Immunoinformatics-Aided Design and Evaluation of a Potential Multi-Epitope Vaccine against Klebsiella Pneumoniae




Abstract
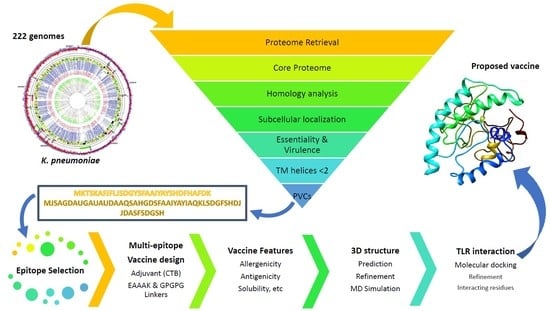
:
1. Introduction
2. Materials and Methods
2.1. Genome and Proteome Retrieval and Pangenome Analysis
2.2. Reverse Vaccinology for Protein Prioritization
2.3. Prediction of B-Cell Epitopes
2.4. Prioritization of B-Cell Derived MHC II Epitopes
2.5. Prioritization of B-Cell Derived MHC I Epitopes
2.6. Multi-Epitope Vaccine Design
2.7. Antigenicity, Allergenicity, Solubility, and Physicochemical Features
2.8. Multi-Epitope Structural Modeling, Refinement, and Validation
2.9. Energy Minimization of Multi-Epitope Vaccine
2.10. Binding Affinity of Poly-Epitope Structure with Toll-Like Receptors
2.11. Reverse Translation and Codon Optimization
3. Results
3.1. Pangenome Analysis of Klebsiella Pneumoniae
3.2. Prioritization of Global Core Antigenic Proteins
3.3. Selection of Epitopes from Global Core Antigenic Proteins
3.4. Multi-Epitope Vaccine Design
3.5. Physicochemical and Other Evaluations of the Multi-Epitope Vaccine
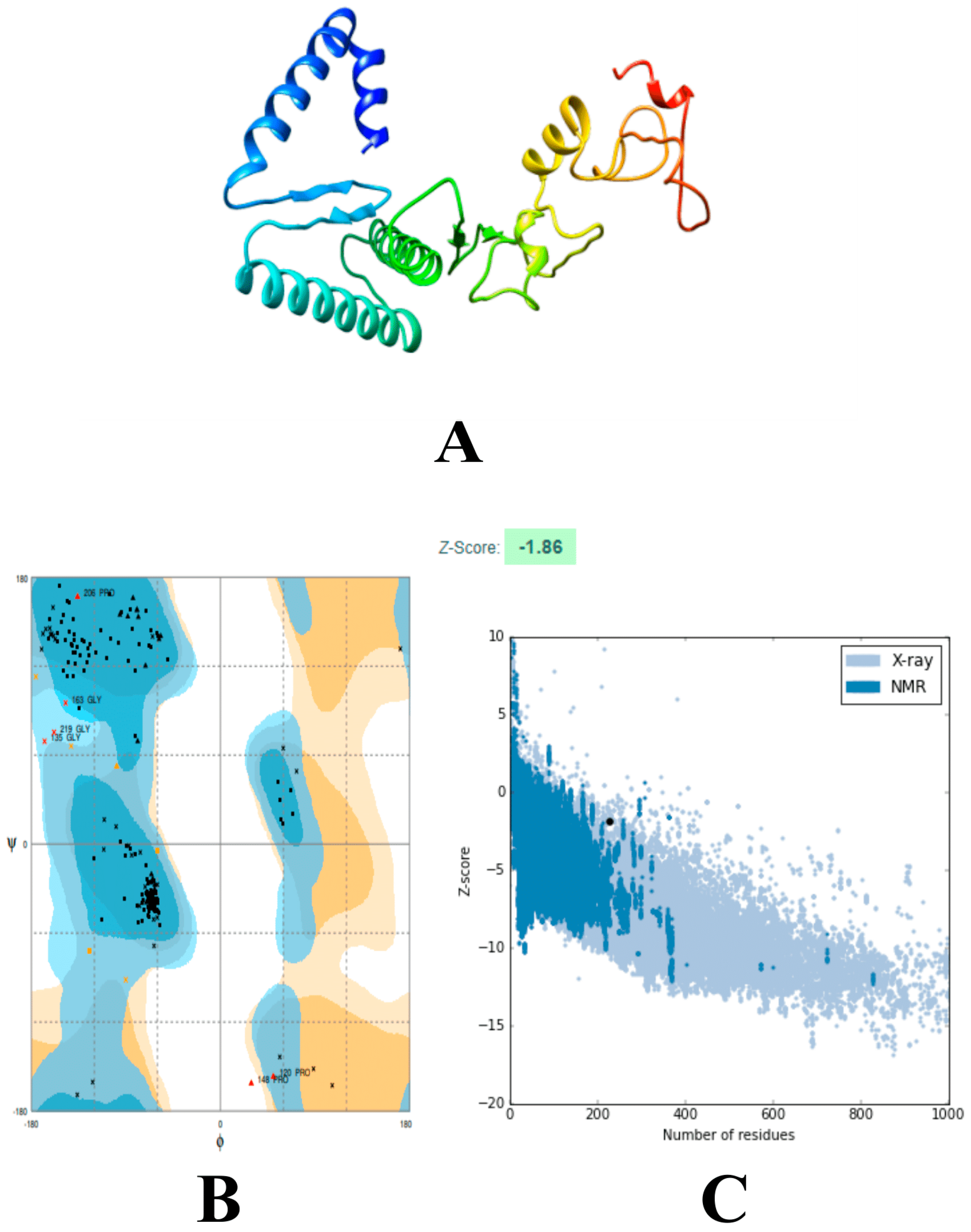
3.6. Modeling and Refinements of the 3D Structure of the Vaccine
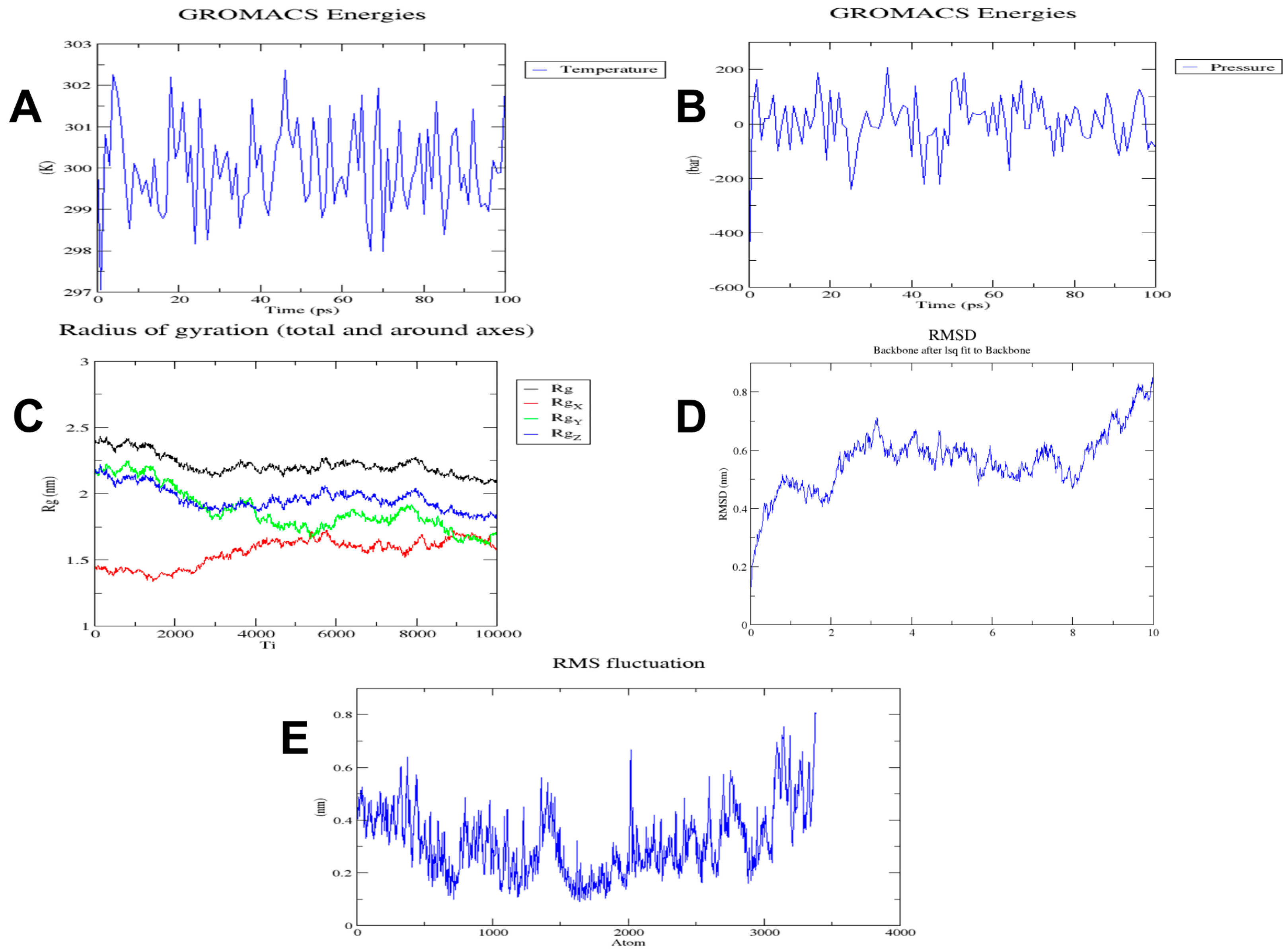
3.7. Molecular Dynamics Simulation of the Multi-Epitope Vaccine
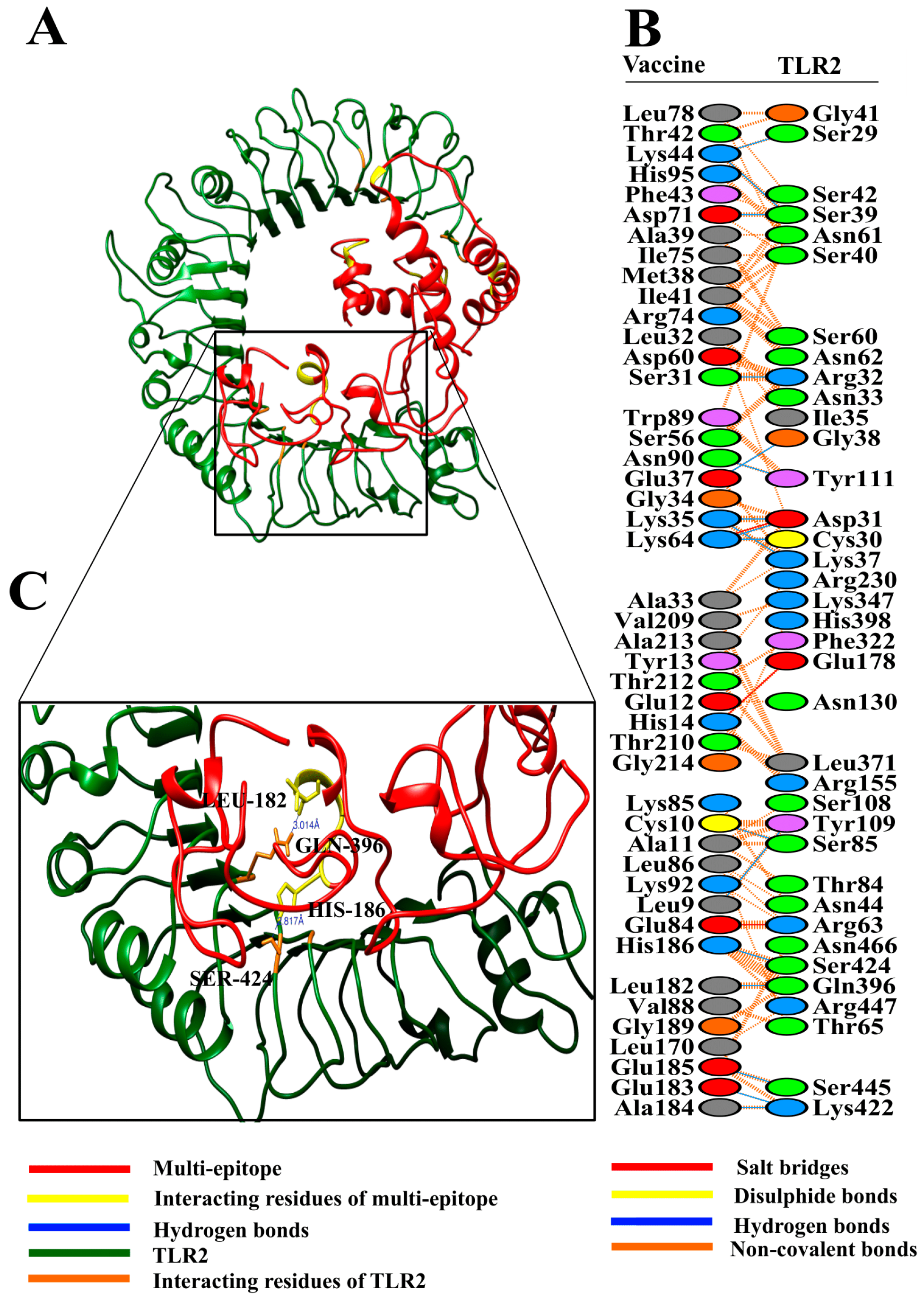
3.8. Molecular Docking of Vaccine with Toll-Like Receptors
3.9. Reverse Translation and Codon Optimization of Vaccine
