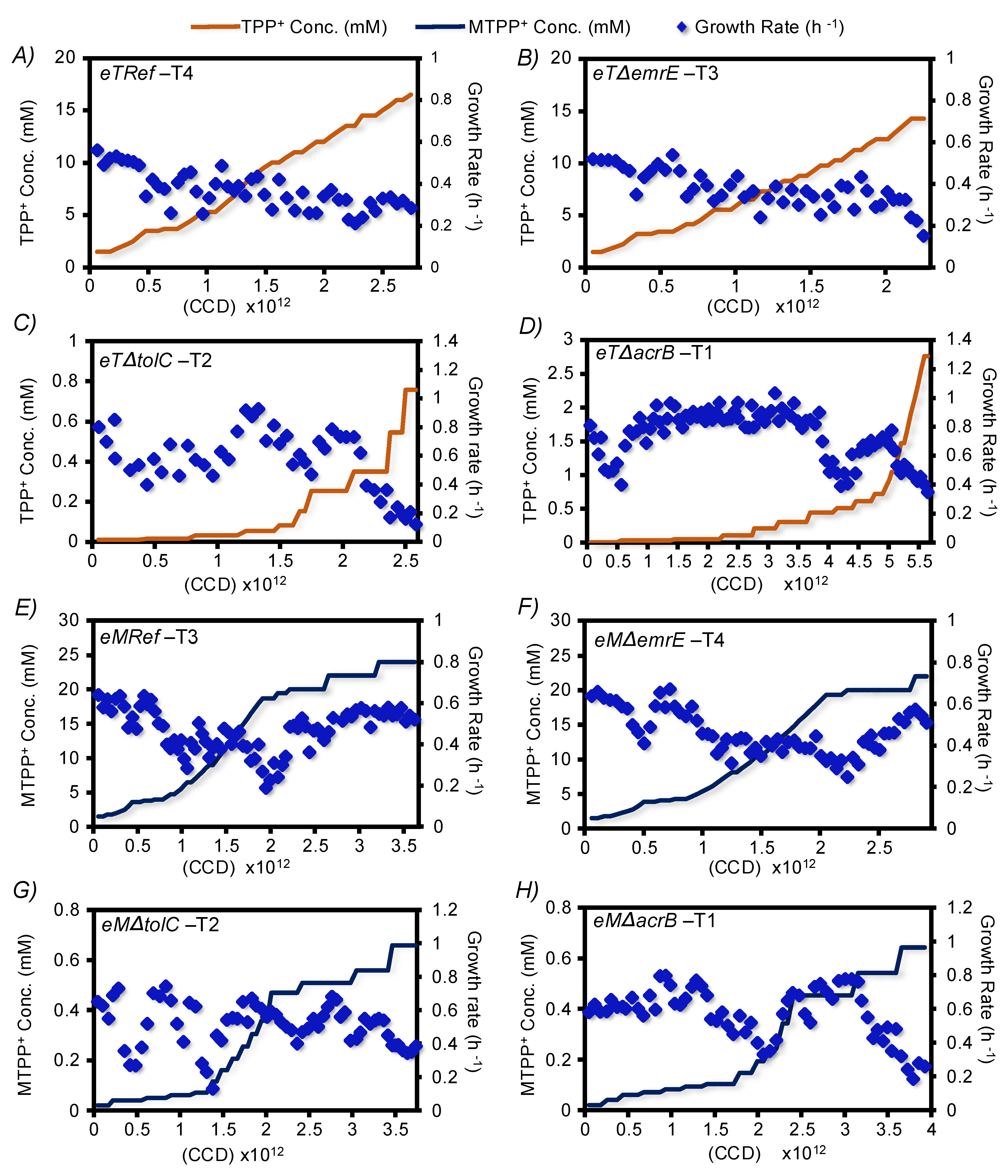
3.1. The TALE Process
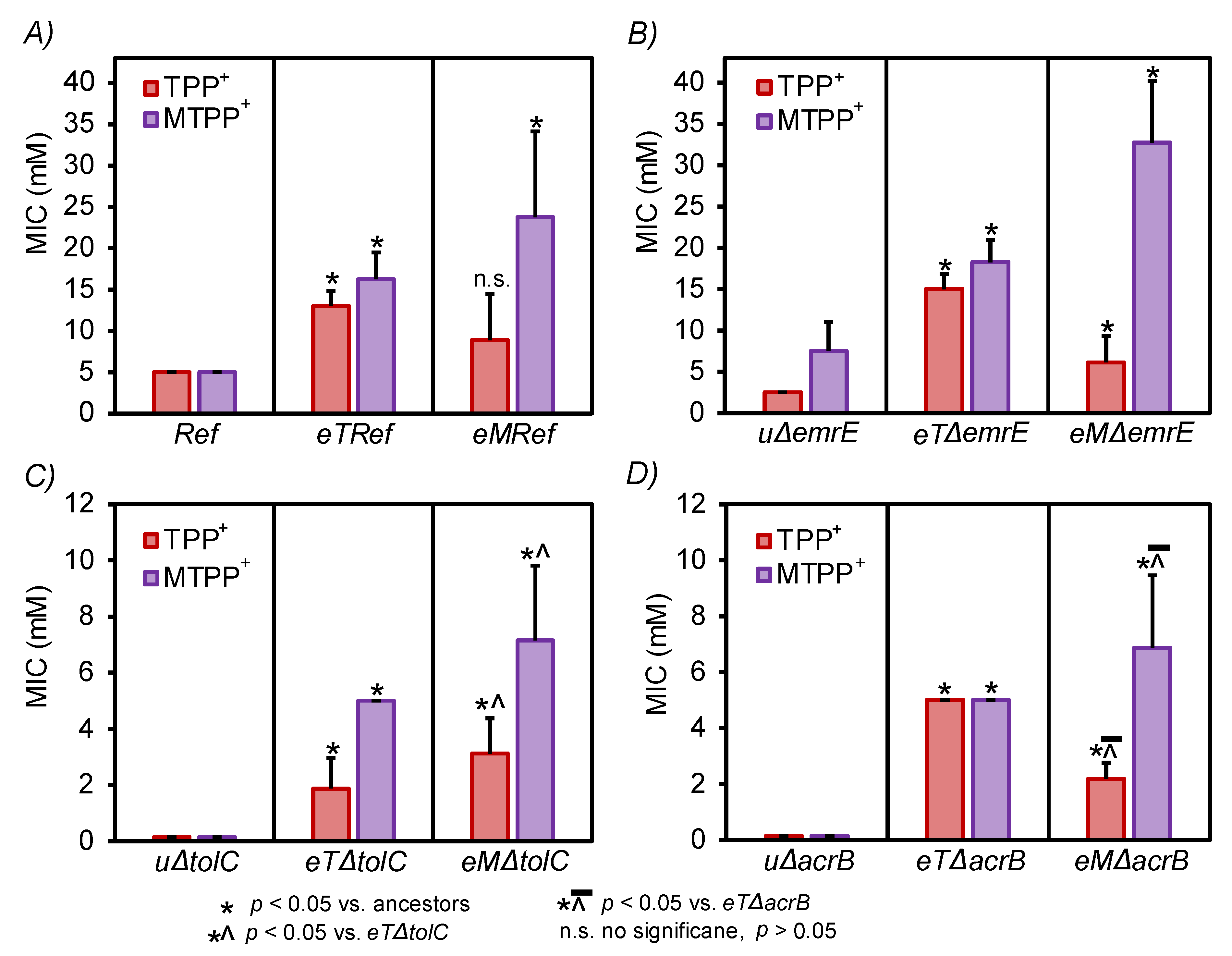
3.2. Validation of Resistant Phenotypes and Cross-Cation Resistance
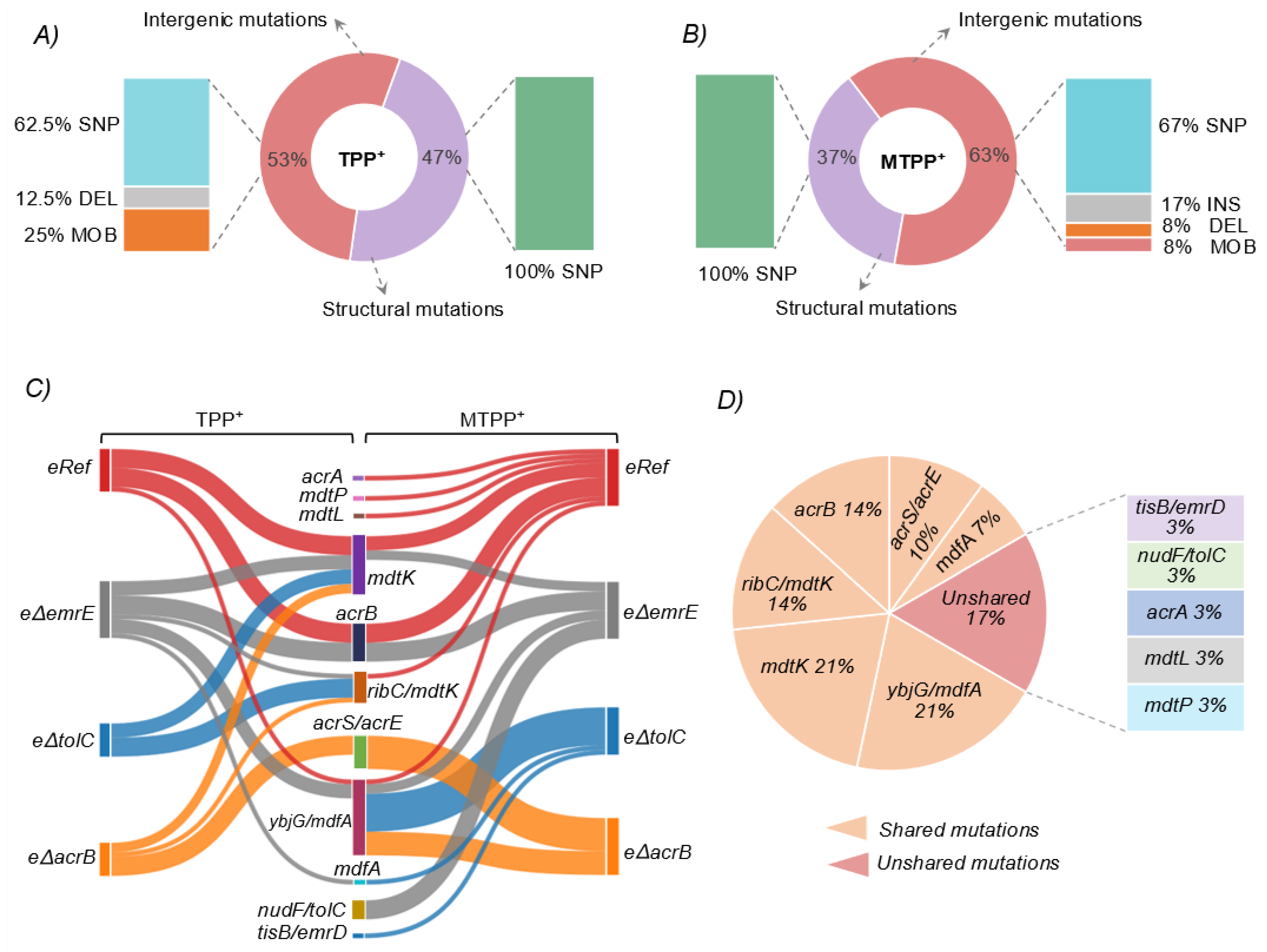
3.3. Whole Genome Re-Sequencing and Mutational Analysis
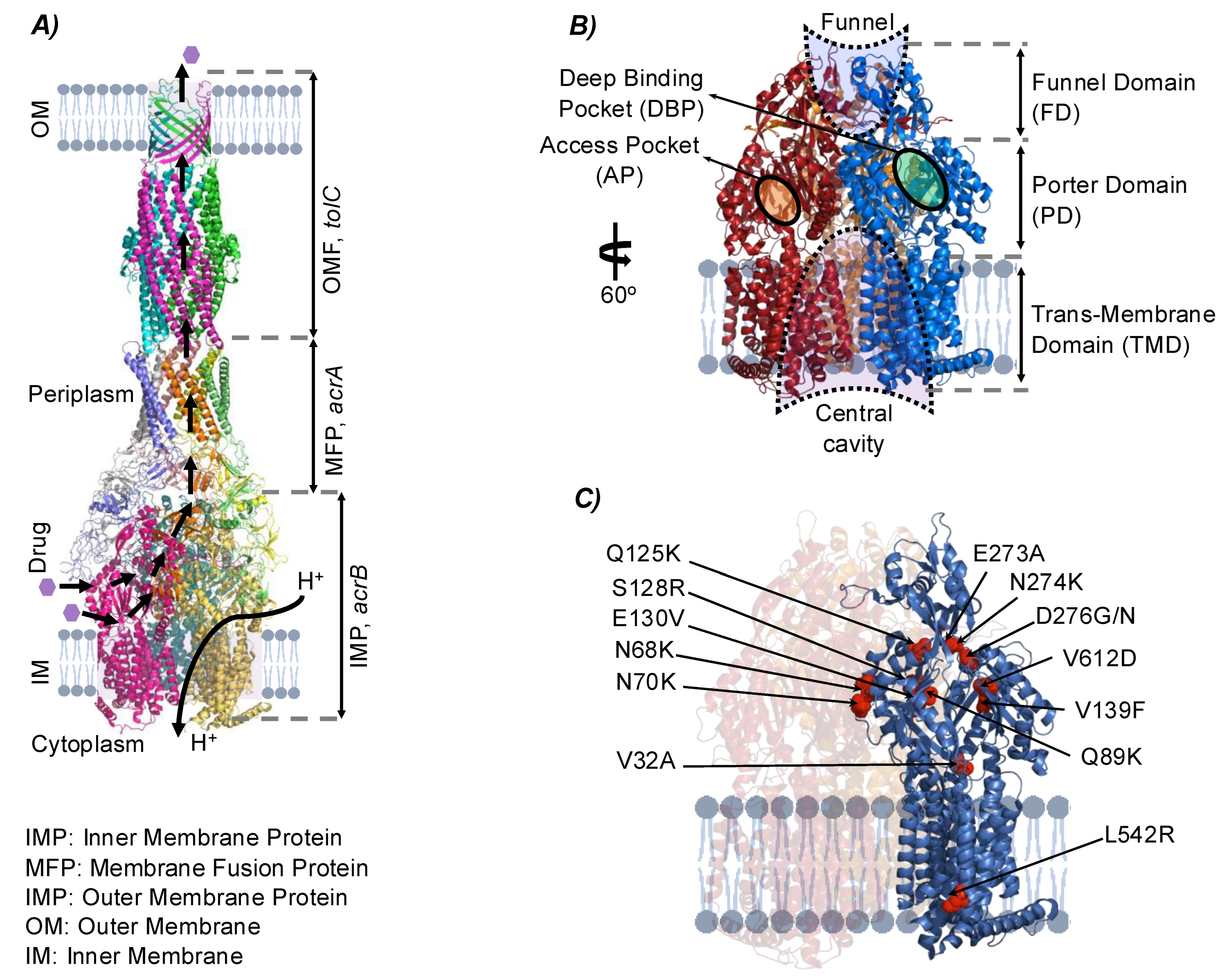
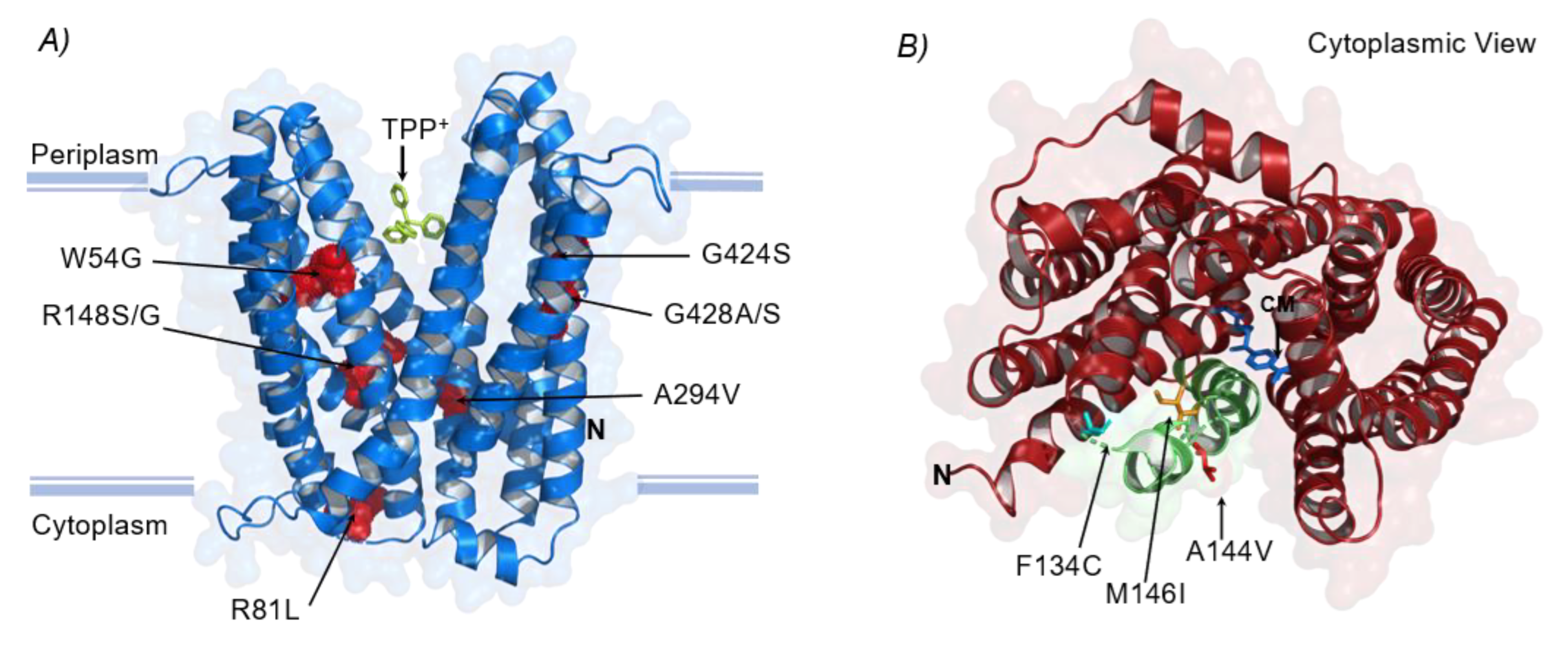
3.3.1. Structural Mutations Affecting MDTs
3.3.2. Intergenic Mutations Affecting MDTs
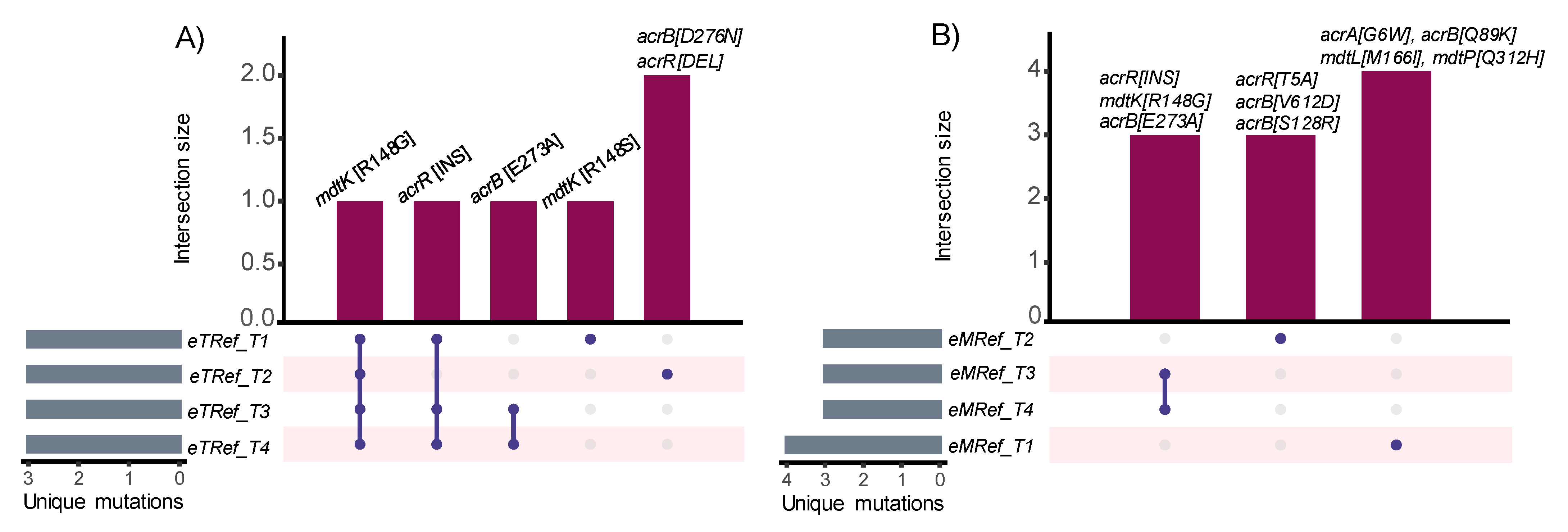
3.4. Estimating Changes in Membrane Transport in Terms of Mutational Convergence
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug Efflux Pumps: Structure, Function and Regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, I.T. Multidrug Efflux Pumps and Resistance: Regulation and Evolution. Curr. Opin. Microbiol. 2003, 6, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic Resistance—The Need for Global Solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, A. Tolerance Engineering in Bacteria for the Production of Advanced Biofuels and Chemicals. Trends Microbiol. 2015, 23, 498–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dyk, T.K. Bacterial Efflux Transport in Biotechnology. Adv. Appl. Microbiol. 2008, 63, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Ma, J.; Rothnie, A.; Biggin, P.C.; Kerr, I.D. Towards Understanding Promiscuity in Multidrug Efflux Pumps. Trends Biochem. Sci. 2014, 39, 8–16. [Google Scholar] [CrossRef]
- Lewinson, O.; Adler, J.; Sigal, N.; Bibi, E. Promiscuity in Multidrug Recognition and Transport: The Bacterial MFS Mdr Transporters. Mol. Microbiol. 2006, 61, 277–284. [Google Scholar] [CrossRef]
- Smith, H.E.; Blair, J.M.A. Redundancy in the Periplasmic Adaptor Proteins AcrA and AcrE Provides Resilience and an Ability to Export Substrates of Multidrug Efflux. J. Antimicrob. Chemother. 2014, 69, 982–987. [Google Scholar] [CrossRef]
- Cudkowicz, N.A.; Schuldiner, S. Deletion of the Major Escherichia coli Multidrug Transporter AcrB Reveals Transporter Plasticity and Redundancy in Bacterial Cells. PLoS ONE 2019, 14, e0218828. [Google Scholar] [CrossRef]
- Lanthaler, K.; Bilsland, E.; Dobson, P.D.; Moss, H.J.; Pir, P.; Kell, D.B.; Oliver, S.G. Genome-Wide Assessment of the Carriers Involved in the Cellular Uptake of Drugs: A Model System in Yeast. BMC Biol. 2011, 9, 70. [Google Scholar] [CrossRef]
- Sandberg, T.E.; Salazar, M.J.; Weng, L.L.; Palsson, B.O.; Feist, A.M. The Emergence of Adaptive Laboratory Evolution as an Efficient Tool for Biological Discovery and Industrial Biotechnology. Metab. Eng. 2019, 56, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Remigi, P.; Masson-Boivin, C.; Rocha, E.P.C. Experimental Evolution as a Tool to Investigate Natural Processes and Molecular Functions. Trends Microbiol. 2019, 27, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawecki, T.J.; Lenski, R.E.; Ebert, D.; Hollis, B.; Olivieri, I.; Whitlock, M.C. Experimental Evolution. Trends Ecol. Evol. 2012, 27, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, A.C.; Kishony, R. Understanding, Predicting and Manipulating the Genotypic Evolution of Antibiotic Resistance. Nat. Rev. Genet. 2013, 14, 243–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, P.J.; Hegreness, M.J.; Aiden, A.P.; Kishony, R. Drug Interactions and the Evolution of Antibiotic Resistance. Nat. Rev. Microbiol. 2009, 7, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Friedman, L.; Alder, J.D.; Silverman, J.A. Genetic Changes That Correlate with Reduced Susceptibility to Daptomycin in Staphylococcus Aureus. Antimicrob. Agents Chemother. 2006, 50, 2137–2145. [Google Scholar] [CrossRef] [Green Version]
- Albert, T.J.; Dailidiene, D.; Dailide, G.; Norton, J.E.; Kalia, A.; Richmond, T.A.; Molla, M.; Singh, J.; Green, R.D.; Berg, D.E. Mutation Discovery in Bacterial Genomes: Metronidazole Resistance in Helicobacter Pylori. Nat. Methods 2005, 2, 951–953. [Google Scholar] [CrossRef]
- Girgis, H.S.; Hottes, A.K.; Tavazoie, S. Genetic Architecture of Intrinsic Antibiotic Susceptibility. PLoS ONE 2009, 4, e5629. [Google Scholar] [CrossRef] [Green Version]
