Self-Assembling Peptide Surfactants A6K and A6D Adopt a-Helical Structures Useful for Membrane Protein Stabilization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Circular Dichroism (CD) Measurements
2.3. Field Emission Scanning Electron Microscopy (FESEM) Studies
3. Results and Discussion
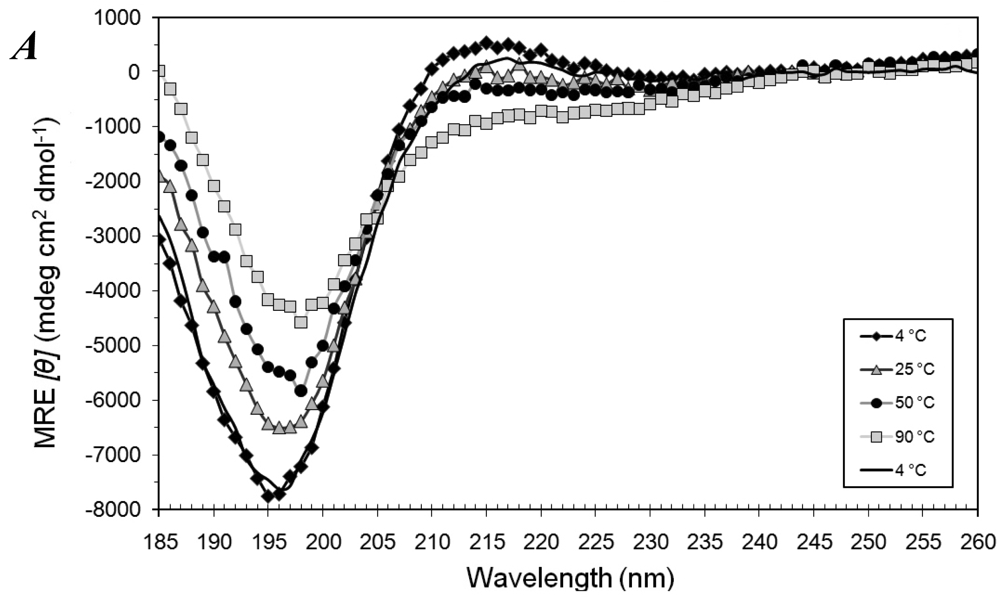
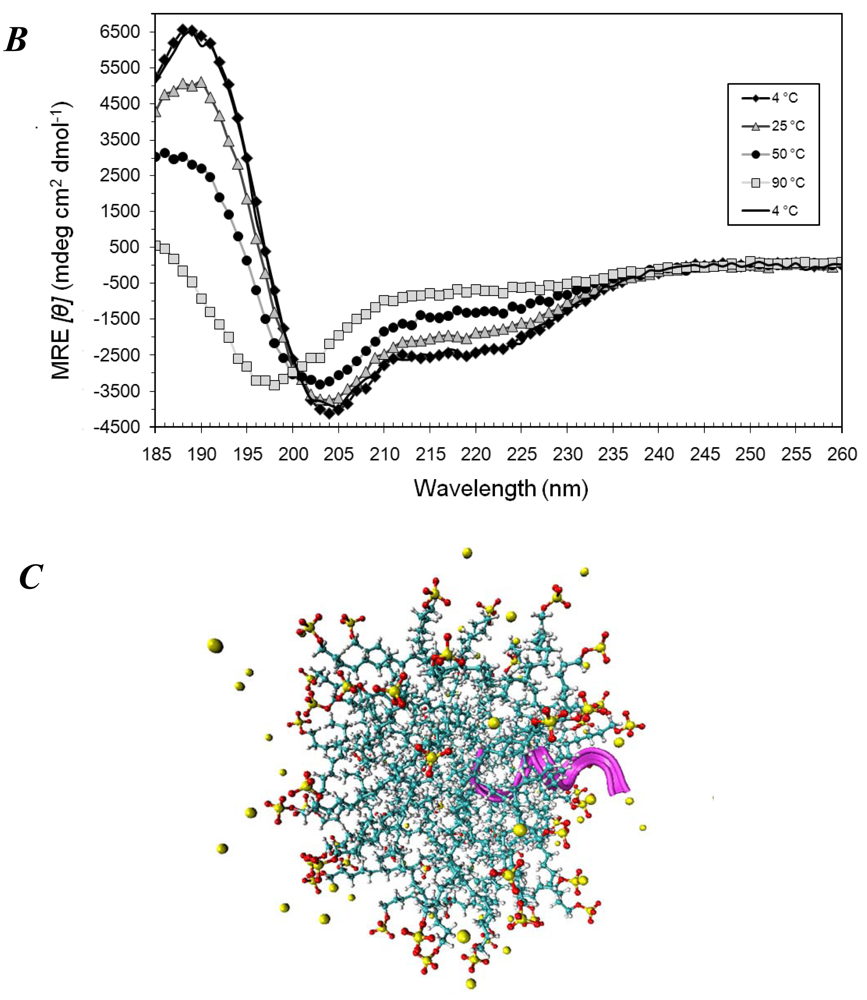
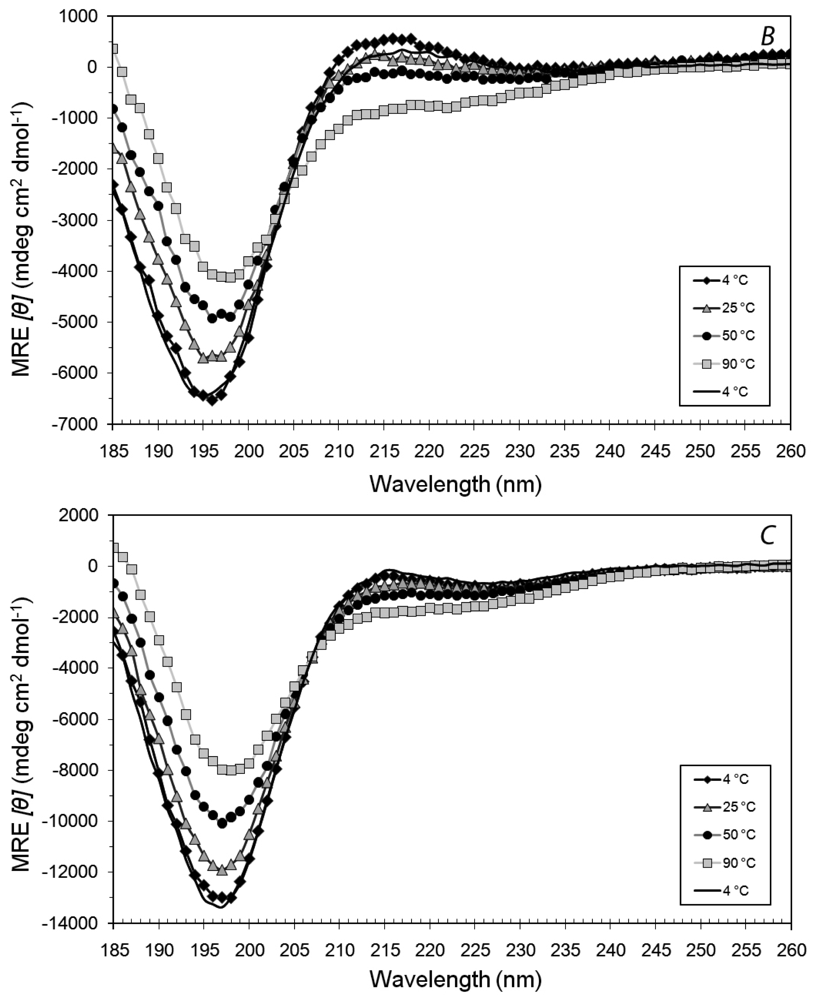
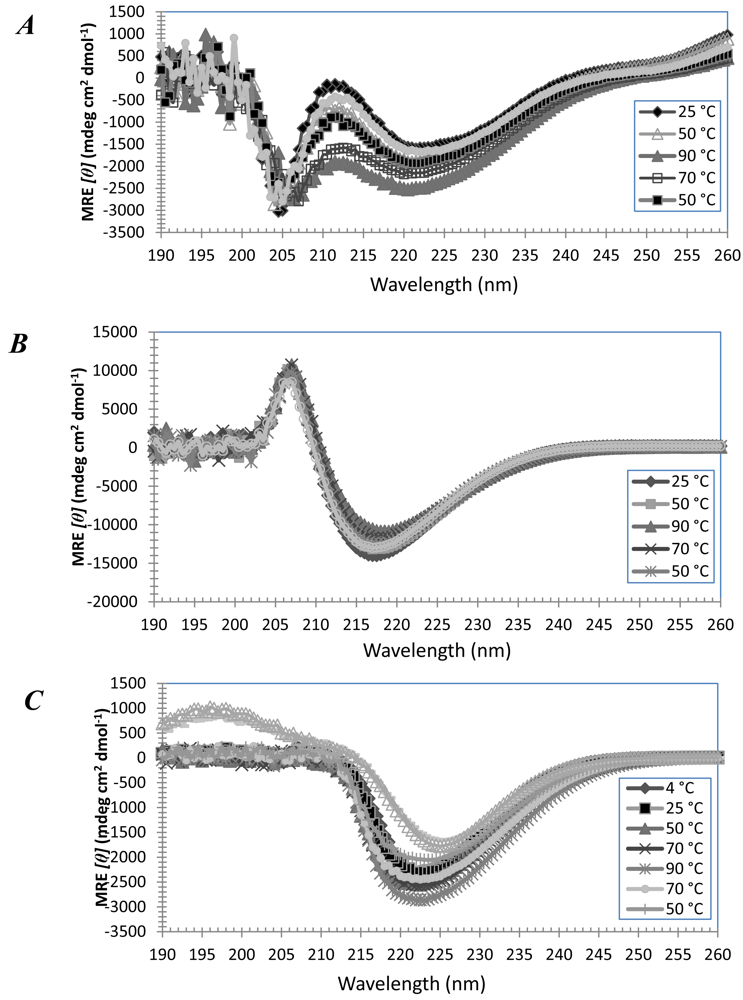
3.1. Secondary Structures of A6D and A6K Peptide Surfactants in SDS
3.2. Secondary Structures of A6D and A6K Peptide Surfactants in Water
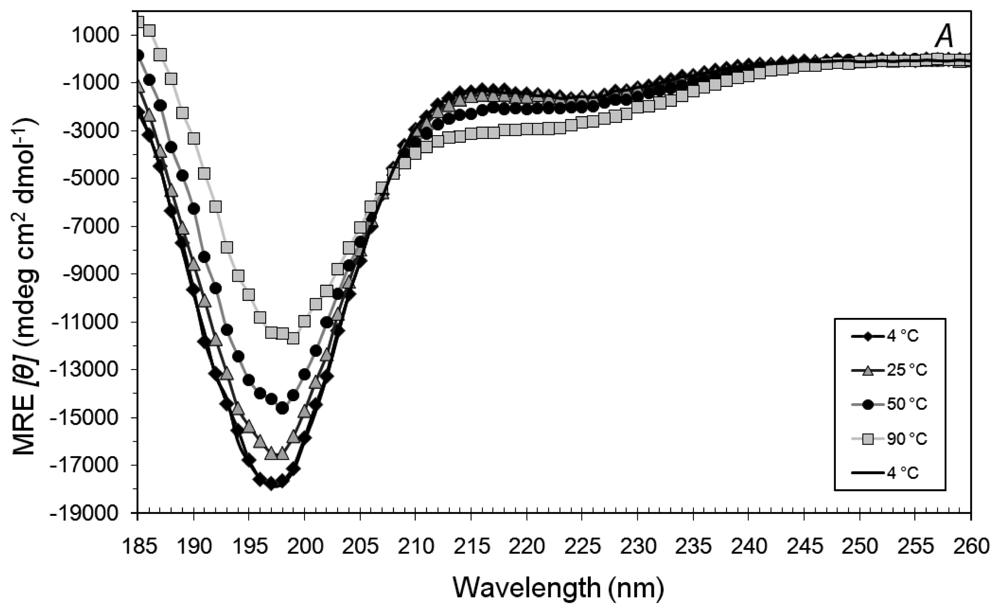
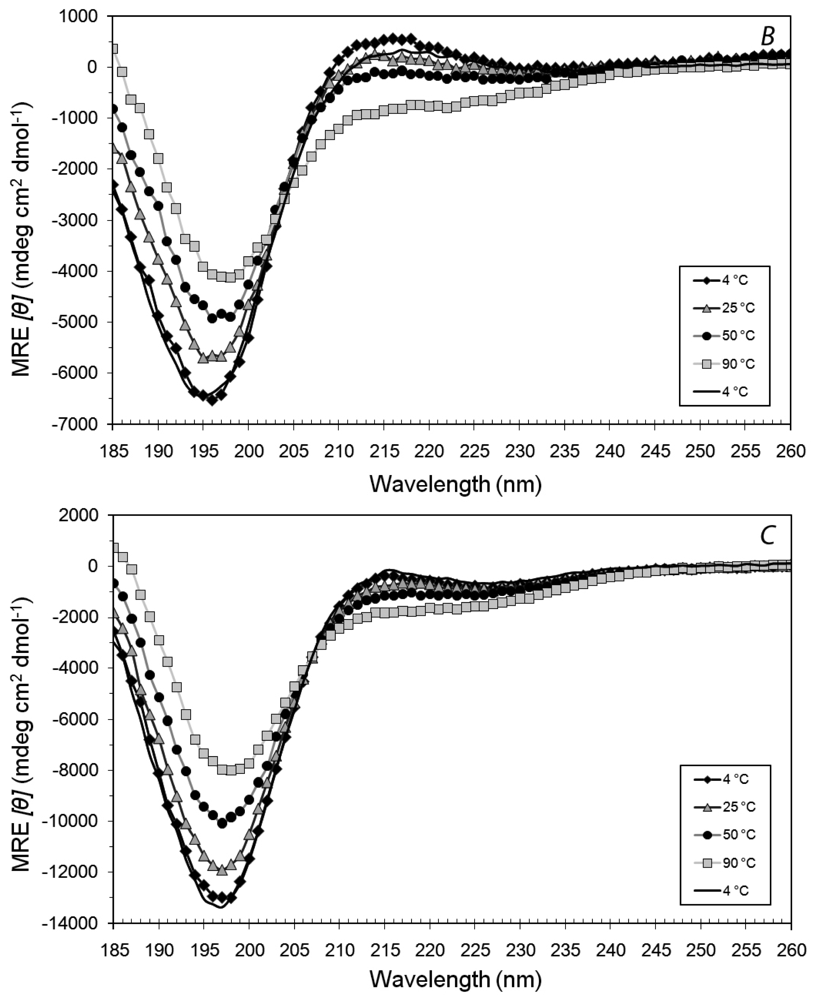
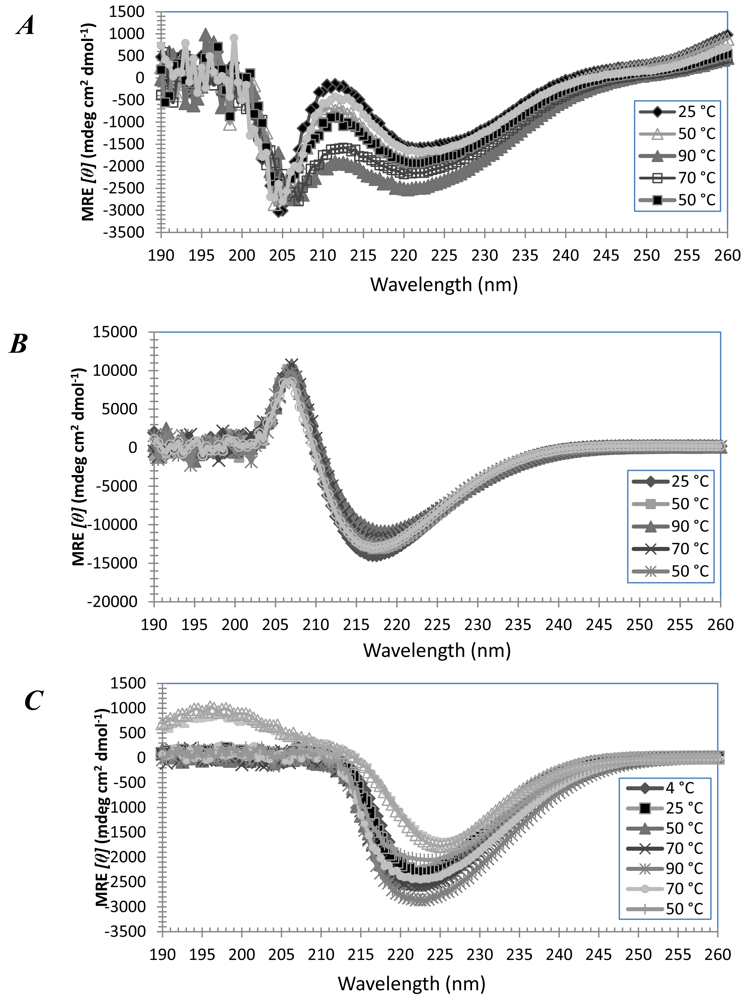

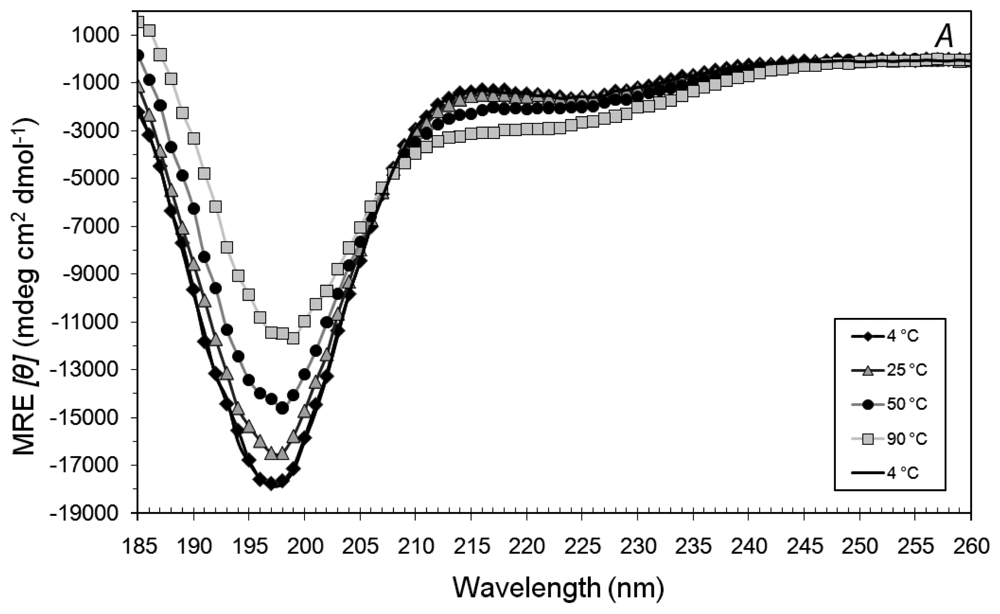
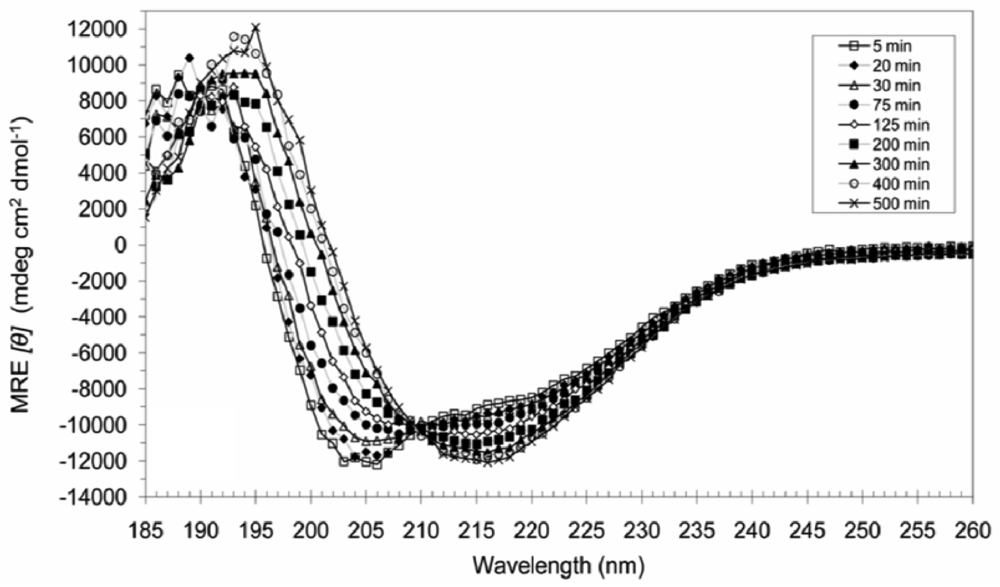
3.3. Secondary Structures of A6D and A6K in Combination, in Water
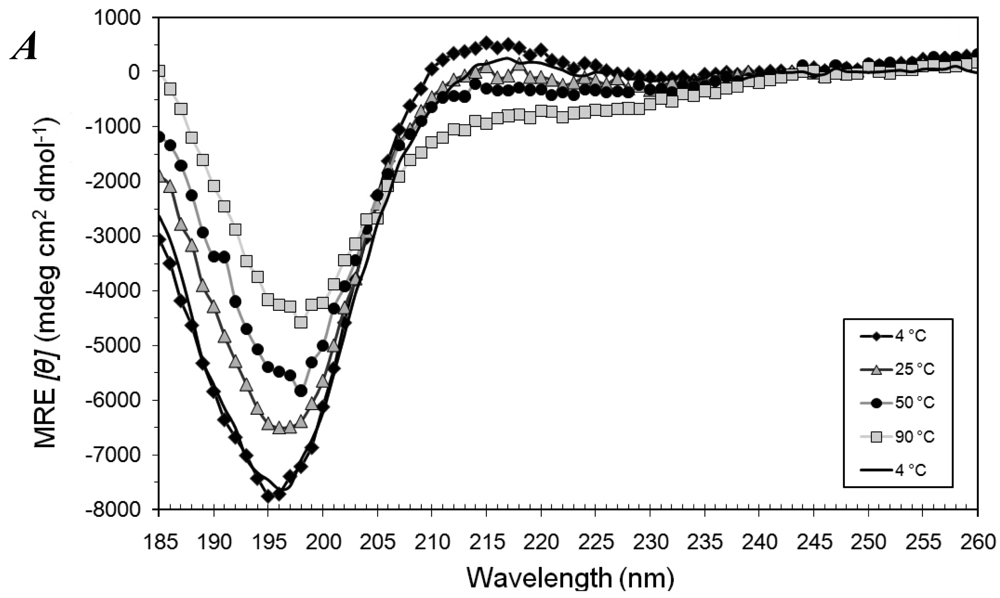
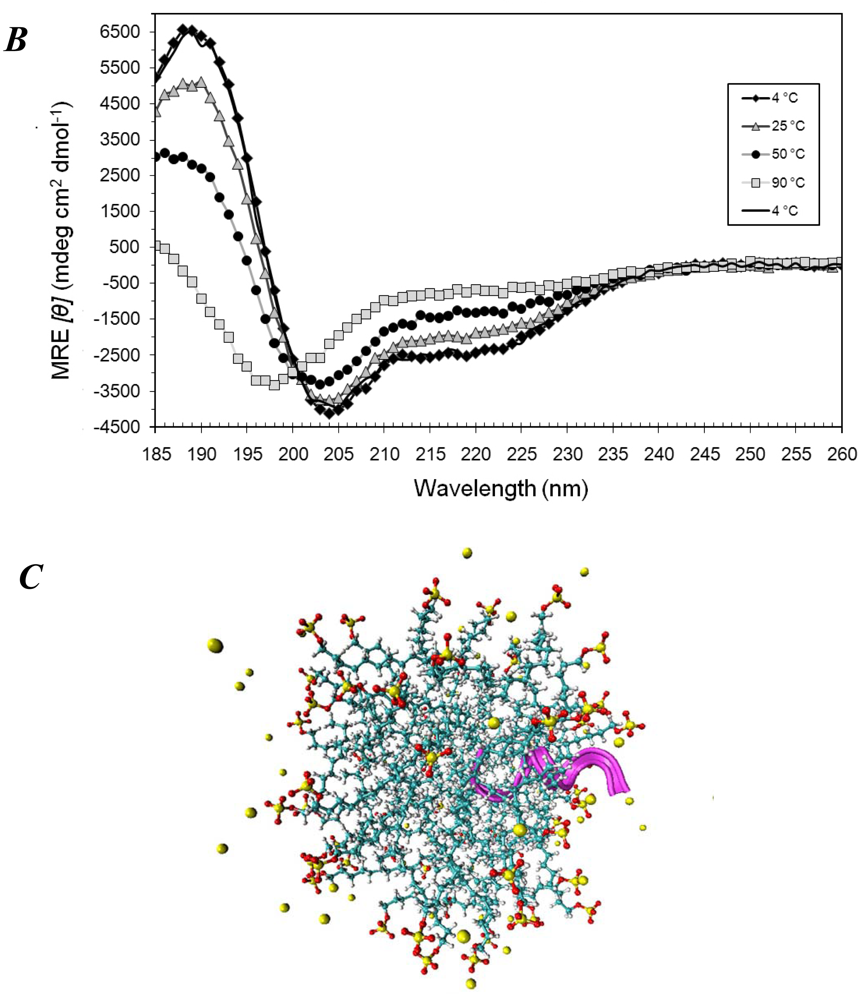
