
Development of a Conserved Chimeric Vaccine for Induction of Strong Immune Response against Staphylococcus aureus Using Immunoinformatics Approaches

 , , ,
, , ,
Abstract
:1. Introduction
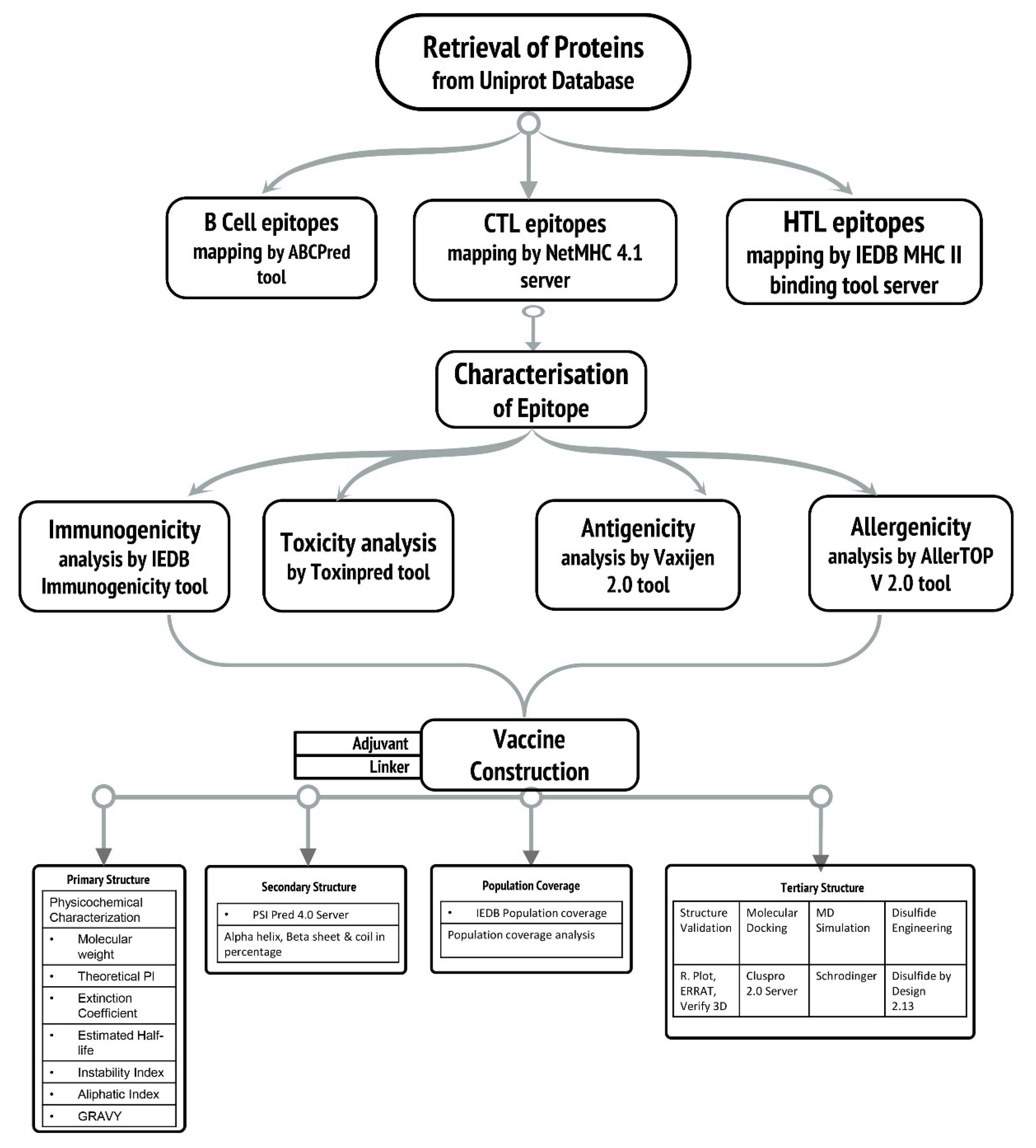
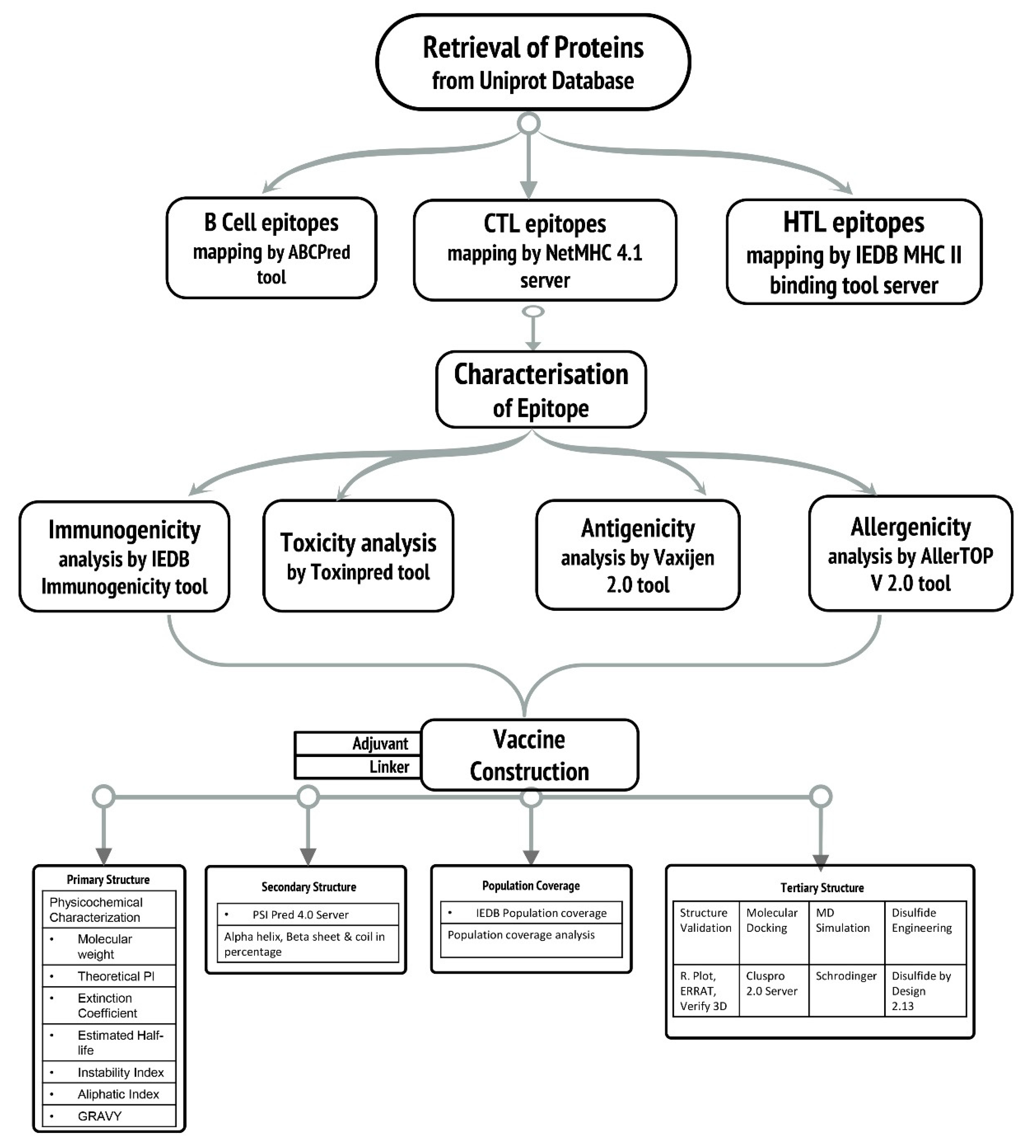
2. Methodology
2.1. Retrieval of Protein Sequences
2.2. Epitopic Region Prediction of B-Cell
2.3. Prediction of Cytotoxic T-Lymphocyte (CTL) and Helper T-Lymphocyte (HTL) Epitopes
2.4. Evaluation of Antigenic, Allergenic, Immunogenicity, and Toxicity
2.5. Epitope Selection Criteria
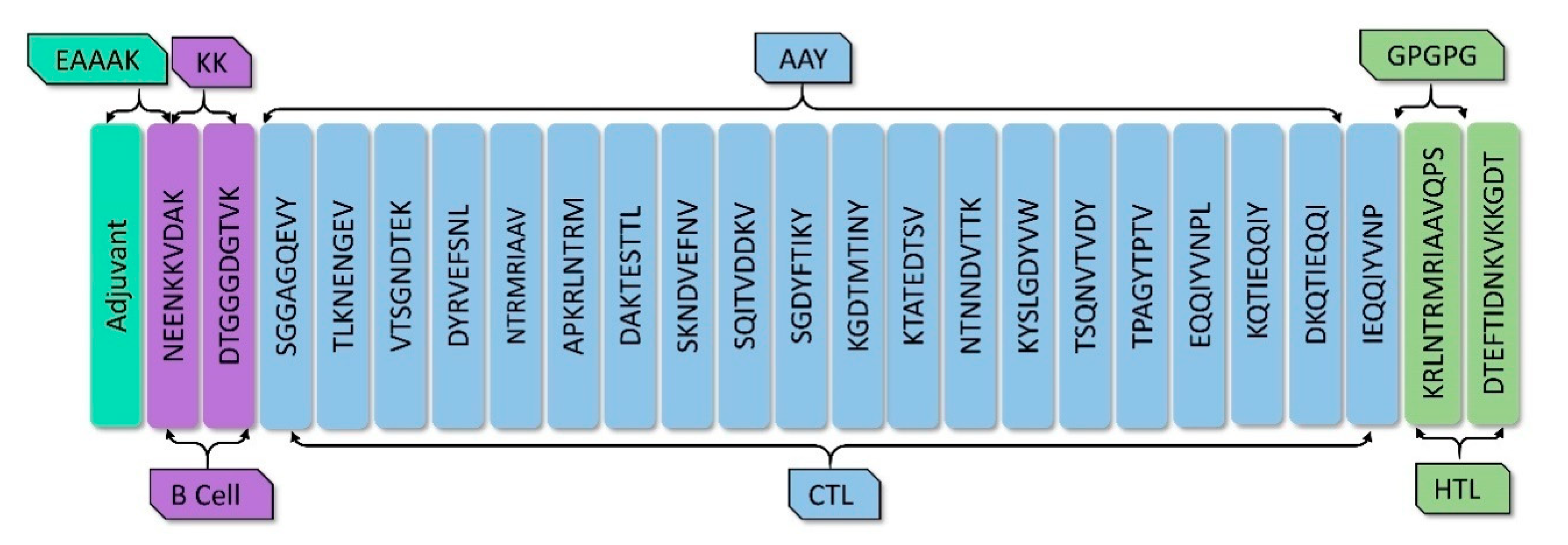
2.6. Vaccine Construction
2.7. Estimation of Population Coverage
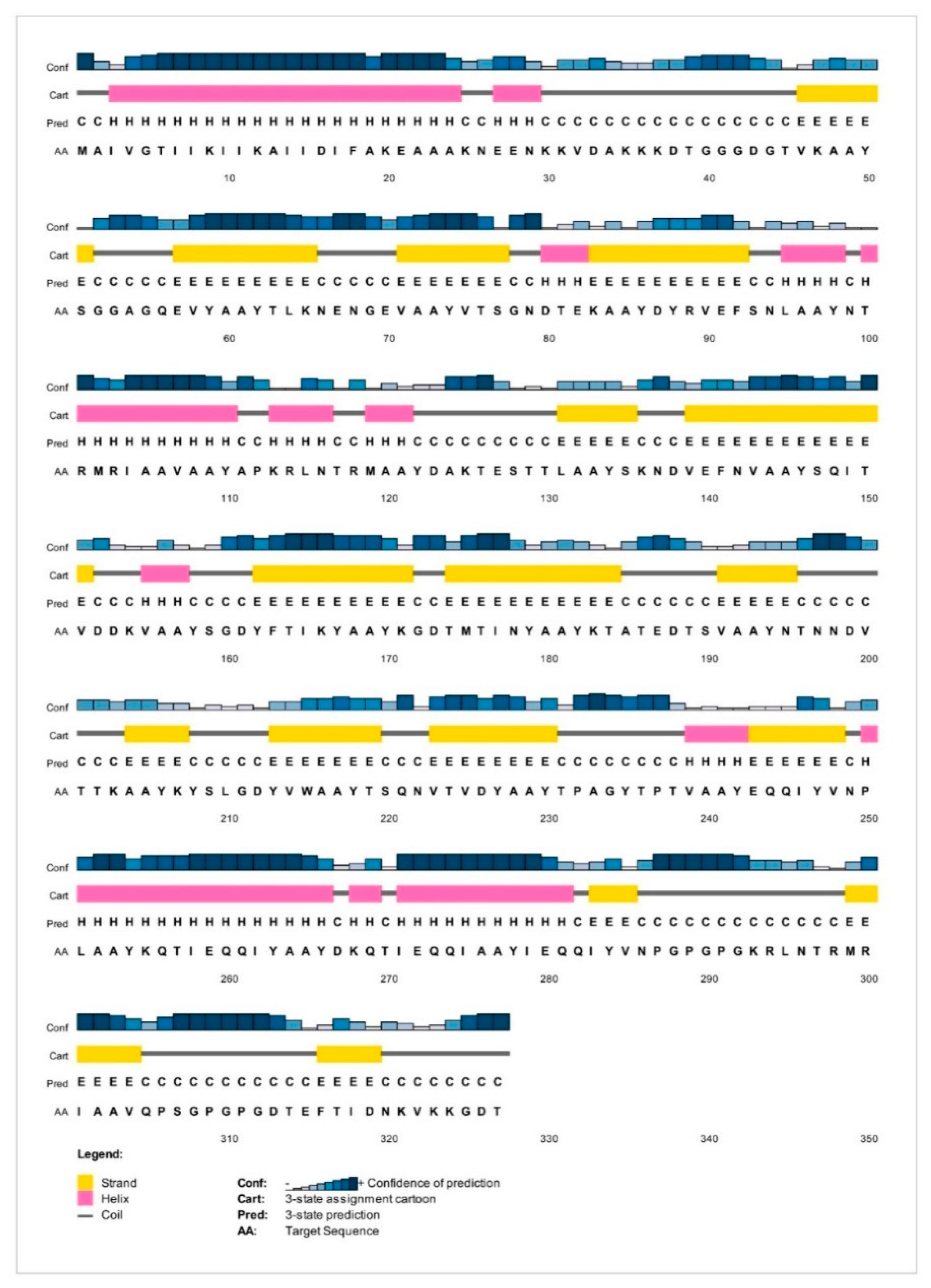
2.8. Analysis of Solubility and Physicochemical Properties and Secondary Structure
2.9. Vaccine Construct’s Antigenicity and Allergenicity Profiling
2.10. Prediction of Interferon-Gamma Inducing Epitopes
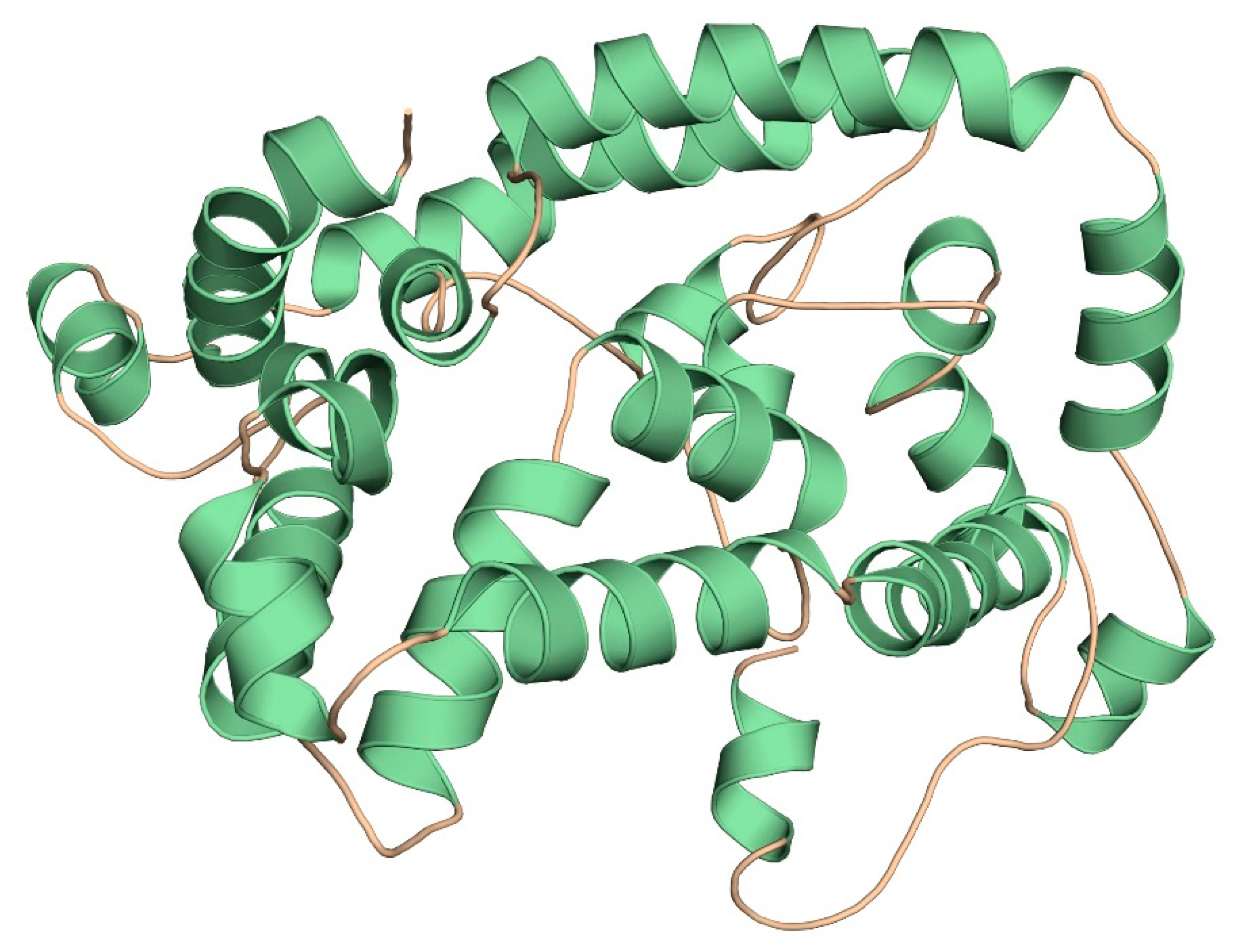
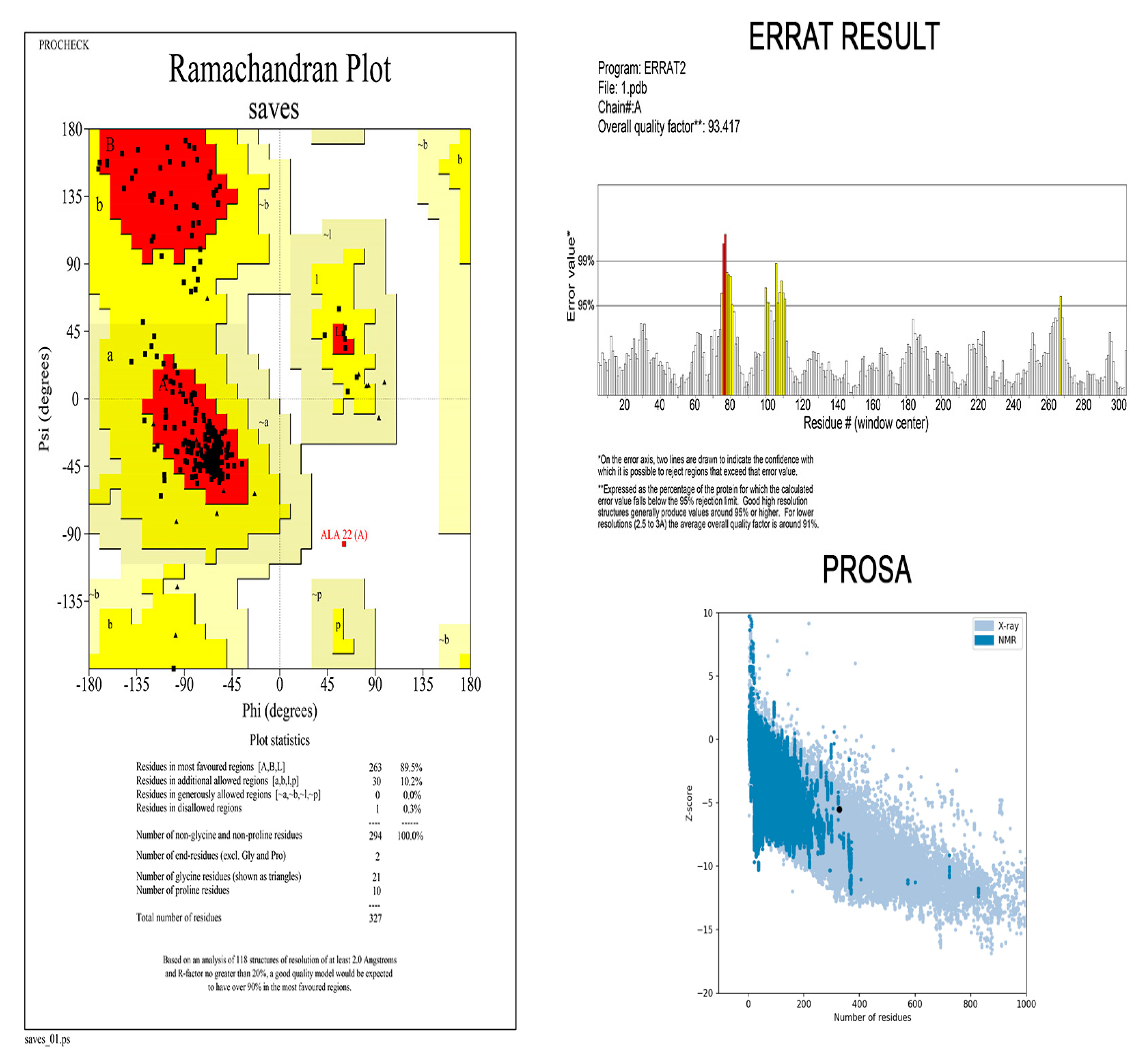
2.11. Three-Dimensional Modelling and Validation
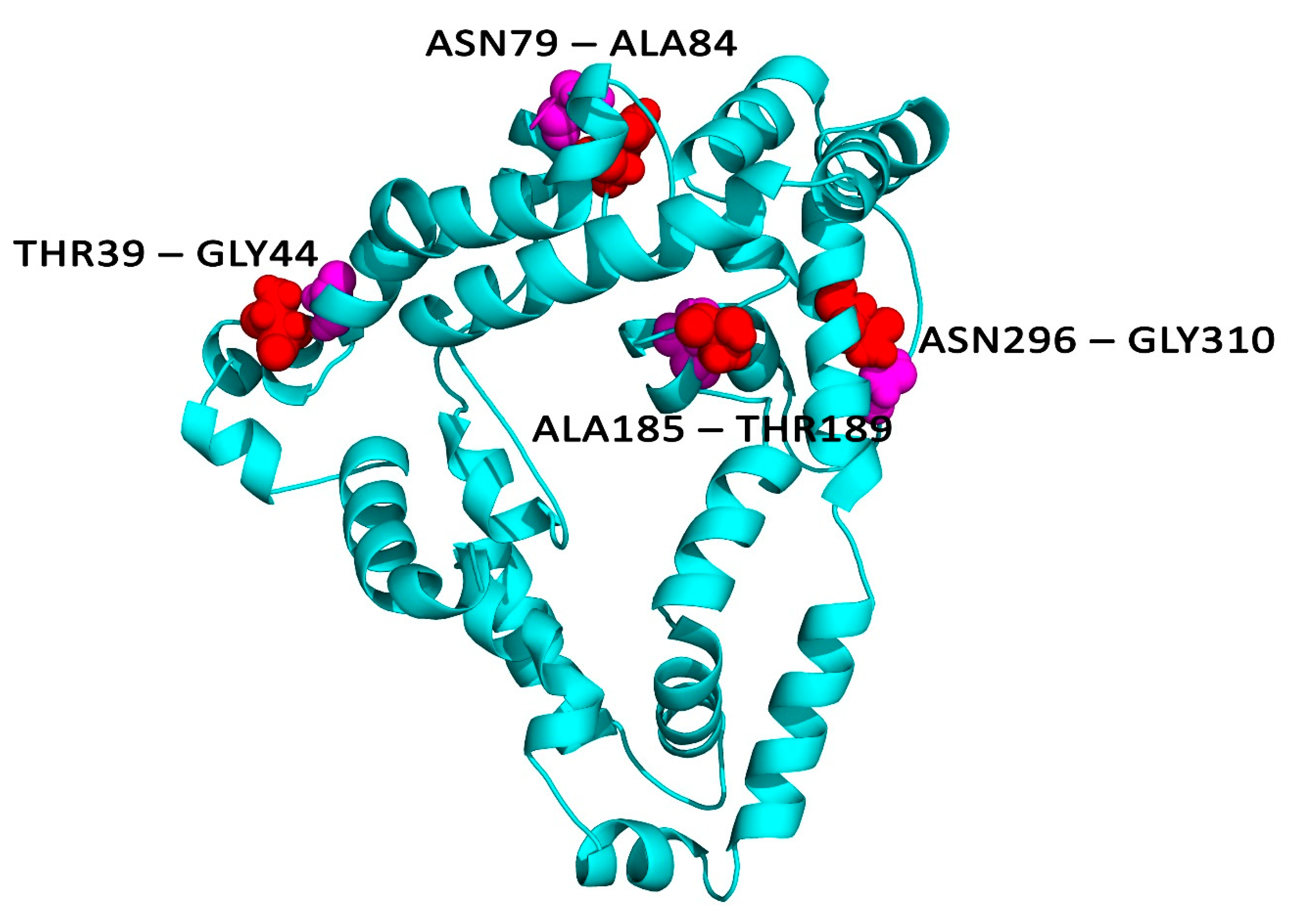
2.12. Disulphide Engineering
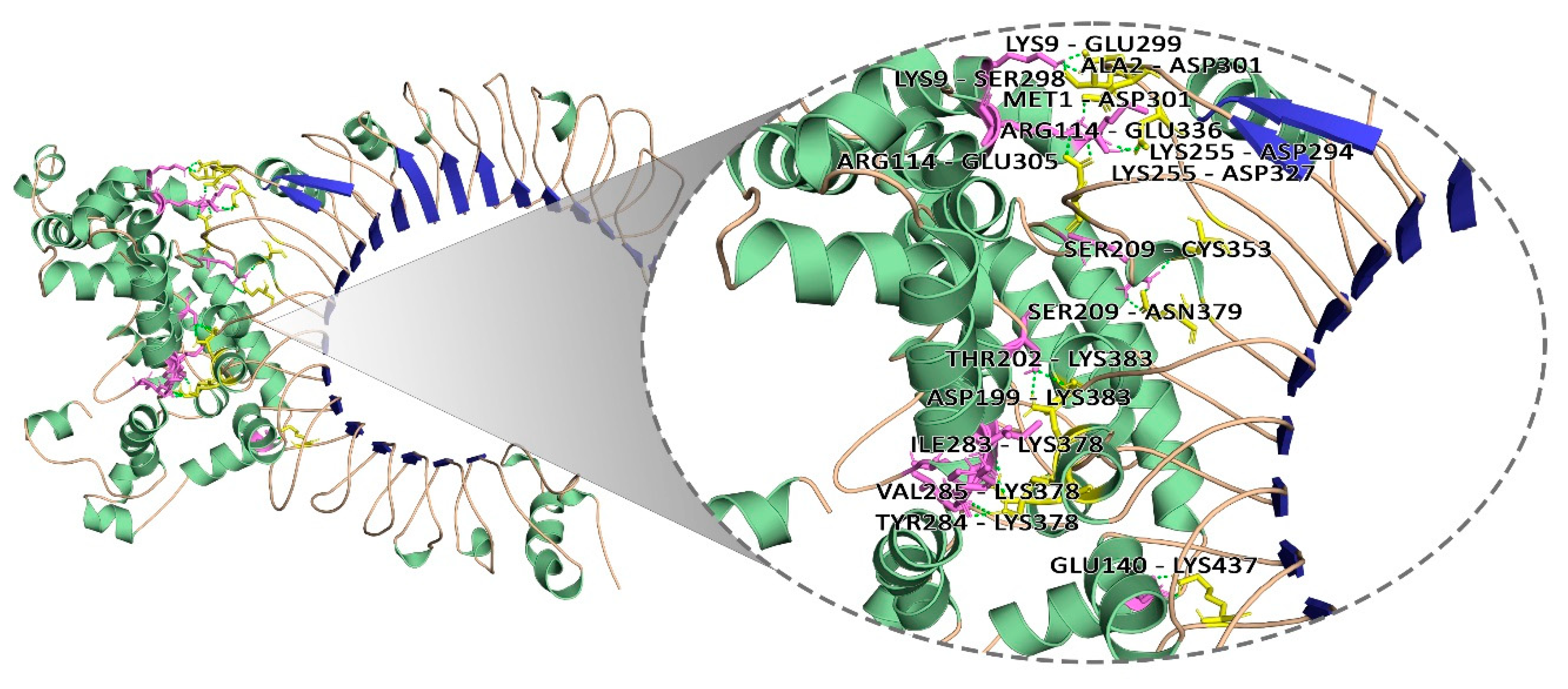
2.13. Molecular Docking Analysis
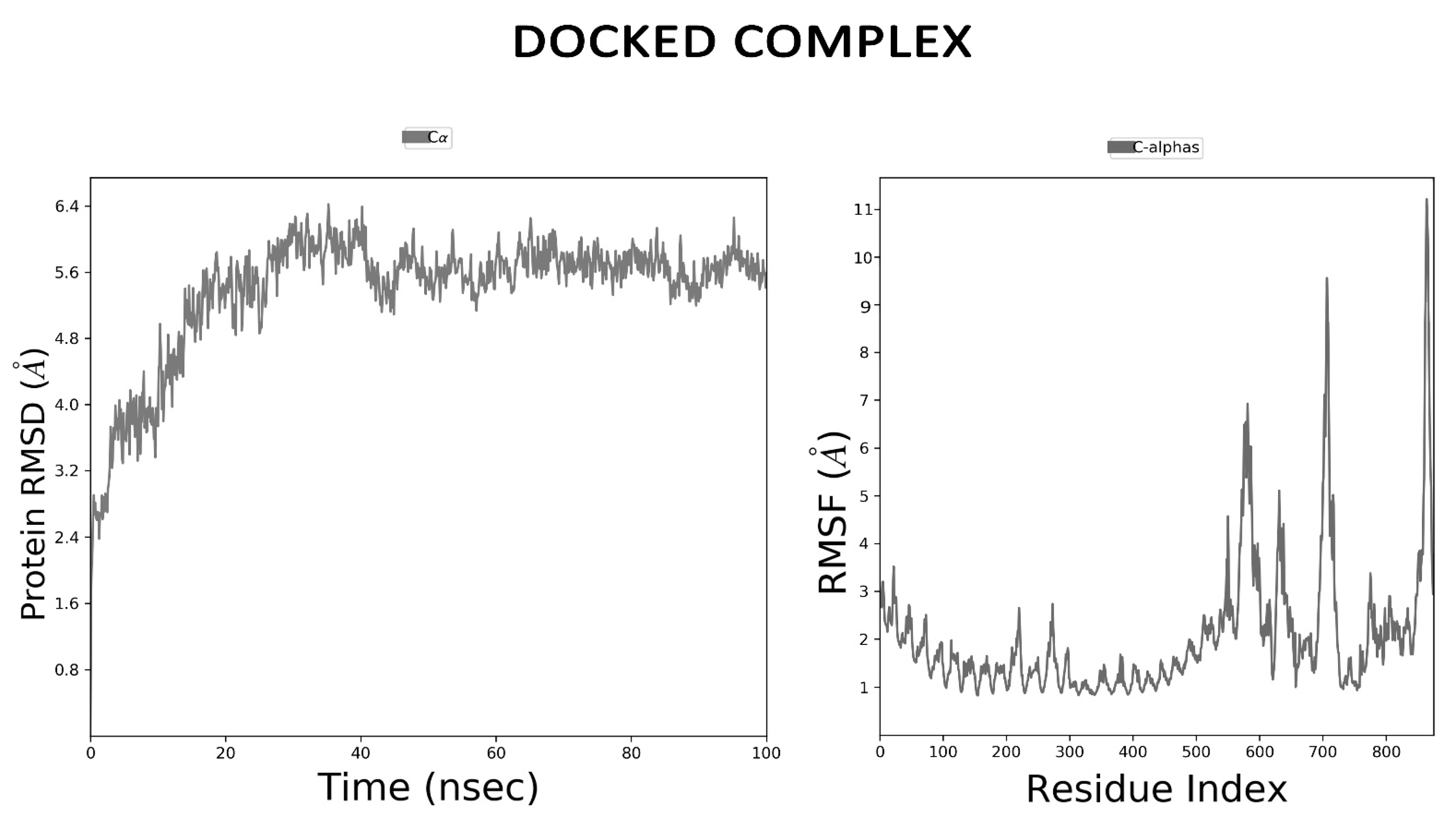
2.14. Dynamics Simulations for Vaccine Stability
2.15. Immune Simulation of the Vaccine Construct
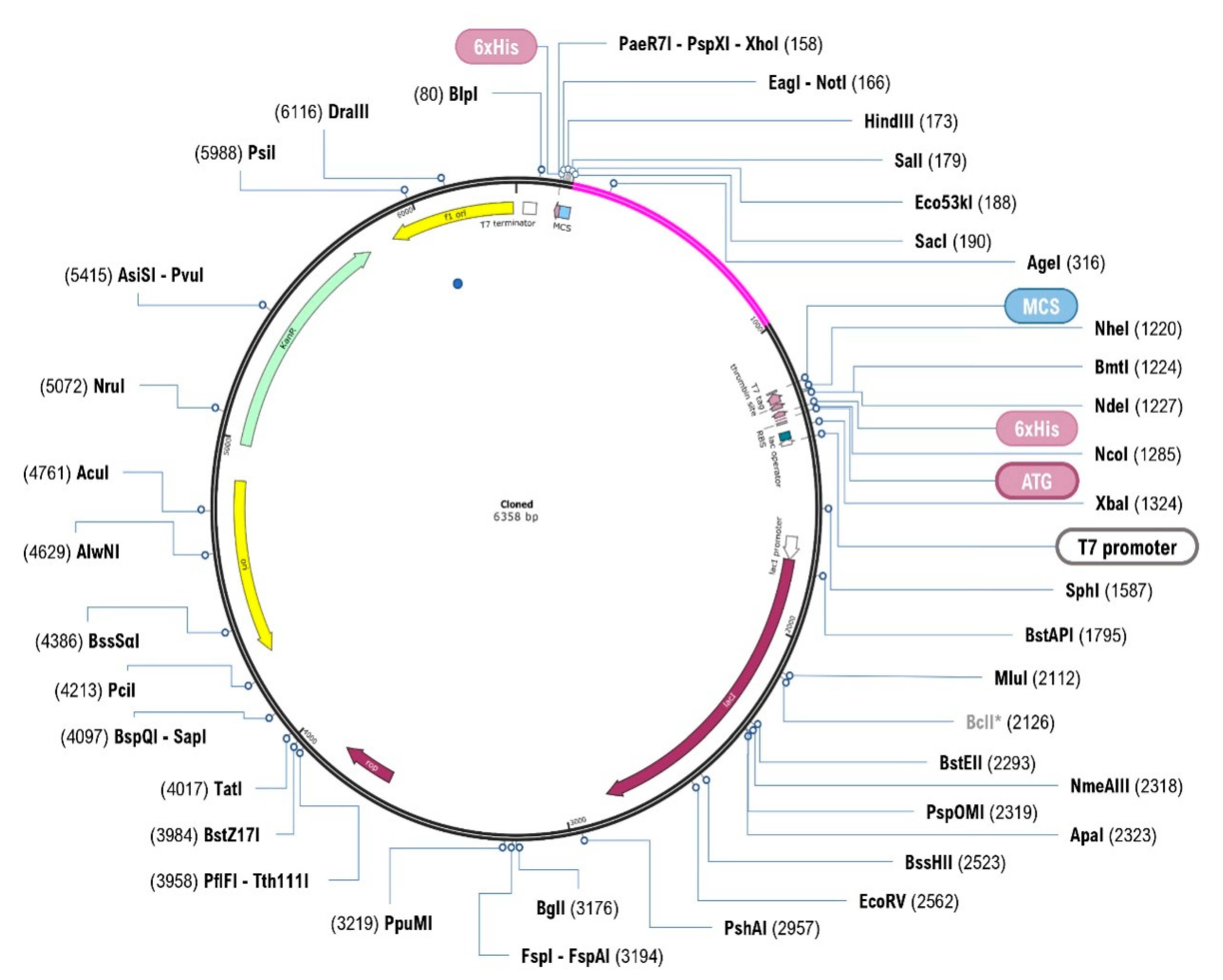
2.16. In Silico Cloning and Optimisation
3. Result
3.1. Epitopic Region Prediction of B-Cell
3.2. Prediction of Cytotoxic T-Lymphocyte (CTL) and Helper T-Lymphocyte (HTL) Epitopes
3.3. Evaluation of Antigenicity, Allergenicity, Toxicity, and Immunogenicity
3.4. Vaccine Construction
3.5. Estimation of Population Coverage
3.6. Analysis of Solubility and Physicochemical Properties and Secondary Structure
3.7. Vaccine’s Antigenicity and Allergenicity Profiling
3.8. Prediction of Interferon-Gamma Inducing Epitopes
3.9. Three-Dimensional Modelling and Validation
3.10. Disulphide Bridging for Vaccine Protein Stability
3.11. Molecular Docking Analysis
3.12. MD Simulation
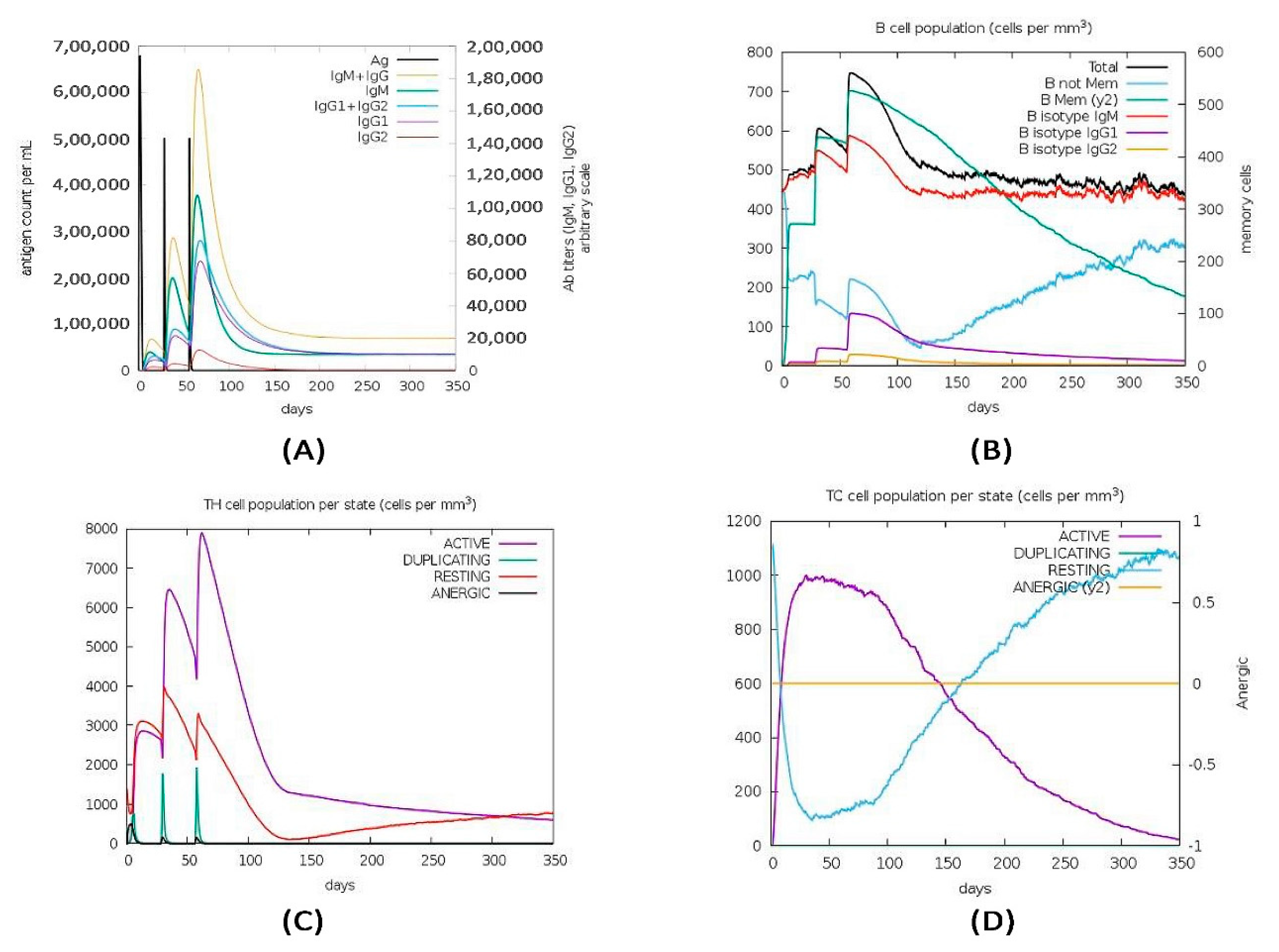
3.13. Immune Simulation of the Vaccine Construct