- Bull, A.T. The Renaissance of Continuous Culture in the Post-Genomics Age. J. Ind. Microbiol. Biotechnol. 2010, 37, 993–1021. [Google Scholar] [CrossRef]
- Zhang, Q.; Lambert, G.; Liao, D.; Kim, H.; Robin, K.; Tung, C.K.; Pourmand, N.; Austin, R.H. Acceleration of Emergence of Bacterial Antibiotic Resistance in Connected Microenvironments. Science 2011, 333, 1764–1767. [Google Scholar] [CrossRef]
- Toprak, E.; Veres, A.; Michel, J.B.; Chait, R.; Hartl, D.L.; Kishony, R. Evolutionary Paths to Antibiotic Resistance under Dynamically Sustained Drug Selection. Nat. Genet. 2011, 44, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.S.; SalcedoSora, J.E.; Kim, S.H.; Sudarsan, S.; Sastry, A.V.; Kell, D.B.; Herrgård, M.J.; Feist, A.M. Membrane Transporter Identification and Modulation via Adaptive Laboratory Evolution. Metab. Eng. 2022, 72, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Mundhada, H.; Seoane, J.M.; Schneider, K.; Koza, A.; Christensen, H.B.; Klein, T.; Phaneuf, P.V.; Herrgard, M.; Feist, A.M.; Nielsen, A.T. Increased Production of L-Serine in Escherichia coli through Adaptive Laboratory Evolution. Metab. Eng. 2017, 39, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, R.; Wei, Y.; Mohamed, E.; Radi, M.; Malina, C.; Herrgård, M.J.; Feist, A.M.; Nielsen, J.; Chen, Y. Adaptive Laboratory Evolution of Tolerance to Dicarboxylic Acids in Saccharomyces Cerevisiae. Metab. Eng. 2019, 56, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Mohamed, E.T.; Radi, M.S.; Herrgård, M.J.; Feist, A.M.; Nielsen, J.; Chen, Y. Elucidating Aromatic Acid Tolerance at Low PH in Saccharomyces Cerevisiae Using Adaptive Laboratory Evolution. Proc. Natl. Acad. Sci. USA 2020, 117, 27954–27961. [Google Scholar] [CrossRef]
- Mohamed, E.T.; Werner, A.Z.; Salvachúa, D.; Singer, C.A.; Szostkiewicz, K.; Rafael Jiménez-Díaz, M.; Eng, T.; Radi, M.S.; Simmons, B.A.; Mukhopadhyay, A.; et al. Adaptive Laboratory Evolution of Pseudomonas Putida KT2440 Improves P-Coumaric and Ferulic Acid Catabolism and Tolerance. Metab. Eng. Commun. 2020, 11, e00143. [Google Scholar] [CrossRef]
- Mohamed, E.T.; Wang, S.; Lennen, R.M.; Herrgård, M.J.; Simmons, B.A.; Singer, S.W.; Feist, A.M. Generation of a Platform Strain for Ionic Liquid Tolerance Using Adaptive Laboratory Evolution. Microb. Cell Fact. 2017, 16, 204. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Radi, M.; Mohamed, E.T.T.; Feist, A.M.; Dragone, G.; Mussatto, S.I. Adaptive Laboratory Evolution of Rhodosporidium Toruloides to Inhibitors Derived from Lignocellulosic Biomass and Genetic Variations behind Evolution. Bioresour. Technol. 2021, 333, 125171. [Google Scholar] [CrossRef]
- Saier, M.H.; Tran, C.V.; Barabote, R.D. TCDB: The Transporter Classification Database for Membrane Transport Protein Analyses and Information. Nucleic Acids Res. 2006, 34, D181–D186. [Google Scholar] [CrossRef] [Green Version]
- Korkhov, V.M.; Tate, C.G. Electron Crystallography Reveals Plasticity within the Drug Binding Site of the Small Multidrug Transporter EmrE. J. Mol. Biol. 2008, 377, 1094–1103. [Google Scholar] [CrossRef]
- Higgins, C.F. Multiple Molecular Mechanisms for Multidrug Resistance Transporters. Nature 2007, 446, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, J.A.; Karamian, B.; Nikaido, H. Optimized Nile Red Efflux Assay of AcrAB-TolC Multidrug Efflux System Shows Competition between Substrates. Antimicrob. Agents Chemother. 2010, 54, 3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kell, D.B. A Protet-Based, Protonic Charge Transfer Model of Energy Coupling in Oxidative and Photosynthetic Phosphorylation. Adv. Microb. Physiol. 2021, 78, 1–177. [Google Scholar] [CrossRef] [PubMed]
- le Rhun, A.; Escalera-Maurer, A.; Bratovič, M.; Charpentier, E. CRISPR-Cas in Streptococcus Pyogenes. RNA Biol. 2019, 16, 380. [Google Scholar] [CrossRef] [Green Version]
- Mehrer, C.R.; Incha, M.R.; Politz, M.C.; Pfleger, B.F. Anaerobic Production of Medium-Chain Fatty Alcohols via a β-Reduction Pathway. Metab. Eng. 2018, 48, 63–71. [Google Scholar] [CrossRef]
