Immune Modulation and Immune-Mediated Pathogenesis of Emerging Tickborne Banyangviruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Tick-Borne Banyangviruses
2.1. Severe Fever with Thrombocytopenia Syndrome Virus (SFTSV)
2.2. Heartland Virus (HRTV)
2.3. Guertu Virus (GTV)
3. Basic Virology of Banyangviruses
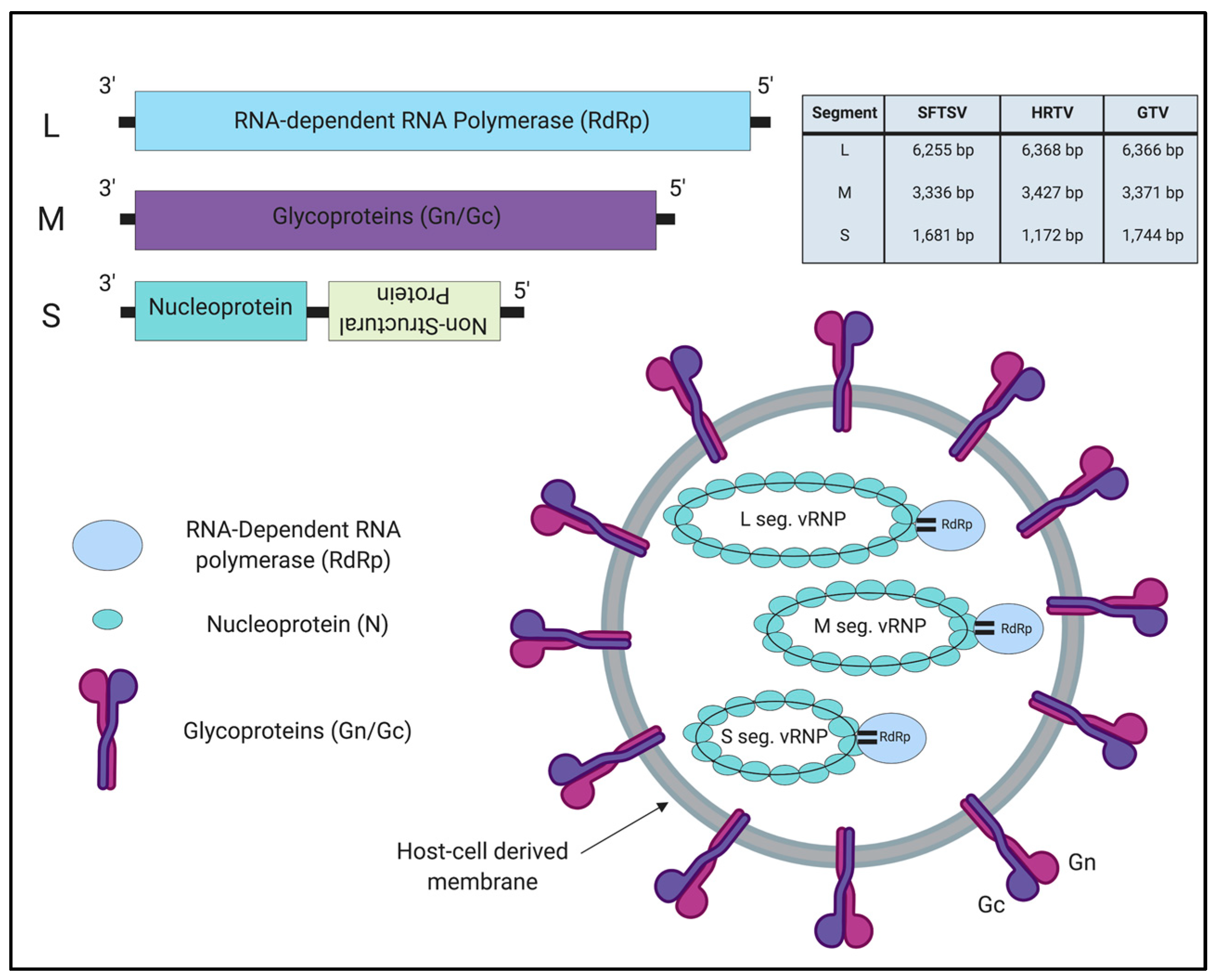
3.1. Genome Organization
3.2. Target Tissues/Cells and Cellular Receptors
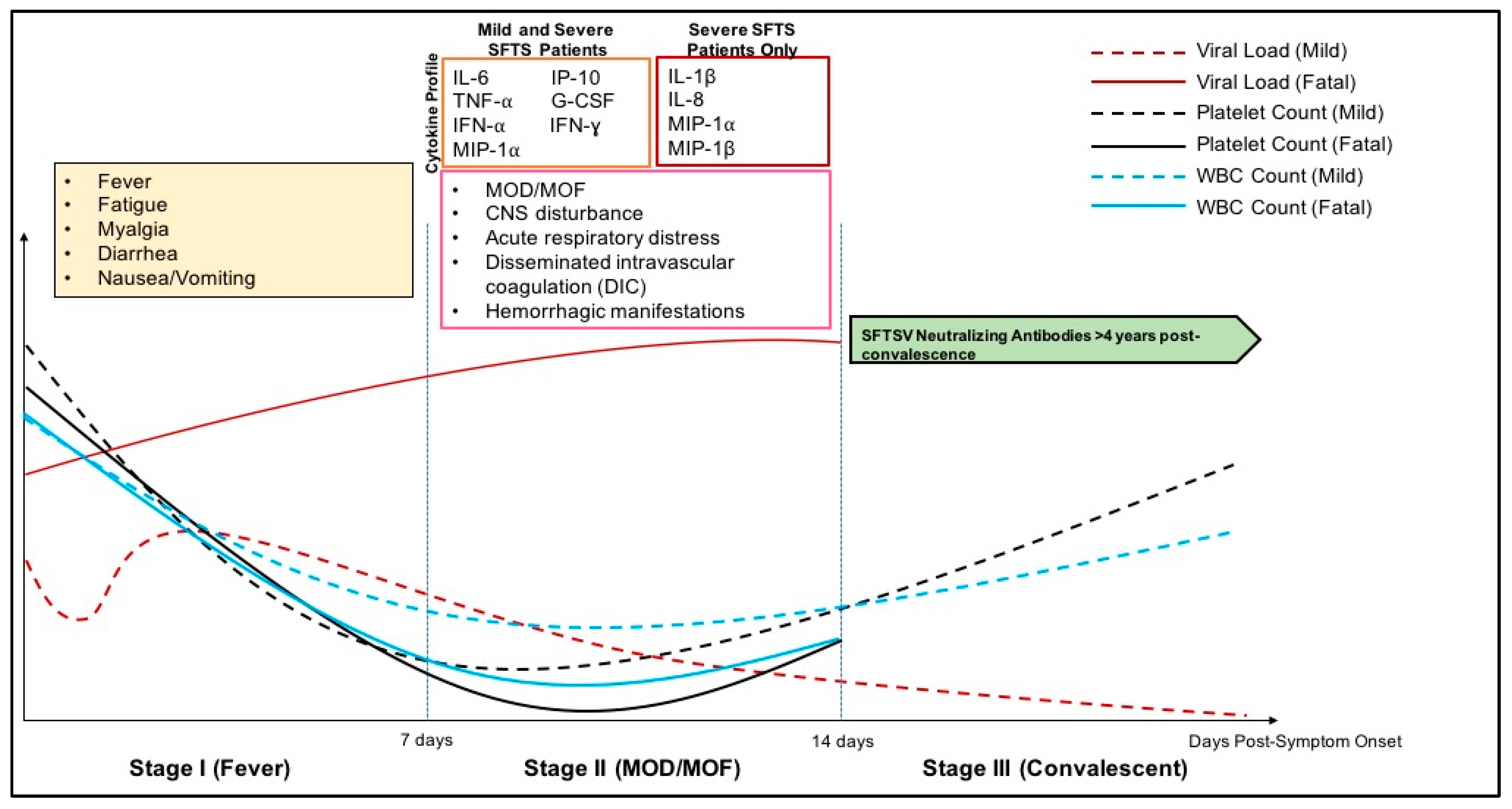
3.3. Clinical Disease Course
3.4. Pathology
3.5. Animal Models of Tickborne Banyangviruses
4. Host Immune Response to Banyangviruses
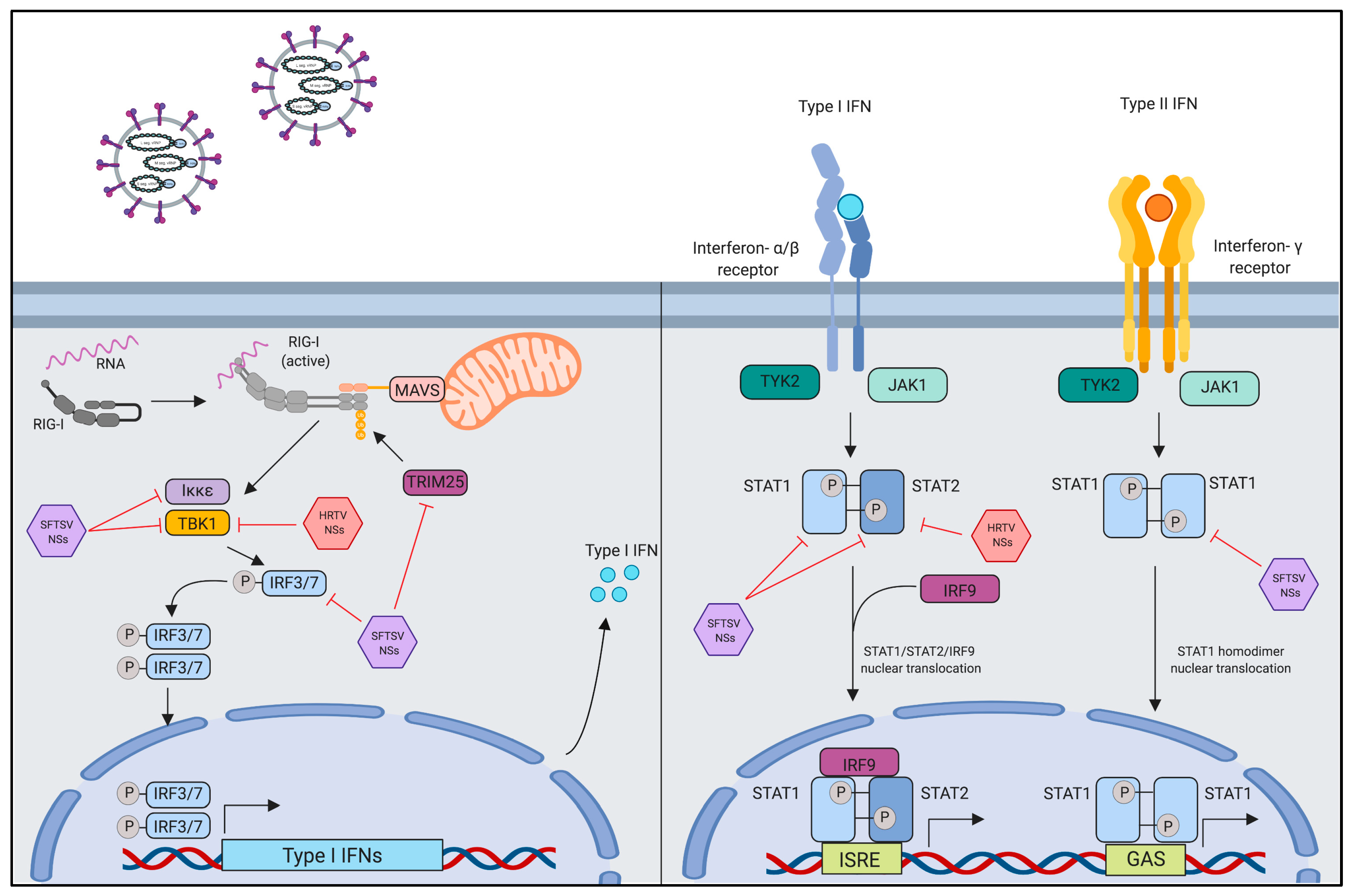
4.1. Innate Immune Evasion by Interferon-Antagonistic Function of Banyangvirus NSs
4.2. Pro-Inflammatory Activation and Suppression by Banyangvirus NSs
4.3. Humoral Immune Response to Banyangviruses
4.4. Cell-Mediated Immune Response to Banyangviruses
4.5. Establishment of Long-Term Immunity
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; Junglen, S.; et al. Changes to virus taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2019). Arch. Virol. 2019, 164, 2417–2429. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-J.; Liang, M.-F.; Zhang, S.-Y.; Liu, Y.; Li, J.-D.; Sun, Y.-L.; Zhang, L.; Zhang, Q.-F.; Popov, V.L.; Li, C.; et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 2011, 364, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-W.; Song, B.G.; Shin, E.-H.; Yun, S.-M.; Han, M.-G.; Park, M.Y.; Park, C.; Ryou, J. Prevalence of severe fever with thrombocytopenia syndrome virus in Haemaphysalis longicornis ticks in South Korea. Ticks Tick-Borne Dis. 2014, 5, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.-M.; Lee, W.-G.; Ryou, J.; Yang, S.-C.; Park, S.-W.; Roh, J.Y.; Lee, Y.