3.14. In Silico Cloning and Optimisation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lehman, S.M.; Mearns, G.; Rankin, D.; Cole, R.A.; Smrekar, F.; Branston, S.D.; Morales, S. Design and preclinical development of a phage product for the treatment of antibiotic-resistant Staphylococcus aureus infections. Viruses 2019, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.J.; Höök, M. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 1998, 6, 484–488. [Google Scholar] [CrossRef]
- Wertheim, H.F.; Melles, D.C.; Vos, M.C.; van Leeuwen, W.; van Belkum, A.; Verbrugh, H.A.; Nouwen, J.L. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis. 2005, 5, 751–762. [Google Scholar] [CrossRef]
- Coates, R.; Moran, J.; Horsburgh, M.J. Staphylococci: Colonizers and pathogens of human skin. Future Microbiol. 2014, 9, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Parlet, C.P.; Brown, M.M.; Horswill, A.R. Commensal staphylococci influence Staphylococcus aureus skin colonization and disease. Trends Microbiol. 2019, 27, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Yang, S.; Rao, X. Vancomycin resistant Staphylococcus aureus infections: A review of case updating and clinical features. J. Adv. Res. 2020, 21, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.O.; Lyon, B.R. Genetics of antimicrobial resistance in Staphylococcus aureus. Future Microbiol. 2009, 4, 565–582. [Google Scholar] [CrossRef]
- Zorzet, A. Overcoming scientific and structural bottlenecks in antibacterial discovery and development. Upsala J. Med. Sci. 2014, 119, 170–175. [Google Scholar] [CrossRef] [Green Version]
- Parker, D. A live vaccine to Staphylococcus aureus infection. Virulence 2018, 9, 700–702. [Google Scholar] [CrossRef] [Green Version]
- Kuklin, N.A.; Clark, D.J.; Secore, S.; Cook, J.; Cope, L.D.; McNeely, T.; Noble, L.; Brown, M.J.; Zorman, J.K.; Wang, X.M.; et al. A novel Staphylococcus aureus vaccine: Iron surface determinant B induces rapid antibody responses in rhesus macaques and specific increased survival in a murine S. aureus sepsis model. Infect. Immun. 2006, 74, 2215–2223. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, D. Veronate (Inhibitex). Curr. Opin. Investig. Drugs 2006, 7, 172–179. [Google Scholar]
- Wardenburg, J.B.; Schneewind, O. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 2008, 205, 287–294. [Google Scholar] [CrossRef]