- Sprouffske, K.; Wagner, A. Growthcurver: An R Package for Obtaining Interpretable Metrics from Microbial Growth Curves. BMC Bioinform. 2016, 17, 172. [Google Scholar] [CrossRef] [Green Version]
- Phaneuf, P.V.; Gosting, D.; Palsson, B.O.; Feist, A.M. Aledb 1.0: A Database of Mutations from Adaptive Laboratory Evolution Experimentation. Nucleic Acids Res. 2019, 47, D1164–D1171. [Google Scholar] [CrossRef] [Green Version]
- Deatherage, D.E.; Barrick, J.E. Identification of Mutations in Laboratory-Evolved Microbes from next-Generation Sequencing Data Using Breseq. Methods Mol. Biol. 2014, 1151, 165–188. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-H.; Feist, A.M.; Barrett, C.L.; Palsson, B.Ø. Cumulative Number of Cell Divisions as a Meaningful Timescale for Adaptive Laboratory Evolution of Escherichia coli. PLoS ONE 2011, 6, e26172. [Google Scholar] [CrossRef] [Green Version]
- Camps, M.; Naukkarinen, J.; Johnson, B.P.; Loeb, L.A. Targeted Gene Evolution in Escherichia coli Using a Highly Error-Prone DNA Polymerase I. Proc. Natl. Acad. Sci. USA 2003, 100, 9727–9732. [Google Scholar] [CrossRef]
- Périchon, B.; Courvalin, P. Antibiotic Resistance. In Encyclopedia of Microbiology, 3rd ed.; Schaechter, M., Ed.; Academic Press: Oxford, UK, 2009; pp. 193–204. [Google Scholar] [CrossRef]
- Wagih, O.; Galardini, M.; Busby, B.P.; Memon, D.; Typas, A.; Beltrao, P. A Resource of Variant Effect Predictions of Single Nucleotide Variants in Model Organisms. Mol. Syst. Biol. 2018, 14, e8430. [Google Scholar] [CrossRef] [PubMed]
- Maneewannakul, K.; Levy, S.B. Identification for Mar Mutants among Quinolone-Resistant Clinical Isolates of Escherichia coli. Antimicrob. Agents Chemother. 1996, 40, 1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, M.A.; Talukder, A.; Piddock, L.J.V. Contribution of Mutation at Amino Acid 45 of AcrR to AcrB Expression and Ciprofloxacin Resistance in Clinical and Veterinary Escherichia coli Isolates. Antimicrob. Agents Chemother. 2005, 49, 4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rau, M.H.; Calero, P.; Lennen, R.M.; Long, K.S.; Nielsen, A.T. Genome-Wide Escherichia coli Stress Response and Improved Tolerance towards Industrially Relevant Chemicals. Microb. Cell Fact. 2016, 15, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.J.; Bang, W.; Drake, M.A. Stress Response of Escherichia coli. Compr Rev. Food Sci. Food Saf. 2006, 5, 52–64. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, K.L.; Yeo, W.S.; Park, S.J.; Roe, J.H. SoxRS-Mediated Lipopolysaccharide Modification Enhances Resistance against Multiple Drugs in Escherichia coli. J. Bacteriol. 2009, 191, 4441–4450. [Google Scholar] [CrossRef] [Green Version]
- Demple, B. Redox Signaling and Gene Control in the Escherichia coli SoxRS Oxidative Stress Regulon—A Review. Gene 1996, 179, 53–57. [Google Scholar] [CrossRef]
- LaCroix, R.A.; Sandberg, T.E.; O’Brien, E.J.; Utrilla, J.; Ebrahim, A.; Guzman, G.I.; Szubin, R.; Palsson, B.O.; Feist, A.M. Use of Adaptive Laboratory Evolution to Discover Key Mutations Enabling Rapid Growth of Escherichia coli K-12 MG1655 on Glucose Minimal Medium. Appl Environ. Microbiol. 2015, 81, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, T.E.; Long, C.P.; Gonzalez, J.E.; Feist, A.M.; Antoniewicz, M.R.; Palsson, B.O. Evolution of E. coli on [U-13C]Glucose Reveals a Negligible Isotopic Influence on Metabolism and Physiology. PLoS ONE 2016, 11, e0151130. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Saier, M.H.; Tam, R.; Reizer, A.; Reizer, J. Two Novel Families of Bacterial Membrane Proteins Concerned with Nodulation, Cell Division and Transport. Mol. Microbiol. 1994, 11, 841–847. [Google Scholar] [CrossRef]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Matsumoto, T.; Yamaguchi, A. Crystal Structures of a Multidrug Transporter Reveal a Functionally Rotating Mechanism. Nature 2006, 443, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Seeger, M.A.; Schiefner, A.; Eicher, T.; Verrey, F.; Diederichs, K.; Pos, K.M. Structural Asymmetry of AcrB Trimer Suggests a Peristaltic Pump Mechanism. Science 2006, 313, 1295–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Yamaguchi, A. Crystal Structure of Bacterial Multidrug Efflux Transporter AcrB. Nature 2002, 419, 587–593. [Google Scholar] [CrossRef]