3.4. Secondary Structural Changes of a Combination of A6D and A6K in SDS
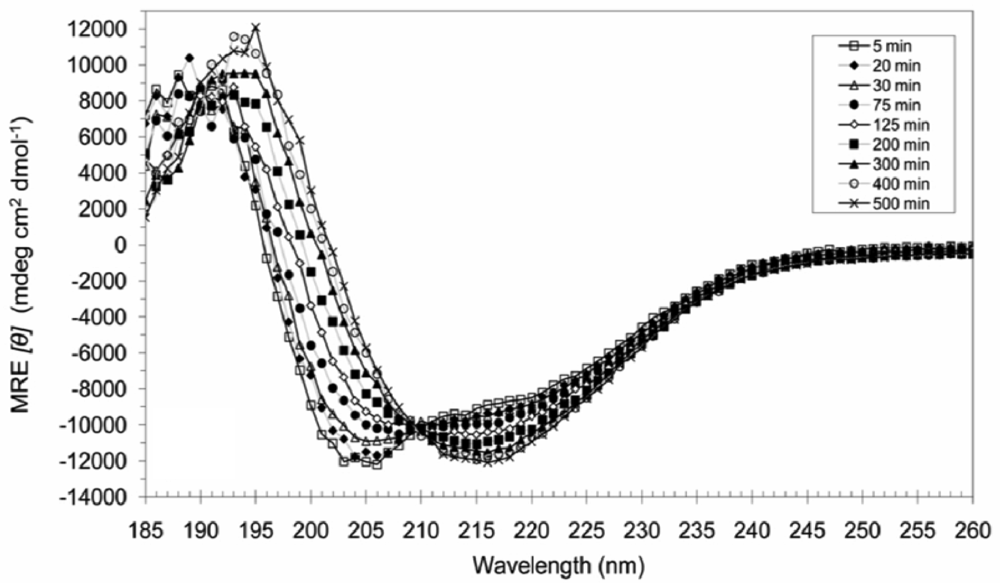
4. Conclusions
Acknowledgments
References
- Hauser, C.A.E.; Zhang, S. Designer self-assembling peptide nanofiber biological materials. Chem. Soc. Rev. 2010, 39, 2780–2790. [Google Scholar]
- Cavalli, S.; Albericio, F.; Kros, A. Amphiphilic peptides and their cross-disciplinary role as building blocks for nanoscience. Chem. Soc. Rev. 2010, 39, 241–263. [Google Scholar]
- Huang, R.; Qi, W.; Feng, L.; Su, R.; He, Z. Self-assembling peptide-polysaccharide hybrid hydrogel as a potential carrier for drug delivery. Soft Matter 2011, 7, 6222–6230. [Google Scholar]
- Woolfson, D.N.; Mahmoud, Z.N. More than just bare scaffolds: Towards multi-component and decorated fibrous biomaterials. Chem. Soc. Rev. 2010, 39, 3464–3479. [Google Scholar]