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tumbarello, M.; Trecarichi, E.M.; De Rosa, F.G.; Giannella, M.; Giacobbe, D.R.; Bassetti, M.; Losito, A.R.; Bartoletti, M.; Del Bono, V.; Corcione, S.; et al. Infections caused by KPC-producing Klebsiella pneumoniae: Differences in therapy and mortality in a multicentre study. J. Antimicrob. Chemother. 2015, 70, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Daikos, G.L.; Markogiannakis, A. Carbapenemase-producing Klebsiella pneumoniae: (When) might we still consider treating with carbapenems? Clin. Microbiol. Infect. 2011, 17, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.B.; Tam, V.H. Detection and treatment options for Klebsiella pneumoniae carbapenemases (KPCs): An emerging cause of multidrug-resistant infection. J. Antimicrob. Chemother. 2010, 65, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, M.; Viale, P.; Viscoli, C.; Trecarichi, E.M.; Tumietto, F.; Marchese, A.; Spanu, T.; Ambretti, S.; Ginocchio, F.; Cristini, F.; et al. Predictors of Mortality in Bloodstream Infections Caused by Klebsiella pneumoniae Carbapenemase-Producing K. pneumoniae: Importance of Combination Therapy. Clin. Infect. Dis. 2012, 55, 943–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, V.; Sharma, S.; Harjai, K.; Mohan, H.; Chhibber, S. Lipopolysaccharide-mediated protection against Klebsiella pneumoniae-induced lobar pneumonia: Intranasal vs. intramuscular route of immunization. Folia Microbiol. 2005, 50, 83–86. [Google Scholar] [CrossRef]