- Brown, E.L.; Dumitrescu, O.; Thomas, D.; Badiou, C.; Koers, E.; Choudhury, P.; Vazquez, V.; Etienne, J.; Lina, G.; Vandenesch, F.; et al. The Panton-Valentine leukocidin vaccine protects mice against lung and skin infections caused by Staphylococcus aureus USA300. Clin. Microbiol. Infect. 2009, 15, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Stranger-Jones, Y.K.; Bae, T.; Schneewind, O. Vaccine assembly from surface proteins of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2006, 103, 16942–16947. [Google Scholar] [CrossRef] [Green Version]
- Daum, R.S.; Spellberg, B. Progress toward a Staphylococcus aureus vaccine. Clin. Infect. Dis. 2012, 54, 560–567. [Google Scholar] [CrossRef] [Green Version]
- Pier, G.B. Will there ever be a universal Staphylococcus aureus vaccine? Hum. Vaccines Immunother. 2013, 9, 1865–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, B.M.; Nielsen, T.B.; Cheng, B.; Pantapalangkoor, P.; Yan, J.; Boyle-Vavra, S.; Daum, R. Vaccines targeting Staphylococcus aureus skin and bloodstream infections require different composition. PLoS ONE 2019, 14, e0217439. [Google Scholar] [CrossRef]
- Cooper, M.D. The early history of B cells. Nat. Rev. Immunol. 2015, 15, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Lu, Y.; Chen, Y.H. Epitope-vaccine as a new strategy against HIV-1 mutation. Immunol. Lett. 2001, 77, 3–6. [Google Scholar] [CrossRef]
- Kuhns, J.J.; Batalia, M.A.; Yan, S.; Collins, E.J. Poor binding of a HER-2/neu epitope (GP2) to HLA-A2. 1 is due to a lack of interactions with the center of the peptide. J. Biol. Chem. 1999, 274, 36422–36427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakib, M.S.; Islam, M.; Hasan, A.K.M.; Nabi, A.H.M. Prediction of epitope-based peptides for the utility of vaccine development from fusion and glycoprotein of nipah virus using in silico approach. Adv. Bioinform. 2014, 2014, 402492. [Google Scholar]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Open Source Drug Discovery Consortium; Raghava, G.P. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadi, M.; Karkhah, A.; Nouri, H.R. Development of a multi-epitope peptide vaccine inducing robust T cell responses against brucellosis using immunoinformatics based approaches. Infect. Genet. Evol. 2017, 51, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, R.C.; Wood, D.H. Computational analysis of part-load flow control for crossflow hydro-turbines. Energy Sustain. Dev. 2018, 45, 38–45. [Google Scholar] [CrossRef]