- Yu, E.W.; McDermott, G.; Zgurskaya, H.I.; Nikaido, H.; Koshland, D.E. Structural Basis of Multiple Drug-Binding Capacity of the AcrB Multidrug Efflux Pump. Science 2003, 300, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.W.; Aires, J.R.; Nikaido, H. AcrB Multidrug Efflux Pump of Escherichia coli: Composite Substrate-Binding Cavity of Exceptional Flexibility Generates Its Extremely Wide Substrate Specificity. J. Bacteriol. 2003, 185, 5657–5664. [Google Scholar] [CrossRef] [Green Version]
- Drew, D.; Klepsch, M.M.; Newstead, S.; Flaig, R.; de Gier, J.W.; Iwata, S.; Beis, K. The Structure of the Efflux Pump AcrB in Complex with Bile Acid. Mol. Membr. Biol. 2008, 25, 677–682. [Google Scholar] [CrossRef]
- Kobylka, J.; Kuth, M.S.; Müller, R.T.; Geertsma, E.R.; Pos, K.M. AcrB: A Mean, Keen, Drug Efflux Machine. Ann. N. Y. Acad. Sci. 2020, 1459, 38–68. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, R.; Sakurai, K.; Yamasaki, S.; Nishino, K.; Yamaguchi, A. Structures of the Multidrug Exporter AcrB Reveal a Proximal Multisite Drug-Binding Pocket. Nature 2011, 480, 565–569. [Google Scholar] [CrossRef]
- Eicher, T.; Cha, H.J.; Seeger, M.A.; Brandstätter, L.; El-Delik, J.; Bohnert, J.A.; Kern, W.V.; Verrey, F.; Grütter, M.G.; Diederichs, K.; et al. Transport of Drugs by the Multidrug Transporter AcrB Involves an Access and a Deep Binding Pocket That Are Separated by a Switch-Loop. Proc. Natl. Acad. Sci. USA 2012, 109, 5687–5692. [Google Scholar] [CrossRef]
- Sjuts, H.; Vargiu, A.V.; Kwasny, S.M.; Nguyen, S.T.; Kim, H.S.; Ding, X.; Ornik, A.R.; Ruggerone, P.; Bowlin, T.L.; Nikaido, H.; et al. Molecular Basis for Inhibition of AcrB Multidrug Efflux Pump by Novel and Powerful Pyranopyridine Derivatives. Proc. Natl. Acad. Sci. USA 2016, 113, 3509–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Fan, G.; Hryc, C.F.; Blaza, J.N.; Serysheva, I.I.; Schmid, M.F.; Chiu, W.; Luisi, B.F.; Du, D. An Allosteric Transport Mechanism for the AcrAB-TolC Multidrug Efflux Pump. Elife 2017, 6, e24905. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Miyashita, N.; Sugita, Y.; Kovalenko, A.; Hirata, F.; Kidera, A. Functionality Mapping on Internal Surfaces of Multidrug Transporter AcrB Based on Molecular Theory of Solvation: Implications for Drug Efflux Pathway. J Phys. Chem. B 2011, 115, 8288–8295. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, V.K.; Vargiu, A.V.; Malloci, G.; Dreier, J.; Ruggerone, P. Molecular Rationale behind the Differential Substrate Specificity of Bacterial RND Multi-Drug Transporters. Sci. Rep. 2017, 7, 8075. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.H.; Paulsen, I.T.; Skurray, R.A. The Multidrug Efflux Protein NorM Is a Prototype of a New Family of Transporters. Mol. Microbiol. 1999, 31, 394–395. [Google Scholar] [CrossRef]
- Lu, M.; Symersky, J.; Radchenko, M.; Koide, A.; Guo, Y.; Nie, R.; Koide, S. Structures of a Na+-Coupled, Substrate-Bound MATE Multidrug Transporter. Proc. Natl. Acad. Sci. USA 2013, 110, 2099–2104. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Radchenko, M.; Symersky, J.; Nie, R.; Guo, Y. Structural Insights into H+-Coupled Multidrug Extrusion by a MATE Transporter. Nat. Struct. Mol. Biol. 2013, 20, 1310. [Google Scholar] [CrossRef] [Green Version]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-Dependent Multidrug Efflux Systems. Microbiol. Rev. 1996, 60, 575–608. [Google Scholar] [CrossRef]