- Cryz, S.J.; Mortimer, P.; Cross, A.S.; Fürer, E.; Germanier, R. Safety and immunogenicity of a polyvalent Klebsiella capsular polysaccharide vaccine in humans. Vaccine 1986, 4, 15–20. [Google Scholar] [CrossRef]
- Kelly, D.F.; Rappuoli, R. Reverse Vaccinology and Vaccines for Serogroup B Neisseria meningitidis. In Hot Topics in Infection and Immunity in Children II; Springer: Boston, MA, USA, 2005; pp. 217–223. [Google Scholar]
- Capecchi, B.; Serruto, D.; Adu-Bobie, J.; Rappuoli, R.; Pizza, M. The genome revolution in vaccine research. Curr. Issues Mol. Biol. 2004, 6, 17–27. [Google Scholar]
- Sette, A.; Rappuoli, R. Reverse vaccinology: Developing vaccines in the era of genomics. Immunity 2010, 33, 530–541. [Google Scholar] [CrossRef]
- Lundberg, U.; Senn, B.M.; Schüler, W.; Meinke, A.; Hanner, M. Identification and characterization of antigens as vaccine candidates against Klebsiella pneumoniae. Hum. Vaccin. Immunother. 2013, 9, 497–505. [Google Scholar] [CrossRef]
- Kumar Jaiswal, A.; Tiwari, S.; Jamal, S.B.; Barh, D.; Azevedo, V.; Soares, S.C. An In Silico Identification of Common Putative Vaccine Candidates against Treponema pallidum: A Reverse Vaccinology and Subtractive Genomics Based Approach. Int. J. Mol. Sci. 2017, 18, 420. [Google Scholar] [CrossRef]