- Bui, H.H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook. Springer Protocols Handbooks; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct. 2013, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Mol. Biol. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Pandey, R.K.; Ojha, R.; Aathmanathan, V.S.; Krishnan, M.; Prajapati, V.K. Immunoinformatics approaches to design a novel multi-epitope subunit vaccine against HIV infection. Vaccine 2018, 36, 2262–2272. [Google Scholar] [CrossRef]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255. [Google Scholar] [CrossRef] [PubMed]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending force field coverage for drug-like small molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]
- Gaafar, B.; Ali, S.A.; Abd-Elrahman, K.A.; Almofti, Y.A. Immunoinformatics approach for multiepitope vaccine prediction from H, M, F, and N proteins of Peste des Petitsruminants virus. J. Immunol. Res. 2019, 2019, 6124030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohanawa, M.; Zhao, S.; Ozaki, M.; Haga, S.; Nan, G.; Kuge, Y.; Tamaki, N. Contribution of toll-like receptor 2 to the innate response against Staphylococcus aureus infection in mice. PLoS ONE 2013, 8, e74287. [Google Scholar]
- Kim, N.H.; Sung, J.Y.; Choi, Y.J.; Choi, S.J.; Ahn, S.; Ji, E.; Kim, M.; Kim, C.J.; Song, K.H.; Choe, P.G.; et al. Toll-like receptor 2 downregulation and cytokine dysregulation predict mortality in patients with Staphylococcus aureus bacteremia. BMC Infect. Dis. 2020, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dziarski, R.; Gupta, D. Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: A reevaluation. Infect. Immun. 2005, 73, 5212–5216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delfani, S.; Mobarez, A.M.; Fooladi, A.A.I.; Amani, J.; Emaneini, M. Protection of mice against Staphylococcus aureus infection by a recombinant protein ClfA–IsdB–Hlg as a vaccine candidate. Med. Microbiol. Immunol. 2016, 205, 47–55. [Google Scholar] [CrossRef]
- Choi, S.J.; Kim, M.H.; Jeon, J.; Kim, O.Y.; Choi, Y.; Seo, J.; Kim, Y.K. Active immunization with extracellular vesicles derived from Staphylococcus aureus effectively protects against staphylococcal lung infections, mainly via Th1 cell-mediated immunity. PLoS ONE 2015, 10, e0136021. [Google Scholar]