- Edgar, R.; Bibi, E. MdfA, an Escherichia coli Multidrug Resistance Protein with an Extraordinarily Broad Spectrum of Drug Recognition. J. Bacteriol. 1997, 179, 2274–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarathinam, K.; Nakada-Nakura, Y.; Parthier, C.; Terada, T.; Juge, N.; Jaenecke, F.; Liu, K.; Hotta, Y.; Miyaji, T.; Omote, H.; et al. Outward Open Conformation of a Major Facilitator Superfamily Multidrug/H+ Antiporter Provides Insights into Switching Mechanism. Nat. Commun. 2018, 9, 4005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Heng, J.; Zhao, Y.; Liu, M.; Liu, Y.; Fan, J.; Wang, X.; Zhang, X.C. Substrate-Bound Structure of the E. Coli Multidrug Resistance Transporter MdfA. Cell Res. 2015, 25, 1060. [Google Scholar] [CrossRef]
- Nilsen, I.W.; Bakke, I.; Vader, A.; Olsvik, Ø.; El-Gewely, M.R. Isolation of Cmr, a Novel Escherichia coli Chloramphenicol Resistance Gene Encoding a Putative Efflux Pump. J. Bacteriol. 1996, 178, 3188–3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shine, J.; Dalgarno, L. Determinant of Cistron Specificity in Bacterial Ribosomes. Nature 1975, 254, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Chapon, C.; Schwartz, M. Expression of MalT, the Regulator Gene of the Maltose Region in Escherichia coli, Is Limited Both at Transcription and Translation. EMBO J. 1982, 1, 369. [Google Scholar] [CrossRef]
- Park, H.S.; Östberg, Y.; Johansson, J.; Wagner, E.G.H.; Uhlin, B.E. Novel Role for a Bacterial Nucleoid Protein in Translation of MRNAs with Suboptimal Ribosome-Binding Sites. Genes Dev. 2010, 24, 1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirumalai, M.R.; Karouia, F.; Tran, Q.; Stepanov, V.G.; Bruce, R.J.; Ott, C.M.; Pierson, D.L.; Fox, G.E. Evaluation of Acquired Antibiotic Resistance in Escherichia coli Exposed to Long-Term Low-Shear Modeled Microgravity and Background Antibiotic Exposure. mBio 2019, 10, e02637-18. [Google Scholar] [CrossRef] [Green Version]
- Dalrymple, B.; Arber, W. Promotion of RNA Transcription on the Insertion Element IS30 of E. Coli K12. EMBO J. 1985, 4, 2687–2693. [Google Scholar] [CrossRef]
- Neuwald, A.F.; Stauffer, G.V. IS30 Activation of an Smp′-LacZ Gene Fusion in Escherichia coli. FEMS Microbiol. Lett. 1990, 68, 13–17. [Google Scholar] [CrossRef]
- Dalrymple, B. Novel Rearrangements of IS30 Carrying Plasmids Leading to the Reactivation of Gene Expression. Mol. Gen. Genet. 1987, 207, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, B.; Caspers, P.; Arber, W. Nucleotide Sequence of the Prokaryotic Mobile Genetic Element IS30. EMBO J. 1984, 3, 2145–2149. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Oshima, T.; Mori, H.; Aono, R.; Yamamoto, K.; Ishihama, A.; Utsumi, R. Transcriptional Regulation of Drug Efflux Genes by EvgAS, a Two-Component System in Escherichia coli. Microbiology 2003, 149, 2819–2828. [Google Scholar] [CrossRef]
- Huerta, A.M.; Collado-Vides, J. Sigma70 Promoters in Escherichia coli: Specific Transcription in Dense Regions of Overlapping Promoter-like Signals. J. Mol. Biol. 2003, 333, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Hong, J.S.J.; Qiu, Y.; Nagarajan, H.; Seo, J.H.; Cho, B.K.; Tsai, S.F.; Palsson, B. Comparative Analysis of Regulatory Elements between Escherichia coli and Klebsiella Pneumoniae by Genome-Wide Transcription Start Site Profiling. PLoS Genet. 2012, 8, e1002867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, N.; van Nimwegen, E. Universal Patterns of Purifying Selection at Noncoding Positions in Bacteria. Genome Res. 2008, 18, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossein Khademi, S.M.; Sazinas, P.; Jelsbak, L. Within-Host Adaptation Mediated by Intergenic Evolution in Pseudomonas Aeruginosa. Genome Biol. Evol. 2019, 11, 1385. [Google Scholar] [CrossRef] [Green Version]
- Morita, Y.; Kodama, K.; Shiota, S.; Mine, T.; Kataoka, A.; Mizushima, T.; Tsuchiya, T. NorM, a Putative Multidrug Efflux Protein, of Vibrio Parahaemolyticus and Its Homolog in Escherichia coli. Antimicrob. Agents Chemother. 1998, 42, 1778. [Google Scholar] [CrossRef] [Green Version]
- Long, F.; Rouquette-Loughlin, C.; Shafer, W.M.; Yu, E.W. Functional Cloning and Characterization of the Multidrug Efflux Pumps NorM from Neisseria Gonorrhoeae and YdhE from Escherichia coli. Antimicrob. Agents Chemother. 2008, 52, 3052–3060. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, T.; Tsuchiya, T. Multidrug Efflux Transporters in the MATE Family. Biochim. Biophys. Acta 2009, 1794, 763–768. [Google Scholar] [CrossRef]