- Vauthey, S.; Santoso, S.; Gong, H.; Watson, N.; Zhang, S. Molecular self-assembly of surfactant-like peptides to form nanotubes and nanovesicles. Proc. Natl. Acad. Sci. USA 2002, 99, 5355–5360. [Google Scholar]
- Von Maltzahn, G.; Vauthey, S.; Santoso, S.; Zhang, S.U. Positively charged surfactant-like peptides self-assemble into nanostructures. Langmuir 2003, 19, 4332–4337. [Google Scholar]
- Yang, S.J.; Zhang, S. Self-assembling behavior of designer lipid-like peptides. Supramol. Chem. 2006, 18, 389–396. [Google Scholar]
- Nagai, A.; Nagai, Y.; Qu, H.; Zhang, S. Dynamic behaviors of lipid-like self-assembling peptide A6D and A6K nanotubes. J. Nanosci. Nanotechnol. 2007, 7, 2246–2252. [Google Scholar]
- Khoe, U.; Yang, Y.; Zhang, S. Synergistic effect and hierarchical nanostructure formation in mixing two designer lipid-like peptide surfactants Ac-A6D-OH and Ac-A6K-NH2. Macromol. Biosci. 2008, 8, 1060–1067. [Google Scholar]
- Wang, X.; Corin, K.; Baaske, P.; Wienken, C.J.; Jerabek-Willemsen, M.; Duhr, S.; Braun, D.; Zhang, S. Peptide surfactants for cell-free production of functional G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 9049–9054. [Google Scholar]
- Yeh, J.I.; Du, S.; Tortajada, A.; Paulo, J.; Zhang, S. Peptergents: Peptide detergents that improve stability and functionality of a membrane protein, glycerol-3-phosphate dehydrogenase. Biochemistry (Mosc) 2005, 44, 16912–16919. [Google Scholar]
- Kiley, P.; Zhao, X.; Vaughn, M.; Baldo, M.A.; Bruce, B.D.; Zhang, S. Self-assembling peptide detergents stabilize isolated Photosystem I on a dry surface for an extended time. PLoS Biol. 2005, 3, e230:1–e230:7. [Google Scholar]
- Zhao, X.; Nagai, Y.; Reeves, P.J.; Kiley, P.; Khorana, H.G.; Zhang, S. Designer short peptide surfactants stabilize G protein-coupled receptor bovine rhodopsin. Proc. Natl. Acad. Sci. USA 2006, 103, 17707–17712. [Google Scholar]