- Lee, B.; Olaniyi, R.; Kwiecinski, J.M.; Wardenburg, J.B. Staphylococcus aureus toxin suppresses antigen-specific T cell responses. J. Clin. Investig. 2020, 130, 1122–1127. [Google Scholar] [CrossRef] [Green Version]
- Spellberg, B.; Ibrahim, A.S.; Yeaman, M.R.; Lin, L.; Fu, Y.; Avanesian, V.; Edwards, J.E., Jr. The antifungal vaccine derived from the recombinant N terminus of Als3p protects mice against the bacterium Staphylococcus aureus. Infect. Immun. 2008, 76, 4574–4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.; Licini, L.; Haelterman, E.; Moris, P.; Lestrate, P.; Damaso, S.; Boutriau, D. Safety and immunogenicity of an investigational 4-component Staphylococcus aureus vaccine with or without AS03B adjuvant: Results of a randomized phase I trial. Hum. Vaccines Immunother. 2015, 11, 620–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karauzum, H.; Venkatasubramaniam, A.; Adhikari, R.P.; Kort, T.; Holtsberg, F.W.; Mukherjee, I.; Aman, M.J. IBT-V02: A Multicomponent Toxoid Vaccine Protects Against Primary and Secondary Skin Infections Caused by Staphylococcus aureus. Front. Immunol. 2021, 12, 475. [Google Scholar] [CrossRef] [PubMed]
- Ikawaty, R.; Brouwer, E.C.; Van Duijkeren, E.; Mevius, D.; Verhoef, J.; Fluit, A.C. Virulence factors of genotyped bovine mastitis Staphylococcus aureus isolates in the Netherlands. Int. J. Dairy Sci. 2010, 5, 60–70. [Google Scholar] [CrossRef]
- Fluit, A.C. Livestock-associated Staphylococcus aureus. Clin. Microbiol. Infect. 2012, 18, 735–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahendran, A.; Vedaldi, A. Visualizing deep convolutional neural networks using natural pre-images. Int. J. Comput. Vis. 2016, 120, 233–255. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, S.R.; Dey, J.; Kushwaha, G.S.; Puhan, P.; Mohakud, N.K.; Panda, S.K.; Suar, M. Immunoinformatic approach employing modeling and simulation to design a novel vaccine construct targeting MDR efflux pumps to confer wide protection against typhoidal Salmonella serovars. J. Biomol. Struct. Dyn. 2021, 1–13. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, S.R.; Sahoo, S.; Dehury, B.; Raina, V.; Patro, S.; Misra, N.; Suar, M. Designing an efficient multi-epitope vaccine displaying interactions with diverse HLA molecules for an efficient humoral and cellular immune response to prevent COVID-19 infection. Expert Rev. Vaccines 2020, 19, 871–885. [Google Scholar] [CrossRef]