-J.; Park, C.; Han, M.G. Severe fever with thrombocytopenia syndrome virus in ticks collected from humans, South Korea, 2013. Emerg. Infect. Dis. 2014, 20, 1358–1361. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Yun, Y.; Bae, S.G.; Park, D.; Kim, S.; Lee, J.M.; Cho, N.-H.; Kim, Y.S.; Lee, K.H. Severe Fever with Thrombocytopenia Syndrome Virus Infection, South Korea, 2010. Emerg. Infect. Dis. 2018, 24, 2103–2105. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Maeda, K.; Suzuki, T.; Ishido, A.; Shigeoka, T.; Tominaga, T.; Kamei, T.; Honda, M.; Ninomiya, D.; Sakai, T.; et al. The first identification and retrospective study of Severe Fever with Thrombocytopenia Syndrome in Japan. J. Infect. Dis. 2014, 209, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Tran, X.C.; Yun, Y.; Van An, L.; Kim, S.-H.; Thao, N.T.P.; Man, P.K.C.; Yoo, J.R.; Heo, S.T.; Cho, N.-H.; Lee, K.H. Endemic Severe Fever with Thrombocytopenia Syndrome, Vietnam. Emerg. Infect. Dis. 2019, 25, 1029–1031. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Lu, L.; Wu, H.; Yang, J.; Ren, J.; Liu, Q. The changing epidemiological characteristics of severe fever with thrombocytopenia syndrome in China, 2011-2016. Sci. Rep. 2017, 7, 9236. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.J.; Park, S.-W.; Bae, I.-G.; Kim, S.-H.; Ryu, S.Y.; Kim, H.A.; Jang, H.-C.; Hur, J.; Jun, J.-B.; Jung, Y.; et al. Severe Fever with Thrombocytopenia Syndrome in South Korea, 2013-2015. PLoS Negl. Trop. Dis. 2016, 10, e0005264. [Google Scholar] [CrossRef]
- National Institute of Infectious Diseases and Tuberculosis and Infectious Diseases, Control Division Severe Fever with Thrombocytopenia Syndrome (SFTS) in Japan, as of June 2019; Infectious Agents Surveillance Report; National Institute of Infectious Diseases: Tokyo, Japan, 2019; pp. 111–112.