- Matsumoto, K.; Vaughn, M.; Bruce, B.D.; Koutsopoulos, S.; Zhang, S. Designer peptide surfactants stabilize functional Photosystem I membrane complex in aqueous solution for extended time. J. Phys. Chem. B 2009, 113, 75–83. [Google Scholar]
- Yaghmur, A.; Laggner, P.; Zhang, S.; Rappolt, M. Tuning curvature and stability of monoolein bilayers by designer lipid-like peptide surfactants. PLoS One 2007, 2, e479:1–e479:10. [Google Scholar]
- Luo, Z.; Akerman, B.; Zhang, S.; Norden, B. Structures of self-assembled amphiphilic peptide-heterodimers: Effects of concentration, pH, temperature and ionic strength. Soft Matter 2010, 6, 2260–2270. [Google Scholar]
- Waterhous, D.V.; Johnson, W.C., Jr. Importance of environment in determining secondary structure in proteins. Biochemistry (Mosc) 1994, 33, 2121–2128. [Google Scholar]
- Hugonin, L.; Barth, A.; Graslund, A.; Peralvarez-Marin, A. Secondary structure transitions and aggregation induced in dynorphin neuropeptides by the detergent sodium dodecyl sulfate. Biochim. Biophys. Acta 2008, 1778, 2580–2587. [Google Scholar]