- Miller, L.S.; Fowler, V.C., Jr.; Shukla, S.K.; Rose, W.E.; Proctor, R.A. Development of a vaccine against Staphylococcus aureus invasive infections: Evidence based on human immunity, genetics and bacterial evasion mechanisms. FEMS Microbiol Rev. 2020, 44, 123–153. [Google Scholar] [CrossRef] [Green Version]
- Pian, Y.; Chen, S.; Hao, H.; Zheng, Y.; Zhu, L.; Xu, B.; Liu, K.; Li, M.; Jiang, H.; Jiang, Y. Phenol-soluble modulin α4 mediates Staphylococcus aureus-associated vascular leakage by stimulating heparin-binding protein release from neutrophils. Sci. Rep. 2016, 6, 1–12. [Google Scholar]
- Guruprasad, K.; Reddy, B.B.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. 1990, 4, 155–161. [Google Scholar] [CrossRef]
- Hajighahramani, N.; Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahmatabadi, S.S.; Ghasemi, Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect. Genet. Evol. 2017, 48, 83–94. [Google Scholar] [CrossRef]
- Sarkar, B.; Ullah, M.A.; Araf, Y.; Islam, N.N.; Zohora, U.S. Immunoinformatics-guided designing and in silico analysis of epitope-based polyvalent vaccines against multiple strains of human coronavirus (HCoV). Expert Rev. Vaccines 2021, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating anopheles salivary protein. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R. Bacterial expression systems for recombinant protein production: E. coli and beyond. Biotechnol. Adv. 2012, 30, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Dey, J.; Mahapatra, S.R.; Singh, P.; Patro, S.; Kushwaha, G.S.; Misra, N.; Suar, M. B and T cell epitope-based peptides predicted from clumping factor protein of Staphylococcus aureus as vaccine targets. Microb. Pathog. 2021, 160, 105171. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot_ID | B-Cell Epitope | Position | Score | Antigenicity Score | Toxicity | Hydrophobicity | Hydropathicity | Hydrophilicity | Charge | Mol wt. |
|---|---|---|---|---|---|---|---|---|---|---|
| O86488 | NEENKKVDAK | 173 | 0.58 | 2.4006 | Non-toxin | −0.57 | −2.32 | 1.64 | 0 | 1174.41 |