- Nishino, K.; Yamaguchi, A. Analysis of a Complete Library of Putative Drug Transporter Genes in Escherichia coli. J. Bacteriol. 2001, 183, 5803–5812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Cook, D.N.; Alberti, M.; Pon, N.G.; Nikaido, H.; Hearst, J.E. Molecular Cloning and Characterization of AcrA and AcrE Genes of Escherichia coli. J. Bacteriol. 1993, 175, 6299–6313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, J.R.; Henrich, B.; Plapp, R. Molecular Analysis and Nucleotide Sequence of the EnvCD Operon of Escherichia coli. Mol. Gen. Genet. 1991, 230, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Multidrug Efflux Pumps of Gram-Negative Bacteria. J. Bacteriol. 1996, 178, 5853–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.; Cook, D.N.; Hearst, J.E.; Nikaido, H. Efflux Pumps and Drug Resistance in Gram-Negative Bacteria. Trends Microbiol. 1994, 2, 489–493. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tsukagoshi, N.; Aono, R. Suppression of Hypersensitivity of Escherichia coli AcrB Mutant to Organic Solvents by Integrational Activation of the AcrEF Operon with the IS1 or IS2 Element. J. Bacteriol. 2001, 183, 2646–2653. [Google Scholar] [CrossRef] [Green Version]
- Robert Klein, J.; Henrich, B.; Plapp, R. Molecular Cloning of TheEnvC Gene OfEscherichia coli. Curr. Microbiol. 1990, 21, 341–347. [Google Scholar] [CrossRef]
- Nielsen, L.E.; Snesrud, E.C.; Onmus-Leone, F.; Kwak, Y.I.; Avilés, R.; Steele, E.D.; Sutter, D.E.; Waterman, P.E.; Lesho, E.P. IS5 Element Integration, a Novel Mechanism for Rapid In Vivo Emergence of Tigecycline Nonsusceptibility in Klebsiella Pneumoniae. Antimicrob. Agents Chemother. 2014, 58, 6151. [Google Scholar] [CrossRef] [Green Version]
- Shuster, Y.; Steiner-Mordoch, S.; Cudkowicz, N.A.; Schuldiner, S. A Transporter Interactome Is Essential for the Acquisition of Antimicrobial Resistance to Antibiotics. PLoS ONE 2016, 11, e0152917. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.Z.; Chang, M.X.; Yang, L.; Liu, Y.Y.; Chen, P.X.; Jiang, H.X. Upregulation of AcrEF in Quinolone Resistance Development in Escherichia coli When AcrAB-TolC Function Is Impaired. Microb. Drug Resist. 2018, 24, 18–23. [Google Scholar] [CrossRef]
- Yang, S.; Clayton, S.R.; Zechiedrich, E.L. Relative Contributions of the AcrAB, MdfA and NorE Efflux Pumps to Quinolone Resistance in Escherichia coli. J. Antimicrob. Chemother. 2003, 51, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, H.; Nishino, K.; Hirata, T.; Yamaguchi, A. Comprehensive Studies of Drug Resistance Mediated by Overexpression of Response Regulators of Two-Component Signal Transduction Systems in Escherichia coli. J. Bacteriol. 2003, 185, 1851–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saier, M.H.; Reddy, V.S.; Tsu, B.V.; Ahmed, M.S.; Li, C.; Moreno-Hagelsieb, G. The Transporter Classification Database (TCDB): Recent Advances. Nucleic Acids Res. 2016, 44, D372–D379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.; Liu, M.; Burgess, R.R. Promoter and Regulon Analysis of Nitrogen Assimilation Factor, Sigma54, Reveal Alternative Strategy for E. Coli MG1655 Flagellar Biosynthesis. Nucleic Acids Res. 2010, 38, 1273–1283. [Google Scholar] [CrossRef] [Green Version]
- Salcedo-Sora, J.E.; Jindal, S.; O’hagan, S.; Kell, D.B. A Palette of Fluorophores That Are Differentially Accumulated by Wild-Type and Mutant Strains of Escherichia coli: Surrogate Ligands for Profiling Bacterial Membrane Transporters. Microbiology 2021, 167, 001016. [Google Scholar] [CrossRef]
- Jindal, S.; Yang, L.; Day, P.J.; Kell, D.B. Involvement of Multiple Influx and Efflux Transporters in the Accumulation of Cationic Fluorescent Dyes by Escherichia coli. BMC Microbiol. 2019, 19, 195. [Google Scholar] [CrossRef] [Green Version]