- Luo, L.-M.; Zhao, L.; Wen, H.-L.; Zhang, Z.-T.; Liu, J.-W.; Fang, L.-Z.; Xue, Z.-F.; Ma, D.-Q.; Zhang, X.-S.; Ding, S.-J.; et al. Haemaphysalis longicornis Ticks as Reservoir and Vector of Severe Fever with Thrombocytopenia Syndrome Virus in China. Emerg. Infect. Dis. 2015, 21, 1770–1776. [Google Scholar] [CrossRef]
- Wang, S.; Li, J.; Niu, G.; Wang, X.; Ding, S.; Jiang, X.; Li, C.; Zhang, Q.; Liang, M.; Bi, Z.; et al. SFTS Virus in Ticks in an Endemic Area of China. Am. J. Trop. Med. Hyg. 2015, 92, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.-M.; Song, B.G.; Choi, W.; Roh, J.Y.; Lee, Y.-J.; Park, W.I.; Han, M.G.; Ju, Y.R.; Lee, W.-J. First Isolation of Severe Fever with Thrombocytopenia Syndrome Virus from Haemaphysalis longicornis Ticks Collected in Severe Fever with Thrombocytopenia Syndrome Outbreak Areas in the Republic of Korea. Vector Borne Zoonotic Dis. 2016, 16, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Li, J.; Liang, M.; Jiang, X.; Jiang, M.; Yin, H.; Wang, Z.; Li, C.; Zhang, Q.; Jin, C.; et al. Severe Fever with Thrombocytopenia Syndrome Virus among Domesticated Animals, China. Emerg. Infect. Dis. 2013, 19, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, H.J.; Byun, J.W.; Lee, M.J.; Kim, N.H.; Kim, D.H.; Kang, H.E.; Nam, H.M. Molecular detection and phylogenetic analysis of severe fever with thrombocytopenia syndrome virus in shelter dogs and cats in the Republic of Korea. Ticks Tick-Borne Dis. 2017, 8, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-G.; Cho, Y.-K.; Jo, Y.-S.; Chae, J.-B.; Oh, S.-S.; Kim, K.-H.; Ko, M.-K.; Yi, J.; Choi, K.-S.; Yu, D.-H.; et al. Prevalence of severe fever with thrombocytopenia syndrome virus in black goats (Capra hircus coreanae) in the Republic of Korea. Ticks Tick-Borne Dis. 2018, 9, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Nonoue, N.; Noda, A.; Kasajima, N.; Noguchi, K.; Takano, A.; Shimoda, H.; Orba, Y.; Muramatsu, M.; Sakoda, Y.; et al. Fatal Tickborne Phlebovirus Infection in Captive Cheetahs, Japan. Emerg. Infect. Dis. 2018, 24, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Guo, X.; Qi, X.; Hu, J.; Zhou, M.; Varma, J.K.; Cui, L.; Yang, H.; Jiao, Y.; Klena, J.D.; et al. A family cluster of infections by a newly recognized bunyavirus in eastern China, 2007: Further evidence of person-to-person transmission. Clin. Infect. Dis. 2011, 53, 1208–1214. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Q.; Hu, W.; Wu, J.; Wang, Y.; Mei, L.; Walker, D.H.; Ren, J.; Wang, Y.; Yu, X.-J. Person-to-person transmission of severe fever with thrombocytopenia syndrome virus. Vector Borne Zoonotic Dis. 2012, 12, 156–160. [Google Scholar] [CrossRef]
- Gai, Z.; Liang, M.; Zhang, Y.; Zhang, S.; Jin, C.; Wang, S.-W.; Sun, L.; Zhou, N.; Zhang, Q.; Sun, Y.; et al. Person-to-Person Transmission of Severe Fever With Thrombocytopenia Syndrome Bunyavirus Through Blood Contact. Clin. Infect. Dis. 2012, 54, 249–252. [Google Scholar] [CrossRef]
- Kim, W.Y.; Choi, W.; Park, S.-W.; Wang, E.B.; Lee, W.-J.; Jee, Y.; Lim, K.S.; Lee, H.-J.; Kim, S.-M.; Lee, S.-O.; et al. Nosocomial Transmission of Severe Fever With Thrombocytopenia Syndrome in Korea. Clin. Infect. Dis. 2015, 60, 1681–1683. [Google Scholar] [CrossRef]
- Hwang, J.; Kang, J.-G.; Oh, S.-S.; Chae, J.-B.; Cho, Y.-K.; Cho, Y.-S.; Lee, H.; Chae, J.-S. Molecular detection of severe fever with thrombocytopenia syndrome virus (SFTSV) in feral cats from Seoul, Korea. Ticks Tick-Borne Dis. 2017, 8, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.Y.; Choi, W.; Kim, J.; Wang, E.; Park, S.-W.; Lee, W.-J.; Choi, J.Y.; Kim, H.Y.; Uh, Y.; Kim, Y.K. Nosocomial person-to-person transmission of severe fever with thrombocytopenia syndrome. Clin. Infect. Dis. 2019, 25, 633.e1–633.e4. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, P.; Li, K.-F.; Wang, H.-L.; Dai, Y.-X.; Cheng, X.; Yan, J.-B. Animals as amplification hosts in the spread of severe fever with thrombocytopenia syndrome virus: A systematic review and meta-analysis. Int. J. Infect. Dis. 2019, 79, 77–84. [Google Scholar] [CrossRef] [PubMed]
- McMullan, L.K.; Folk, S.M.; Kelly, A.J.; MacNeil, A.; Goldsmith, C.S.; Metcalfe, M.G.; Batten, B.C.; Albariño, C.G.; Zaki, S.R.; Rollin, P.E.; et al. A New Phlebovirus Associated with Severe Febrile Illness in Missouri. N. Engl. J. Med. 2012, 367, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Savage, H.M.; Godsey, M.S.; Lambert, A.; Panella, N.A.; Burkhalter, K.L.; Harmon, J.R.; Lash, R.R.; Ashley, D.C.; Nicholson, W.L. First Detection of Heartland Virus (Bunyaviridae: Phlebovirus) from Field Collected Arthropods. Am. J. Trop. Med. Hyg. 2013, 89, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Godsey, M.S.; Savage, H.M.; Burkhalter, K.L.; Bosco-Lauth, A.M.; Delorey, M.J. Transmission of Heartland Virus (Bunyaviridae: Phlebovirus) by Experimentally Infected Amblyomma americanum (Acari: Ixodidae). J. Med. Entomol. 2016, 53, 1226–1233. [Google Scholar] [CrossRef]
- Riemersma, K.K.; Komar, N. Heartland Virus Neutralizing Antibodies in Vertebrate Wildlife, United States, 2009-2014. Emerg. Infect. Dis. 2015, 21, 1830–1833. [Google Scholar] [CrossRef]
- Statistics & Maps- Heartland virus- CDC. Available online: https://www.cdc.gov/heartland-virus/statistics/index.html (accessed on 19 August 2019).