| O86489 | DTGGGDGTVK | 593 | 0.58 | 4.1781 | Non-toxin | −0.17 | −0.97 | 0.67 | −1 | 906.06 |
| Uniprot_ID | CTL Epitope | Alleles | Position | Score | Antigencity Score | Immunogenicity | Toxicity |
|---|---|---|---|---|---|---|---|
| O86488 | SGGAGQEVY | HLA-A*01:01 | 562 | 1.841 | 2.6468 | 0.08295 | Non-toxin |
| TLKNENGEV | HLA-A*02:01 | 822 | 4.4391 | 1.4625 | 0.102 | Non-toxin | |
| VTSGNDTEK | HLA-A*03:01 | 977 | 1.3119 | 2.012 | 0.08624 | Non-toxin | |
| DYRVEFSNL | HLA-A*24:02 | 629 | 0.9136 | 1.2237 | 0.12264 | Non-toxin | |
| NTRMRIAAV | HLA-A*26:01 | 229 | 1.307 | 0.7456 | 0.07166 | Non-toxin | |
| APKRLNTRM | HLA-B*07:02 | 224 | 0.0577 | 1.3117 | 0.02819 | Non-toxin | |
| DAKTESTTL | HLA-B*08:01 | 180 | 0.1135 | 1.9205 | 0.03373 | Non-toxin | |
| SKNDVEFNV | HLA-B*27:05 | 370 | 4.146 | 1.2062 | 0.24969 | Non-toxin | |
| SQITVDDKV | HLA-B*40:01 | 279 | 1.829 | 0.8785 | 0.03606 | Non-toxin | |
| SGDYFTIKY | HLA-B*58:01 | 289 | 3.063 | 1.6238 | 0.14034 | Non-toxin | |
| O86489 | KGDTMTINY | HLA-A*01:01 | 323 | 0.311 | 1.8025 | 0.02034 | Non-toxin |
| KTATEDTSV | HLA-A*02:01 | 142 | 3.039 | 1.4951 | 0.10624 | Non-toxin | |
| NTNNDVTTK | HLA-A*03:01 | 160 | 1.096 | 2.0046 | 0.10729 | Non-toxin | |
| KYSLGDYVW | HLA-A*24:02 | 830 | 0.105 | 0.4525 | 0.01002 | Non-toxin | |
| TSQNVTVDY | HLA-A*26:01 | 415 | 1.098 | 1.3552 | 0.08043 | Non-toxin | |
| TPAGYTPTV | HLA-B*07:02 | 786 | 0.194 | 0.9088 | 0.09306 | Non-toxin | |
| EQQIYVNPL | HLA-B*08:01 | 449 | 1.332 | 0.7245 | 0.11964 | Non-toxin | |
| KQTIEQQIY | HLA-B*27:05 | 445 | 4.415 | 0.8527 | 0.11498 | Non-toxin | |
| DKQTIEQQI | HLA-B*39:01 | 444 | 1.591 | 1.2024 | 0.05987 | Non-toxin | |
| IEQQIYVNP | HLA-B*40:01 | 448 | 2.472 | 0.7436 | 0.00302 | Non-toxin |
| Uniprot_ID | MHC II Epitope | Alleles | Position | IC50 Value | Percentile Rank | Antigenicity Score | Toxicity | Hydrophobicity | Hydropathicity | Hydrophilicity | Charge | Mol. wt. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O86488 | KRLNTRMRIAAVQPS | HLA-DQA1*01:02, HLA-DQB1*06:02, HLA-DPB1*01:01, HLA-DRB1*01:01, HLA-DRB1*09:01,HLA-DRB3*02:02, HLA-DRB1*13:02, HLA-DRB1*11:01, HLA-DRB1*04:01, HLA-DRB1*12:01, HLA-DPA1*03:01, HLA-DPB1*04:02, HLA-DRB1*04:05, HLA-DRB1*15:01, HLA-DQA1*01:01, HLA-DQB1*05:01, HLA-DRB1*08:02, HLA-DPA1*02:01, HLA-DPB1*14:01, HLA-DPA1*01:03, HLA-DPB1*04:01, HLA-DQA1*05:01, HLA-DQB1*03:01, HLA-DQA1*04:01, HLA-DQB1*04:02, HLA-DPA1*02:01, HLA-DPA1*02:01, HLA-DPB1*05:01, HLA-DPA1*01:03, HLA-DPB1*02:01, HLA-DQA1*05:01, HLA-DQB1*02:01, HLA-DQA1*03:01, HLA-DQB1*03:02, HLA-DRB3*01:01, HLA-DRB5*01:01, HLA-DRB1*07:01, HLA-DRB4*01:01, HLA-DRB1*03:01 | 226–240 | 30 | 0.11 | 0.9801 | Non-toxin | −0.38 | −0.63 | 0.33 | 4 | 1741.3 |
| O86489 | DTEFTIDNKVKKGDT | HLA-DRB1*03:01, HLA-DRB3*01:01, HLA-DPA1*03:01, HLA-DPB1*04:02, HLA-DPA1*01:03, HLA-DPB1*02:01,HLA-DRB1*01:01, HLA-DRB1*09:01,HLA-DRB3*02:02, HLA-DRB1*13:02, HLA-DRB1*11:01, HLA-DRB1*04:01, HLA-DRB1*12:01,HLA-DRB1*04:05, HLA-DRB1*15:01, HLA-DQA1*01:01, HLA-DQB1*05:01, HLA-DRB1*08:02, HLA-DPA1*02:01, HLA-DPB1*14:01, HLA-DPA1*01:03, HLA-DPB1*04:01, HLA-DQA1*05:01, HLA-DQB1*03:01, HLA-DQA1*04:01, HLA-DQB1*04:02, HLA-DPA1*02:01, HLA-DPB1*01:01, HLA-DPA1*02:01, HLA-DPB1*05:01,HLA-DQA1*05:01, HLA-DQB1*02:01, HLA-DQA1*03:01, HLA-DQB1*03:02, HLA-DQA1*01:02, HLA-DQB1*06:02, HLA-DRB5*01:01, HLA-DRB1*07:01, HLA-DRB4*01:01, | 312–326 | 88 | 0.88 | 1.2192 | Non-toxin | −0.35 | −1.35 | 0.95 | −1 | 1711.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, R.; Sahoo, P.; Mahapatra, S.R.; Dey, J.; Ghosh, M.; Kushwaha, G.S.; Misra, N.; Suar, M.; Raina, V.; Son, Y.-O. Development of a Conserved Chimeric Vaccine for Induction of Strong Immune Response against Staphylococcus aureus Using Immunoinformatics Approaches. Vaccines 2021, 9, 1038. https://doi.org/10.3390/vaccines9091038
Chatterjee R, Sahoo P, Mahapatra SR, Dey J, Ghosh M, Kushwaha GS, Misra N, Suar M, Raina V, Son Y-O. Development of a Conserved Chimeric Vaccine for Induction of Strong Immune Response against Staphylococcus aureus Using Immunoinformatics Approaches. Vaccines. 2021; 9(9):1038. https://doi.org/10.3390/vaccines9091038
Chicago/Turabian StyleChatterjee, Rahul, Panchanan Sahoo, Soumya Ranjan Mahapatra, Jyotirmayee Dey, Mrinmoy Ghosh, Gajraj Singh Kushwaha, Namrata Misra, Mrutyunjay Suar, Vishakha Raina, and Young-Ok Son. 2021. "Development of a Conserved Chimeric Vaccine for Induction of Strong Immune Response against Staphylococcus aureus Using Immunoinformatics Approaches" Vaccines 9, no. 9: 1038. https://doi.org/10.3390/vaccines9091038