- Tenaillon, O.; Rodríguez-Verdugo, A.; Gaut, R.L.; McDonald, P.; Bennett, A.F.; Long, A.D.; Gaut, B.S. The Molecular Diversity of Adaptive Convergence. Science 2012, 335, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Weinreich, D.M.; Watson, R.A.; Chao, L. Perspective: Sign Epistasis and Genetic Costraint on Evolutionary Trajectories. Evolution 2005, 59, 1165–1174. [Google Scholar] [CrossRef]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R Package for the Visualization of Intersecting Sets and Their Properties. Bioinformatics 2017, 33, 2938. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Cation | Gene | Mutation Types (Unique Counts) | Product | Number of Occurrences across Replicates (n = 4) a,b,c |
|---|---|---|---|---|---|
| eTRef | TPP+ | ybjG/mdfA | SNP (2) | undecaprenyl pyrophosphate phosphatase/multidrug efflux system protein | 1 * |
| acrB | SNP (4) | multidrug efflux system protein | 4 | ||
| mdtK | SNP (3) | multidrug efflux system transporter | 4 | ||
| eTΔemrE | mdtK | SNP (3) | multidrug efflux system transporter | 3 | |
| acrB | SNP (6) | multidrug efflux system protein | 4 | ||
| ybjG/mdfA | MOB (1) | undecaprenyl pyrophosphate phosphatase/multidrug efflux system protein | 2 | ||
| SNP (1) | 1 a | ||||
| ribC/mdtK | SNP (1) | riboflavin synthase, alpha subunit/multidrug efflux system transporter | 1 c | ||
| mdfA | SNP (1) | multidrug efflux system protein | 1 b | ||
| eTΔtolC | mdtK | SNP (2) | multidrug efflux system transporter | 3 | |
| ribC/mdtK | DEL (1) | riboflavin synthase, alpha subunit/multidrug efflux system transporter | 2 | ||
| SNP (1) | 2 | ||||
| eTΔacrB | ribC/mdtK | SNP (1) | riboflavin synthase, alpha subunit/multidrug efflux system transporter | 1 b | |
| mdtK | SNP (4) | multidrug efflux system transporter | 2 | ||
| acrS/acrE | MOB (3) | acrAB operon transcriptional repressor/cytoplasmic membrane lipoprotein | 4 | ||
| eMRef | MTPP+ | acrB | SNP (8) | multidrug efflux system protein | 4 |
| acrA | SNP (2) | multidrug efflux system protein | 1 c | ||
| ribC/mdtK | SNP (1) | riboflavin synthase, alpha subunit/multidrug efflux system transporter | 1 *,b | ||
| mdtK | SNP (2) | multidrug efflux system transporter | 3 | ||
| mdtL | SNP (1) | multidrug efflux system protein | 1 *,b | ||
| mdtP | SNP (1) | outer membrane factor of efflux pump | 1 *,b | ||
| ybjG/mdfA | SNP (1) | undecaprenyl pyrophosphate phosphatase/multidrug efflux system protein | 1 b | ||
| eMΔemrE | nudF/tolC | SNP (1) | ADP-ribose pyrophosphatase/transport channel | 1 c | |
| DEL (1) | 3 | ||||
| acrB | SNP (7) | multidrug efflux system protein | 4 | ||
| mdtK | SNP (1) | multidrug efflux system transporter | 2 | ||
| ybjG/mdfA | SNP (2) | undecaprenyl pyrophosphate phosphatase/multidrug efflux system protein | 2 | ||
| eMΔtolC | ybjG/mdfA | SNP (2) | undecaprenyl pyrophosphate phosphatase/multidrug efflux system protein | 4 | |
| INS (2) | 4 | ||||
| mdfA | SNP (2) | multidrug efflux system protein | 1 a | ||
| tisB/emrD | SNP (1) | toxic membrane persister formation peptide, LexA-regulated/multidrug efflux system protein | 1 b | ||
| eMΔacrB | ybjG/mdfA | SNP (1) | undecaprenyl pyrophosphate phosphatase/multidrug efflux system protein | 3 | |
| INS (2) | 2 | ||||
| acrS/acrE | MOB (3) | acrAB operon transcriptional repressor/cytoplasmic membrane lipoprotein | 3 | ||
| SNP (2) | 4 |
| MDTs | Strain | Mutations | Occurrence (n = 4) | Hydropathy Index/Polarity/Acidity Change |
|---|---|---|---|---|
| acrB | eTRef | D276G PD Asp → Gly | (2) | −3.5/Polar/Acidic → −0.4/Nonpolar/Neutral |
| D276N PD Asp → Asn | (2) | −3.5/Polar/Acidic → −3.5/Polar/Neutral | ||
| E273A PD Glu → Ala | (2) | −3.5/Polar/Acidic → 1.8/Nonpolar/Neutral | ||
| N68K PD Asn → Lys | (1) | −3.5/Polar/Neutral → −3.5/Polar/Basic | ||
| eMRef | V612D PD Val → Asp | (1) | 4.2/Nonpolar/Neutral → −3.5/Polar/Acidic | |
| E273A PD Glu → Ala | (2) | −3.5/Polar/Acidic → 1.8/Nonpolar/Neutral | ||
| D276G PD Asp → Gly | (2) | −3.5/Polar/Acidic → −0.4/Nonpolar/Neutral | ||
| N274K PD Asn → Lys | (1) | −3.5/Polar/Neutral → −3.9/Polar/Basic | ||