- Lindsey, N.P.; Menitove, J.E.; Biggerstaff, B.J.; Turabelidze, G.; Parton, P.; Peck, K.; Basile, A.J.; Kosoy, O.I.; Fischer, M.; Staples, J.E. Seroprevalence of Heartland Virus Antibodies in Blood Donors, Northwestern Missouri, USA. Emerg. Infect. Dis. 2019, 25, 358–360. [Google Scholar] [CrossRef]
- Shen, S.; Duan, X.; Wang, B.; Zhu, L.; Zhang, Y.; Zhang, J.; Wang, J.; Luo, T.; Kou, C.; Liu, D.; et al. A novel tick-borne phlebovirus, closely related to severe fever with thrombocytopenia syndrome virus and Heartland virus, is a potential pathogen. Emerg. Microbes Infect. 2018, 7, 95. [Google Scholar] [CrossRef]
- Zhu, L.; Yin, F.; Moming, A.; Zhang, J.; Wang, B.; Gao, L.; Ruan, J.; Wu, Q.; Wu, N.; Wang, H.; et al. First case of laboratory-confirmed severe fever with thrombocytopenia syndrome disease revealed the risk of SFTSV infection in Xinjiang, China. Emerg. Microbes Infect. 2019, 8, 1122–1125. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org//taxonomy/p/taxonomy-history?taxnode_id=201856224 (accessed on 18 August 2019).
- Elliott, R.M.; Brennan, B. Emerging phleboviruses. Curr. Opin. Virol. 2014, 5, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, M.; Plegge, T.; Pöhlmann, S. The Role of Phlebovirus Glycoproteins in Viral Entry, Assembly and Release. Viruses 2016, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Sun, Y.; Wang, Y.; Liu, M.; Liu, C.; Wang, W.; Liu, X.; Li, L.; Deng, F.; Wang, H.; et al. The nucleoprotein of severe fever with thrombocytopenia syndrome virus processes a stable hexameric ring to facilitate RNA encapsidation. Protein Cell 2013, 4, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M.; Weber, F. Bunyaviruses and the Type I Interferon System. Viruses 2009, 1, 1003–1021. [Google Scholar] [CrossRef] [PubMed]
- Wuerth, J.D.; Weber, F. Phleboviruses and the Type I Interferon Response. Viruses 2016, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Hiraki, T.; Yoshimitsu, M.; Suzuki, T.; Goto, Y.; Higashi, M.; Yokoyama, S.; Tabuchi, T.; Futatsuki, T.; Nakamura, K.; Hasegawa, H.; et al. Two autopsy cases of severe fever with thrombocytopenia syndrome (SFTS) in Japan: A pathognomonic histological feature and unique complication of SFTS. Pathol. Int. 2014, 64, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Muehlenbachs, A.; Fata, C.R.; Lambert, A.J.; Paddock, C.D.; Velez, J.O.; Blau, D.M.; Staples, J.E.; Karlekar, M.B.; Bhatnagar, J.; Nasci, R.S.; et al. Heartland virus-associated death in tennessee. Clin. Infect. Dis. 2014, 59, 845–850. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Wang, Q.; Yu, X.; Liu, M.; Xie, H.; Qian, L.; Ye, L.; Yang, Z.; Zhang, J.; et al. Multiple organ involvement in severe fever with thrombocytopenia syndrome: An immunohistochemical finding in a fatal case. J. Virol. 2018, 15, 97. [Google Scholar] [CrossRef]
- Saijo, M. Pathophysiology of severe fever with thrombocytopenia syndrome and development of specific antiviral therapy. J. Infect. Chemother. 2018, 24, 773–781. [Google Scholar] [CrossRef]
- Fill, M.M.A.; Compton, M.L.; McDonald, E.C.; Moncayo, A.C.; Dunn, J.R.; Schaffner, W.; Bhatnagar, J.; Zaki, S.R.; Jones, T.F.; Shieh, W.J. Novel clinical and pathologic findings in a heartland virus-associated death. Clin. Infect. Dis. 2017, 64, 510–512. [Google Scholar]
- Lozach, P.-Y.; Kühbacher, A.; Meier, R.; Mancini, R.; Bitto, D.; Bouloy, M.; Helenius, A. DC-SIGN as a receptor for phleboviruses. Cell Host Microbe 2011, 10, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Li, X.; Zhang, X.; Liu, W.; Kühl, A.; Kaup, F.; Soldan, S.S.; González-Scarano, F.; Weber, F.; He, Y.; et al. Severe fever with thrombocytopenia virus glycoproteins are targeted by neutralizing antibodies and can use DC-SIGN as a receptor for pH-dependent entry into human and animal cell lines. J. Virol. 2013, 87, 4384–4394. [Google Scholar] [CrossRef] [PubMed]
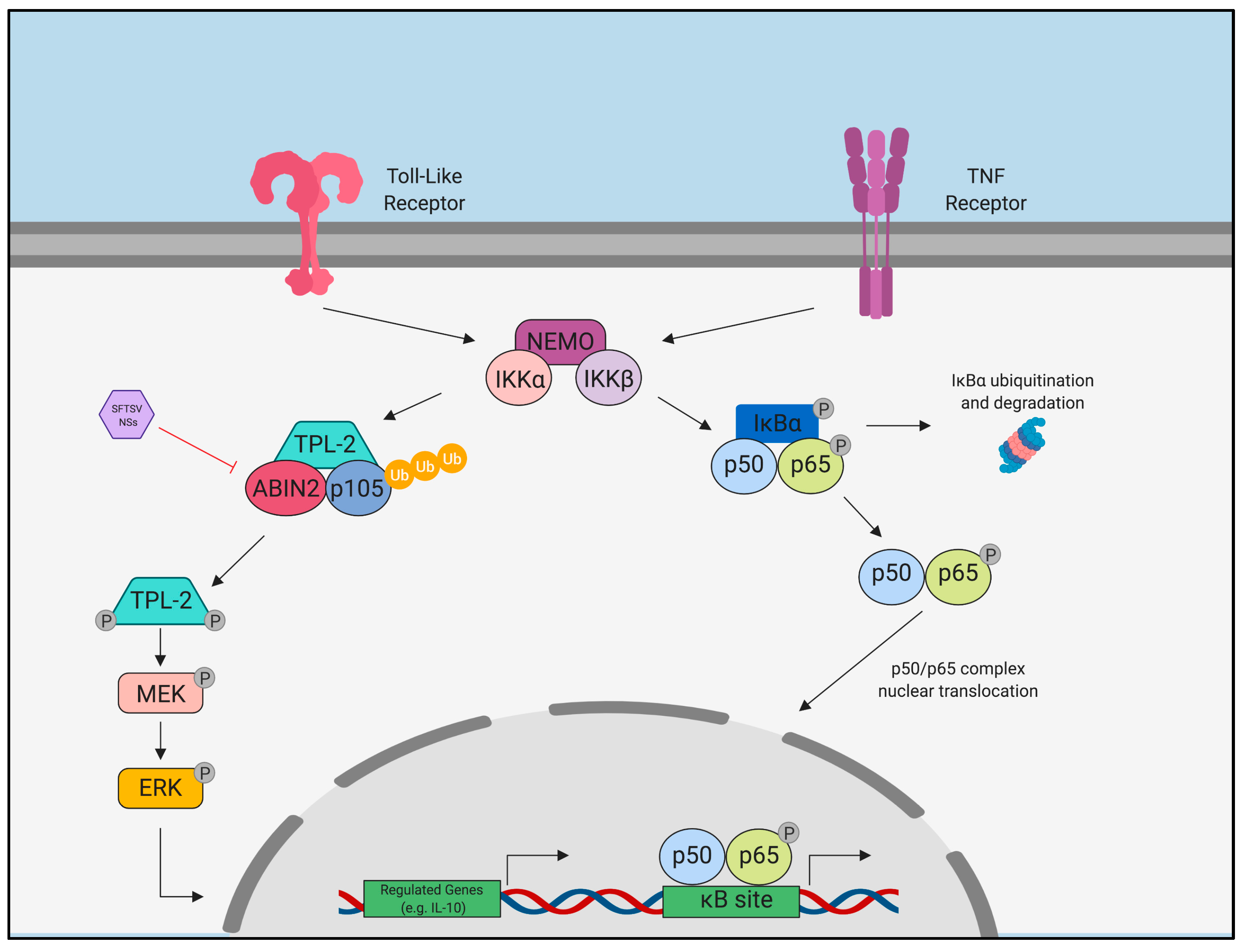
- Qu, B.; Qi, X.; Wu, X.; Liang, M.; Li, C.; Cardona, C.J.; Xu, W.; Tang, F.; Li, Z.; Wu, B.; et al. Suppression of the Interferon and NF-κB Responses by Severe Fever with Thrombocytopenia Syndrome Virus. J. Virol. 2012, 86, 8388–8401. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Park, S.-J.; Sun, Y.; Yoo, J.-S.; Pudupakam, R.S.; Foo, S.-S.; Shin, W.-J.; Chen, S.B.; Tsichlis, P.N.; Lee, W.-J.; et al. Severe fever with thrombocytopenia syndrome phlebovirus non-structural protein activates TPL2 signalling pathway for viral immunopathogenesis. Nat. Microbiol. 2019, 4, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Fu, Y.; Wang, H.; Guan, Y.; Zhu, W.; Guo, M.; Zheng, N.; Wu, Z. Severe Fever With Thrombocytopenia Syndrome Virus-Induced Macrophage Differentiation Is Regulated by miR-146. Front. Immunol. 2019, 10, 1095. [Google Scholar] [CrossRef]
- Tani, H.; Shimojima, M.; Fukushi, S.; Yoshikawa, T.; Fukuma, A.; Taniguchi, S.; Morikawa, S.; Saijo, M. Characterization of Glycoprotein-Mediated Entry of Severe Fever with Thrombocytopenia Syndrome Virus. J. Virol. 2016, 90, 5292–5301. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Qi, Y.; Liu, C.; Gao, W.; Chen, P.; Fu, L.; Peng, B.; Wang, H.; Jing, Z.; Zhong, G.; et al. Nonmuscle Myosin Heavy Chain IIA Is a Critical Factor Contributing to the Efficiency of Early Infection of Severe Fever with Thrombocytopenia Syndrome Virus. J. Virol. 2014, 88, 237–248. [Google Scholar] [CrossRef]
- Liu, S.; Chai, C.; Wang, C.; Amer, S.; Lv, H.; He, H.; Sun, J.; Lin, J. Systematic review of severe fever with thrombocytopenia syndrome:virology, epidemiology, and clinical characteristics. Rev. Med. Virol. 2014, 24, 90–102. [Google Scholar] [CrossRef]