- Montserret, R.; McLeish, M.J.; Bockmann, A.; Geourjon, C.; Penin, F. Involvement of electrostatic interactions in the mechanism of peptide folding induced by sodium dodecyl sulfate binding. Biochemistry (Mosc) 2000, 39, 8362–8373. [Google Scholar]
- Wu, C.S.; Ikeda, K.; Yang, J.T. Ordered conformation of polypeptides and proteins in acidic dodecyl sulfate solution. Biochemistry (Mosc) 1981, 20, 566–570. [Google Scholar]
- Lin, J.-M.; Lin, T.-L.; Jeng, U.S.; Huang, Z.-H.; Huang, Y.-S. Aggregation structure of Alzheimer amyloid-b(1–40) peptide with sodium dodecyl sulfate as revealed by small-angle X-ray and neutron scattering. Soft Matter 2009, 5, 3913–3919. [Google Scholar]
- Wahlstrom, A.; Hugonin, L.; Peralvarez-Marin, A.; Jarvet, J.; Graslund, A. Secondary structure conversions of Alzheimer's Abeta(1–40) peptide induced by membrane-mimicking detergents. FEBS J. 2008, 275, 5117–5128. [Google Scholar]
- Hatakeyama, Y.; Sawada, T.; Kawano, M.; Fujita, M. Conformational preferences of short peptide fragments. Angew. Chem. Int. Ed. Engl. 2009, 48, 8695–8698. [Google Scholar]
- Chakrabartty, A.; Doig, A.J.; Baldwin, R.L. Helix capping propensities in peptides parallel those in proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 11332–11336. [Google Scholar]
- Kelly, S.M.; Jess, T.J.; Price, N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta 2005, 1751, 119–139. [Google Scholar]
- Hauser, C.A.; Deng, R.; Mishra, A.; Loo, Y.; Khoe, U.; Zhuang, F.; Cheong, D.W.; Accardo, A.; Sullivan, M.B.; Riekel, C.; et al. Natural tri- to hexapeptides self-assemble in water to amyloid beta-type fiber aggregates by unexpected alpha-helical intermediate structures. Proc. Natl. Acad. Sci. USA 2011, 108, 1361–1366. [Google Scholar]
- Schuch, M.; Gross, G.A.; Koehler, J.M. Formation and fluorimetric characterization of micelles in a micro-flow through system with static micro mixer. Sensors 2007, 7, 2499–2509. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhuang, F.; Oglęcka, K.; Hauser, C.A.E. Self-Assembling Peptide Surfactants A6K and A6D Adopt a-Helical Structures Useful for Membrane Protein Stabilization. Membranes 2011, 1, 314-326. https://doi.org/10.3390/membranes1040314
Zhuang F, Oglęcka K, Hauser CAE. Self-Assembling Peptide Surfactants A6K and A6D Adopt a-Helical Structures Useful for Membrane Protein Stabilization. Membranes. 2011; 1(4):314-326. https://doi.org/10.3390/membranes1040314
Chicago/Turabian StyleZhuang, Furen, Kamila Oglęcka, and Charlotte A. E. Hauser. 2011. "Self-Assembling Peptide Surfactants A6K and A6D Adopt a-Helical Structures Useful for Membrane Protein Stabilization" Membranes 1, no. 4: 314-326. https://doi.org/10.3390/membranes1040314