| V139F PD Val → Phe | (1) | 4.2/Nonpolar/Neutral → −2.8/Nonpolar/Neutral | ||
| Q125K PD Gln → Lys | (1) | −3.5/Polar/Neutral → −3.9/Polar/Basic | ||
| S128R PD Ser → Arg | (1) | −0.8/Polar/Neutral → −4.5/Polar/Basic(strongly) | ||
| Q89K PD Gln → Lys | (1) | −3.5/Polar/Neutral → −3.9/Polar/Basic | ||
| eTΔemrE | D276G PD Asp → Gly | (2) | −3.5/Polar/Acidic → −0.4/Nonpolar/Neutral | |
| N274K PD Asn → Lys | (1) | −3.5/Polar/Neutral → −3.9/Polar/Basic | ||
| E273A PD Glu → Ala | (1) | −3.5/Polar/Acidic → 1.8/Nonpolar/Neutral | ||
| E130V PD Glu → Val | (1) | −3.5/Polar/Acidic → 4.2/Nonpolar/Neutral | ||
| Q125K PD Gln → Lys | (1) | −3.5/Polar/Neutral → −3.9/Polar/Basic | ||
| N70K PD Asn → Lys | (1) | −3.5/Polar/Neutral → −3.9/Polar/Basic | ||
| eMΔemrE | V612D PD Val → Asp | (1) | 4.2/Nonpolar/Neutral → −3.5/Polar/Acidic | |
| L542R TMD Leu → Arg | (1) | 3.8/Nonpolar/Neutral → −4.5/Polar/Basic(strongly) | ||
| D276G PD Asp → Gly | (1) | −3.5/Polar/Acidic → −0.4/Nonpolar/Neutral | ||
| E273A PD Glu → Ala | (1) | −3.5/Polar/Acidic → 1.8/Nonpolar/Neutral | ||
| V139F PD Val → Phe | (2) | 4.2/Nonpolar/Neutral → −2.8/Nonpolar/Neutral | ||
| Q89K PD Gln → Lys | (1) | −3.5/Polar/Neutral → −3.9/Polar/Basic | ||
| V32A PD Val → Ala | (2) | 4.2/Nonpolar/Neutral → 1.8/Nonpolar/Neutral | ||
| mdtK | eTRef | R148S Arg → Ser | (2) | −4.5/Polar/Basic(strongly) → −0.8/Polar/Neutral |
| M1M Met → Met | (1) | 1.9/Nonpolar/neutral → 1.9/Nonpolar/neutral (synonymous) | ||
| R148G Arg → Gly | (4) | −4.5/Polar/Basic(strongly) → −0.4/Nonpolar/neutral | ||
| eMRef | R81C Arg → Cys | (2) | −4.5/Polar/Basic(strongly) → 2.5/Polar/Neutral | |
| R148G Arg → Gly | (2) | −4.5/Polar/Basic(strongly) → −0.4/Nonpolar/neutral | ||
| eTΔemrE | R81C Arg → Cys | (1) | −4.5/Polar/Basic(strongly) → 2.5/Polar/Neutral | |
| R81L Arg → Leu | (1) | −4.5/Polar/Basic(strongly) → 3.8/Nonpolar/Neutral | ||
| R148G Arg → Gly | (3) | −4.5/Polar/Basic(strongly) → −0.4/Nonpolar/neutral | ||
| eMΔemrE | R148G Arg → Gly | (2) | −4.5/Polar/Basic(strongly) → −0.4/Nonpolar/neutral | |
| eTΔtolC | M1M b Met → Met | (1) | 1.9/Nonpolar/Neutral → 1.9/Nonpolar/Neutral (No change) | |
| G428A Gly → Ala | (1) | −0.4/Nonpolar/Neutral → 1.8/Nonpolar/Neutral | ||
| eMΔacrB | W54G Trp → Gly | (1) | −0.9/Nonpolar/Neutral → −0.4/Nonpolar/Neutral | |
| A294V Ala → Val | (1) | 1.8/Nonpolar/Neutral → 4.2/Nonpolar/Neutral | ||
| G424S Gly → Ser | (1) | −0.4/Nonpolar/Neutral → −0.8/polar/Neutral | ||
| G428S Gly → Ser | (1) | −0.4/Nonpolar/Neutral → −0.8/polar/Neutral | ||
| mdfA | eTΔemrE | A144V Ala → Val | (1) | 1.8/Nonpolar/Neutral → 4.2/Nonpolar/Neutral |
| eMΔtolC | F134C Phe → Cys | (1) | −2.8/Nonpolar/Neutral → 2.5/Polar/Neutral | |
| M146I Met → Ile | (1) | 1.9/Nonpolar/neutral → 4.5/Nonpolar/neutral | ||
| acrA | eMRef | T177T Thr → Thr | (1) | −0.7/Polar/neutral → −0.7/Polar/neutral (synonymous) |
| G6W (1) Gly→ Trp | (1) | −0.4/Nonpolar/Neutral → −0.9/Nonpolar/neutral | ||
| mdtL | eMRef | M166I Met → Ile | (1) | 1.9/Nonpolar/neutral → 4.5/Nonpolar/neutral |
| mdtP | eMRef | Q312H Gln → His | (1) | −3.5/Polar/Neutral → −3.2/Polar/Basic (Weakly) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radi, M.S.; Munro, L.J.; Salcedo-Sora, J.E.; Kim, S.H.; Feist, A.M.; Kell, D.B. Understanding Functional Redundancy and Promiscuity of Multidrug Transporters in E. coli under Lipophilic Cation Stress. Membranes 2022, 12, 1264. https://doi.org/10.3390/membranes12121264
Radi MS, Munro LJ, Salcedo-Sora JE, Kim SH, Feist AM, Kell DB. Understanding Functional Redundancy and Promiscuity of Multidrug Transporters in E. coli under Lipophilic Cation Stress. Membranes. 2022; 12(12):1264. https://doi.org/10.3390/membranes12121264
Chicago/Turabian StyleRadi, Mohammad S., Lachlan J. Munro, Jesus E. Salcedo-Sora, Se Hyeuk Kim, Adam M. Feist, and Douglas B. Kell. 2022. "Understanding Functional Redundancy and Promiscuity of Multidrug Transporters in E. coli under Lipophilic Cation Stress" Membranes 12, no. 12: 1264. https://doi.org/10.3390/membranes12121264