- Li, D.X. Severe fever with thrombocytopenia syndrome: A newly discovered emerging infectious disease. Clin. Microbiol. Infect. 2015, 21, 614–620. [Google Scholar] [CrossRef]
- Robles, N.J.C.; Han, H.J.; Park, S.-J.; Choi, Y.K. Epidemiology of severe fever and thrombocytopenia syndrome virus infection and the need for therapeutics for the prevention. Clin. Exp. Vaccine Res. 2018, 7, 43–50. [Google Scholar] [CrossRef]
- Kwon, J.-S.; Kim, M.-C.; Kim, J.Y.; Jeon, N.-Y.; Ryu, B.-H.; Hong, J.; Kim, M.-J.; Chong, Y.P.; Lee, S.-O.; Choi, S.-H.; et al. Kinetics of viral load and cytokines in severe fever with thrombocytopenia syndrome. J. Clin. Virol. 2018, 101, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wu, H.; Gao, J.; Zhou, X.; Zhu, R.; Zhang, C.; Bai, H.; Abdullah, A.S.; Pan, H. Two confirmed cases of severe fever with thrombocytopenia syndrome with pneumonia: Implication for a family cluster in East China. BMC Infect. Dis. 2017, 17, 537. [Google Scholar] [CrossRef]
- Uehara, N.; Yano, T.; Ishihara, A.; Saijou, M.; Suzuki, T. Fatal Severe Fever with Thrombocytopenia Syndrome: An Autopsy Case Report. Intern. Med. Tokyo Jpn. 2016, 55, 831–838. [Google Scholar] [CrossRef]
- Nakano, A.; Ogawa, H.; Nakanishi, Y.; Fujita, H.; Mahara, F.; Shiogama, K.; Tsutsumi, Y.; Takeichi, T. Hemophagocytic Lymphohistiocytosis in a Fatal Case of Severe Fever with Thrombocytopenia Syndrome. Intern. Med. 2017, 56, 1597–1602. [Google Scholar] [CrossRef]
- Kaneko, M.; Shikata, H.; Matsukage, S.; Maruta, M.; Shinomiya, H.; Suzuki, T.; Hasegawa, H.; Shimojima, M.; Saijo, M. A patient with severe fever with thrombocytopenia syndrome and hemophagocytic lymphohistiocytosis-associated involvement of the central nervous system. J. Infect. Chemother. 2018, 24, 292–297. [Google Scholar] [CrossRef]
- Carlson, A.L.; Pastula, D.M.; Lambert, A.J.; Staples, J.E.; Muehlenbachs, A.; Turabelidze, G.; Eby, C.S.; Keller, J.; Hess, B.; Buller, R.S.; et al. Heartland Virus and Hemophagocytic Lymphohistiocytosis in Immunocompromised Patient, Missouri, USA. Emerg. Infect. Dis. 2018, 24, 893–897. [Google Scholar] [CrossRef]
- Miyamoto, S.; Ito, T.; Terada, S.; Eguchi, T.; Furubeppu, H.; Kawamura, H.; Yasuda, T.; Kakihana, Y. Fulminant myocarditis associated with severe fever with thrombocytopenia syndrome: A case report. BMC Infect. Dis. 2019, 19, 266. [Google Scholar] [CrossRef]
- Nakamura, S.; Iwanaga, N.; Hara, S.; Shimada, S.; Kashima, Y.; Hayasaka, D.; Abe, K.; Izumikawa, K.; Yanagihara, K.; Miyazaki, Y.; et al. Viral load and inflammatory cytokine dynamics associated with the prognosis of severe fever with thrombocytopenia syndrome virus infection: An autopsy case. J. Infect. Chemother. 2019, 25, 480–484. [Google Scholar] [CrossRef]
- Kim, K.-H.; Lee, M.J.; Ko, M.K.; Lee, E.Y.; Yi, J. Brief Communication Severe Fever with Thrombocytopenia Syndrome Patients with Hemophagocytic Lymphohistiocytosis Retrospectively Identified in Korea. J. Korean Med. Sci. 2018, 33, 319. [Google Scholar] [CrossRef]
- Deng, B.; Zhang, S.; Geng, Y.; Zhang, Y.; Wang, Y.; Yao, W.; Wen, Y.; Cui, W.; Zhou, Y.; Gu, Q.; et al. Cytokine and chemokine levels in patients with severe fever with thrombocytopenia syndrome virus. PLoS ONE 2012, 7, e41365. [Google Scholar] [CrossRef]
- Jin, C.; Liang, M.; Ning, J.; Gu, W.; Jiang, H.; Wu, W.; Zhang, F.; Li, C.; Zhang, Q.; Zhu, H.; et al. Pathogenesis of emerging severe fever with thrombocytopenia syndrome virus in C57/BL6 mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 10053–10058. [Google Scholar] [CrossRef] [PubMed]
- Bosco-Lauth, A.M.; Calvert, A.E.; Root, J.J.; Gidlewski, T.; Bird, B.H.; Bowen, R.A.; Muehlenbachs, A.; Zaki, S.R.; Brault, A.C. Vertebrate host susceptibility to Heartland Virus. Emerg. Infect. Dis. 2016, 22, 2070–2077. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Orba, Y.; Maede-White, K.; Scott, D.; Feldmann, F.; Liang, M.; Ebihara, H. Animal models of emerging tick-borne phleboviruses: Determining target cells in a lethal model of SFTSV infection. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Westover, J.B.; Rigas, J.D.; Van Wettere, A.J.; Li, R.; Hickerson, B.T.; Jung, K.H.; Miao, J.; Reynolds, E.S.; Conrad, B.L.; Nielson, S.; et al. Heartland virus infection in hamsters deficient in type I interferon signaling: Protracted disease course ameliorated by favipiravir. Virology 2017, 511, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, B.; Paessler, S.; Walker, D.H.; Tesh, R.B.; Yu, X.-J. The Pathogenesis of Severe Fever with Thrombocytopenia Syndrome Virus Infection in Alpha/Beta Interferon Knockout Mice: Insights into the Pathologic Mechanisms of a New Viral Hemorrhagic Fever. J. Virol. 2014, 88, 1781–1786. [Google Scholar] [CrossRef] [PubMed]
- Gowen, B.B.; Westover, J.B.; Miao, J.; Van Wettere, A.J.; Rigas, J.D.; Hickerson, B.T.; Jung, K.-H.; Li, R.; Conrad, B.L.; Nielson, S.; et al. Modeling Severe Fever with Thrombocytopenia Syndrome Virus Infection in Golden Syrian Hamsters: Importance of STAT2 in Preventing Disease and Effective Treatment with Favipiravir. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Jiang, H.; Liang, M.; Han, Y.; Gu, W.; Zhang, F.; Zhu, H.; Wu, W.; Chen, T.; Li, C.; et al. SFTS Virus Infection in Nonhuman Primates. J. Infect. Dis. 2015, 211, 915–925. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, Y.-I.; Park, A.; Kwon, H.-I.; Kim, E.-H.; Si, Y.-J.; Song, M.-S.; Lee, C.-H.; Jung, K.; Shin, W.-J.; et al. Ferret animal model of severe fever with thrombocytopenia syndrome phlebovirus for human lethal infection and pathogenesis. Nat. Microbiol. 2019, 4, 438–446. [Google Scholar] [CrossRef]
- Park, E.-S.; Shimojima, M.; Nagata, N.; Ami, Y.; Yoshikawa, T.; Iwata-Yoshikawa, N.; Fukushi, S.; Watanabe, S.; Kurosu, T.; Kataoka, M.; et al. Severe Fever with Thrombocytopenia Syndrome Phlebovirus causes lethal viral hemorrhagic fever in cats. Sci. Rep. 2019, 9, 11990. [Google Scholar] [CrossRef]
- Koyama, S.; Ishii, K.J.; Coban, C.; Akira, S. Innate immune response to viral infection. Cytokine 2008, 43, 336–341. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.S.; Mossman, K.L. Viral Evasion Strategies in Type I IFN Signaling - A Summary of Recent Developments. Front. Immunol. 2016, 7, 498. [Google Scholar] [CrossRef] [PubMed]
- García-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Engelmayer, J.; Larsson, M.; Subklewe, M.; Chahroudi, A.; Cox, W.I.; Steinman, R.M.; Bhardwaj, N. Vaccinia virus inhibits the maturation of human dendritic cells: A novel mechanism of immune evasion. J. Immunol. 1999, 163, 6762–6768. [Google Scholar] [PubMed]
- Yen, B.; Mulder, L.C.F.; Martinez, O.; Basler, C.F. Molecular basis for ebolavirus VP35 suppression of human dendritic cell maturation. J. Virol. 2014, 88, 12500–12510. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yen, B.C.; Basler, C.F. Effects of Filovirus Interferon Antagonists on Responses of Human Monocyte-Derived Dendritic Cells to RNA Virus Infection. J. Virol. 2016, 90, 5108–5118. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F. Molecular pathogenesis of viral hemorrhagic fever. Semin. Immunopathol. 2017, 39, 551–561. [Google Scholar] [CrossRef]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef]
- Gough, D.J.; Messina, N.L.; Hii, L.; Gould, J.A.; Sabapathy, K.; Robertson, A.P.S.; Trapani, J.A.; Levy, D.E.; Hertzog, P.J.; Clarke, C.J.P.; et al. Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol. 2010, 8, e1000361. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Wang, X.; Mühlberger, E.; Volchkov, V.; Paragas, J.; Klenk, H.D.; García-Sastre, A.; Palese, P. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. USA 2000, 97, 12289–12294. [Google Scholar] [CrossRef] [PubMed]
- Luthra, P.; Ramanan, P.; Mire, C.E.; Weisend, C.; Tsuda, Y.; Yen, B.; Liu, G.; Leung, D.W.; Geisbert, T.W.; Ebihara, H.; et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 2013, 14, 74–84. [Google Scholar] [CrossRef]
- Dalrymple, N.A.; Cimica, V.; Mackow, E.R. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. mBio 2015, 6, e00553-15. [Google Scholar] [CrossRef]
- Didcock, L.; Young, D.F.; Goodbourn, S.; Randall, R.E. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 1999, 73, 9928–9933. [Google Scholar] [PubMed]
- Didcock, L.; Young, D.F.; Goodbourn, S.; Randall, R.E. Sendai virus and simian virus 5 block activation of interferon-responsive genes: Importance for virus pathogenesis. J. Virol. 1999, 73, 3125–3133. [Google Scholar]
- Kubota, T.; Yokosawa, N.; Yokota, S.; Fujii, N. C terminal CYS-RICH region of mumps virus structural V protein correlates with block of interferon alpha and gamma signal transduction pathway through decrease of STAT 1-alpha. Biochem. Biophys. Res. Commun. 2001, 283, 255–259. [Google Scholar] [CrossRef]
- Nishio, M.; Tsurudome, M.; Ito, M.; Kawano, M.; Komada, H.; Ito, Y. High resistance of human parainfluenza type 2 virus protein-expressing cells to the antiviral and anti-cell proliferative activities of alpha/beta interferons: Cysteine-rich V-specific domain is required for high resistance to the interferons. J. Virol. 2001, 75, 9165–9176. [Google Scholar] [CrossRef]
- Andrejeva, J.; Poole, E.; Young, D.F.; Goodbourn, S.; Randall, R.E. The p127 subunit (DDB1) of the UV-DNA damage repair binding protein is essential for the targeted degradation of STAT1 by the V protein of the paramyxovirus simian virus 5. J. Virol. 2002, 76, 11379–11386. [Google Scholar] [CrossRef]
- Park, M.-S.; García-Sastre, A.; Cros, J.F.; Basler, C.F.; Palese, P. Newcastle disease virus V protein is a determinant of host range restriction. J. Virol. 2003, 77, 9522–9532. [Google Scholar] [CrossRef]
- Palosaari, H.; Parisien, J.-P.; Rodriguez, J.J.; Ulane, C.M.; Horvath, C.M. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J. Virol. 2003, 77, 7635–7644. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Tsurudome, M.; Ito, M.; Garcin, D.; Kolakofsky, D.; Ito, Y. Identification of paramyxovirus V protein residues essential for STAT protein degradation and promotion of virus replication. J. Virol. 2005, 79, 8591–8601. [Google Scholar] [CrossRef] [PubMed]
- Devaux, P.; Priniski, L.; Cattaneo, R. The measles virus phosphoprotein interacts with the linker domain of STAT1. Virology 2013, 444, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Ly, H.J.; Ikegami, T. Rift Valley fever virus NSs protein functions and the similarity to other bunyavirus NSs proteins. J. Virol. 2016, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Rezelj, V.V.; Överby, A.K.; Elliott, R.M. Generation of mutant Uukuniemi viruses lacking the nonstructural protein NSs by reverse genetics indicates that NSs is a weak interferon antagonist. J. Virol. 2015, 89, 4849–4856. [Google Scholar] [CrossRef]
- Rezelj, V.V.; Li, P.; Chaudhary, V.; Elliott, R.M.; Jin, D.-Y.; Brennan, B. Differential Antagonism of Human Innate Immune Responses by Tick-Borne Phlebovirus Nonstructural Proteins. mSphere 2017, 2, e00234-17. [Google Scholar] [CrossRef] [PubMed]
- Billecocq, A.; Spiegel, M.; Vialat, P.; Kohl, A.; Weber, F.; Bouloy, M.; Haller, O. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J. Virol. 2004, 78, 9798–9806. [Google Scholar] [CrossRef]
- Copeland, A.M.; Van Deusen, N.M.; Schmaljohn, C.S. Rift Valley fever virus NSS gene expression correlates with a defect in nuclear mRNA export. Virology 2015, 486, 88–93. [Google Scholar] [CrossRef]
- Ning, Y.-J.; Wang, M.; Deng, M.; Shen, S.; Liu, W.; Cao, W.-C.; Deng, F.; Wang, Y.-Y.; Hu, Z.; Wang, H. Viral suppression of innate immunity via spatial isolation of TBK1/IKKε from mitochondrial antiviral platform. J. Mol. Cell Biol. 2014, 6, 324–337. [Google Scholar] [CrossRef]
- Santiago, F.W.; Covaleda, L.M.; Sanchez-Aparicio, M.T.; Silvas, J.A.; Diaz-Vizarreta, A.C.; Patel, J.R.; Popov, V.; Yu, X.; García-Sastre, A.; Aguilar, P.V. Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J. Virol. 2014, 88, 4572–4585. [Google Scholar] [CrossRef]
- Wu, X.; Qi, X.; Liang, M.; Li, C.; Cardona, C.J.; Li, D.; Xing, Z. Roles of viroplasm-like structures formed by nonstructural protein NSs in infection with severe fever with thrombocytopenia syndrome virus. FASEB J. 2014, 28, 2504–2516. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, M.; Igarashi, M.; Koshiba, T.; Irie, T.; Takada, A.; Ichinohe, T. Two Conserved Amino Acids within the NSs of Severe Fever with Thrombocytopenia Syndrome Phlebovirus Are Essential for Anti-interferon Activity. J. Virol. 2018, 92, e00706-18. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Bai, M.; Qi, X.; Li, C.; Liang, M.; Li, D.; Cardona, C.J.; Xing, Z. Suppression of the IFN-α and -β Induction through Sequestering IRF7 into Viral Inclusion Bodies by Nonstructural Protein NSs in Severe Fever with Thrombocytopenia Syndrome Bunyavirus Infection. J. Immunol. 2019, 202, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, V.; Zhang, S.; Yuen, K.-S.; Li, C.; Lui, P.-Y.; Fung, S.-Y.; Wang, P.-H.; Chan, C.-P.; Li, D.; Kok, K.-H.; et al. Suppression of type I and type III IFN signalling by NSs protein of severe fever with thrombocytopenia syndrome virus through inhibition of STAT1 phosphorylation and activation. J. Gen. Virol. 2015, 96, 3204–3211. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.-J.; Feng, K.; Min, Y.-Q.; Cao, W.-C.; Wang, M.; Deng, F.; Hu, Z.; Wang, H. Disruption of type I interferon signaling by the nonstructural protein of severe fever with thrombocytopenia syndrome virus via the hijacking of STAT2 and STAT1 into inclusion bodies. J. Virol. 2015, 89, 4227–4236. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, Y.; Sakai, M.; Shimojima, M.; Saijo, M.; Itoh, M.; Gotoh, B. Nonstructural protein of severe fever with thrombocytopenia syndrome phlebovirus targets STAT2 and not STAT1 to inhibit type I interferon-stimulated JAK-STAT signaling. Microbes Infect. 2018, 20, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, R.; Sakabe, S.; Urata, S.; Yasuda, J. Species-Specific Pathogenicity of Severe Fever with Thrombocytopenia Syndrome Virus Is Determined by Anti-STAT2 Activity of NSs. J. Virol. 2019, 93, e02226-18. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ye, H.; Li, S.; Jiao, B.; Wu, J.; Zeng, P.; Chen, L. Severe fever with thrombocytopenia syndrome virus inhibits exogenous Type i IFN signaling pathway through its NSs in vitro. PLoS ONE 2017, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.-J.; Mo, Q.; Feng, K.; Min, Y.-Q.; Li, M.; Hou, D.; Peng, C.; Zheng, X.; Deng, F.; Hu, Z.; et al. Interferon-γ-Directed Inhibition of a Novel High-Pathogenic Phlebovirus and Viral Antagonism of the Antiviral Signaling by Targeting STAT1. Front. Immunol. 2019, 10, 1182. [Google Scholar] [CrossRef]
- Ning, Y.-J.; Feng, K.; Min, Y.-Q.; Deng, F.; Hu, Z.; Wang, H. Heartland virus NSs protein disrupts host defenses by blocking the TBK1 kinase-IRF3 transcription factor interaction and signaling required for interferon induction. J. Biol. Chem. 2017, 292, 16722–16733. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Deng, F.; Hu, Z.; Wang, H.; Ning, Y.-J. Heartland virus antagonizes type I and III interferon antiviral signaling by inhibiting phosphorylation and nuclear translocation of STAT2 and STAT1. J. Biol. Chem. 2019, 294, 9503–9517. [Google Scholar] [CrossRef] [PubMed]
- Gentile, L.F.; Moldawer, L.L. DAMPs, PAMPs, and the origins of SIRS in bacterial sepsis. Shock Augusta Ga 2013, 39, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; McGuire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of cytokines as a double-edged sword in sepsis. Vivo Athens Greece 2013, 27, 669–684. [Google Scholar]
- Oberholzer, A.; Oberholzer, C.; Moldawer, L.L. Sepsis syndromes: Understanding the role of innate and acquired immunity. Shock Augusta Ga 2001, 16, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, H.; Rockx, B.; Marzi, A.; Feldmann, F.; Haddock, E.; Brining, D.; LaCasse, R.A.; Gardner, D.; Feldmann, H. Host response dynamics following lethal infection of rhesus macaques with Zaire ebolavirus. J. Infect. Dis. 2011, 204, S991–S999. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-M.; Lei, X.-Y.; Yu, H.; Zhang, J.-Z.; Yu, X.-J. Correlation of cytokine level with the severity of severe fever with thrombocytopenia syndrome. Virol. J. 2017, 14, 6. [Google Scholar] [CrossRef]
- Sun, Y.; Jin, C.; Zhan, F.; Wang, X.; Liang, M.; Zhang, Q.; Ding, S.; Guan, X.; Huo, X.; Li, C.; et al. Host cytokine storm is associated with disease severity of severe fever with thrombocytopenia syndrome. J. Infect. Dis. 2012, 206, 1085–1094. [Google Scholar] [CrossRef]
- Zhao, J.; He, S.; Minassian, A.; Li, J.; Feng, P. Recent advances on viral manipulation of NF-κB signaling pathway. Curr. Opin. Virol. 2015, 15, 103–111. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Dudziak, D.; Dirmeier, U.; Hobom, G.; Riedel, A.; Schlee, M.; Staudt, L.M.; Rosenwald, A.; Behrends, U.; Bornkamm, G.W.; et al. Active NF-kappaB signalling is a prerequisite for influenza virus infection. J. Gen. Virol. 2004, 85, 2347–2356. [Google Scholar] [CrossRef]
- Luco, S.; Delmas, O.; Vidalain, P.-O.; Tangy, F.; Weil, R.; Bourhy, H. RelAp43, a member of the NF-κB family involved in innate immune response against Lyssavirus infection. PLoS Pathog. 2012, 8, e1003060. [Google Scholar] [CrossRef]
- Besson, B.; Sonthonnax, F.; Duchateau, M.; Ben Khalifa, Y.; Larrous, F.; Eun, H.; Hourdel, V.; Matondo, M.; Chamot-Rooke, J.; Grailhe, R.; et al. Regulation of NF-κB by the p105-ABIN2-TPL2 complex and RelAp43 during rabies virus infection. PLoS Pathog. 2017, 13, e1006697. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Jin, C.; Zhu, L.; Liang, M.; Li, C.; Cardona, C.J.; Li, D.; Xing, Z. Host Responses and Regulation by NFκB Signaling in the Liver and Liver Epithelial Cells Infected with A Novel Tick-borne Bunyavirus. Sci. Rep. 2015, 5, 11816. [Google Scholar] [CrossRef] [PubMed]
- Sang, Y.; Miller, L.C.; Blecha, F. Macrophage Polarization in Virus-Host Interactions. J. Clin. Cell. Immunol. 2015, 6, 311. [Google Scholar] [PubMed]
- Ward, N.S.; Casserly, B.; Ayala, A. The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin. Chest Med. 2008, 29, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Kometani, K.; Ise, W. Memory B cells. Nat. Rev. Immunol. 2015, 15, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Zheng, N.; Liu, Y.; Tian, C.; Wu, X.; Ma, X.; Chen, D.; Zou, X.; Wang, G.; Wang, H.; et al. Deficient humoral responses and disrupted B-cell immunity are associated with fatal SFTSV infection. Nat. Commun. 2018, 9, 3328. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Feng, Z.; Geng, D.; Sun, Y.; Yuan, G. Dynamic changes of laboratory parameters and peripheral blood lymphocyte subsets in severe fever with thrombocytopenia syndrome patients. Int. J. Infect. Dis. 2017, 58, 45–51. [Google Scholar] [CrossRef]
- Takahashi, T.; Suzuki, T.; Hiroshige, S.; Nouno, S.; Matsumura, T.; Tominaga, T.; Yujiri, T.; Katano, H.; Sato, Y.; Hasegawa, H. Transient Appearance of Plasmablasts in the Peripheral Blood of Japanese Patients With Severe Fever With Thrombocytopenia Syndrome. J. Infect. Dis. 2019, 220, 23–27. [Google Scholar] [CrossRef]
- Bradfute, S.B.; Braun, D.R.; Shamblin, J.D.; Geisbert, J.B.; Paragas, J.; Garrison, A.; Hensley, L.E.; Geisbert, T.W. Lymphocyte death in a mouse model of Ebola virus infection. J. Infect. Dis. 2007, 196, S296–S304. [Google Scholar] [CrossRef]
- Reed, D.S.; Hensley, L.E.; Geisbert, J.B.; Jahrling, P.B.; Geisbert, T.W. Depletion of peripheral blood T lymphocytes and NK cells during the course of ebola hemorrhagic Fever in cynomolgus macaques. Viral Immunol. 2004, 17, 390–400. [Google Scholar] [CrossRef]
- Li, M.M.; Zhang, W.J.; Liu, J.; Li, M.Y.; Zhang, Y.F.; Xiong, Y.; Xiong, S.E.; Zou, C.C.; Xiong, L.Q.; Liang, B.Y.; et al. Dynamic changes in the immunological characteristics of T lymphocytes in surviving patients with severe fever with thrombocytopenia syndrome (SFTS). Int. J. Infect. Dis. 2018, 70, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, M.; Xiong, S.; Wang, H.; Xiong, Y.; Li, M.; Lu, M.; Yang, D.; Peng, C.; Zheng, X. Decreased myeloid dendritic cells indicate a poor prognosis in patients with severe fever with thrombocytopenia syndrome. Int. J. Infect. Dis. 2017, 54, 113–120. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, L.; Wen, H.; Yang, Y.; Yu, H.; Yu, X. Neutralizing Antibodies to Severe Fever with Thrombocytopenia Syndrome Virus 4 Years after Hospitalization, China. Emerg. Infect. Dis. 2016, 22, 1985–1987. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shao, L.; Bi, Y.; Niu, G. Neutralizing antibodies to Severe Fever with Thrombocytopenia Syndrome Virus in general population, Shandong Province, China. Sci. Rep. 2018, 8, 15401. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Li, D.; Wen, D.; Li, S.; Zhao, C.; Qi, Y.; Jangra, R.K.; Wu, C.; Xia, D.; Zhang, X.; et al. Single dose of a rVSV-based vaccine elicits complete protection against severe fever with thrombocytopenia syndrome virus. NPJ Vaccines 2019, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Huang, D.-D.; Bai, J.-Y.; Zhuang, L.; Lu, Q.-B.; Zhang, X.-A.; Liu, W.; Wang, J.-Y.; Cao, W.-C. Immunization with Recombinant SFTSV/NSs Protein Does Not Promote Virus Clearance in SFTSV-Infected C57BL/6J Mice. Viral Immunol. 2015, 28, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.-E.; Kim, Y.-I.; Park, S.-J.; Yu, M.-A.; Kwon, H.-I.; Eo, S.; Kim, T.-S.; Seok, J.; Choi, W.-S.; Jeong, J.H.; et al. Development of a SFTSV DNA vaccine that confers complete protection against lethal infection in ferrets. Nat. Commun. 2019, 10, 3836. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendoza, C.A.; Ebihara, H.; Yamaoka, S. Immune Modulation and Immune-Mediated Pathogenesis of Emerging Tickborne Banyangviruses. Vaccines 2019, 7, 125. https://doi.org/10.3390/vaccines7040125
Mendoza CA, Ebihara H, Yamaoka S. Immune Modulation and Immune-Mediated Pathogenesis of Emerging Tickborne Banyangviruses. Vaccines. 2019; 7(4):125. https://doi.org/10.3390/vaccines7040125
Chicago/Turabian StyleMendoza, Crystal A., Hideki Ebihara, and Satoko Yamaoka. 2019. "Immune Modulation and Immune-Mediated Pathogenesis of Emerging Tickborne Banyangviruses" Vaccines 7, no. 4: 125. https://doi.org/10.3390/vaccines7040125
APA StyleMendoza, C. A., Ebihara, H., & Yamaoka, S. (2019). Immune Modulation and Immune-Mediated Pathogenesis of Emerging Tickborne Banyangviruses. Vaccines, 7(4), 125. https://doi.org/10.3390/vaccines7040125