- Hasan, M.A.; Khan, M.A.; Sharmin, T.; Hasan Mazumder, M.H.; Chowdhury, A.S. Identification of putative drug targets in Vancomycin-resistant Staphylococcus aureus (VRSA) using computer aided protein data analysis. Gene 2016, 575, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Hou, B.; Wang, B.; Lin, X.; Gong, W.; Dong, H.; Zhu, S.; Chen, S.; Xue, X.; Zhao, K.-N.; et al. A multi-epitope vaccine based on Chlamydia trachomatis major outer membrane protein induces specific immunity in mice. Acta Biochim. Biophys. Sin. 2014, 46, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Kaumaya, P.T.P.; Kobs-Conrad, S.; Seo, Y.H.; Lee, H.; Vanbuskirk, A.M.; Feng, N.; Sheridan, J.F.; Stevens, V. Peptide vaccines incorporating a “promiscuous” T-cell epitope bypass certain haplotype restricted immune responses and provide broad spectrum immunogenicity. J. Mol. Recognit. 1993, 6, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Calle, J.M.; Oliveira, G.A.; Watta, C.O.; Soverow, J.; Parra-Lopez, C.; Nardin, E.H. A linear peptide containing minimal T- and B-cell epitopes of Plasmodium falciparum circumsporozoite protein elicits protection against transgenic sporozoite challenge. Infect. Immun. 2006, 74, 6929–6939. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Fikes, J. Epitope-based vaccines: An update on epitope identification, vaccine design and delivery. Curr. Opin. Immunol. 2003, 15, 461–470. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Rizwan, M.; Naz, A.; Ahmad, J.; Naz, K.; Obaid, A.; Parveen, T.; Ahsan, M.; Ali, A. VacSol: A high throughput in silico pipeline to predict potential therapeutic targets in prokaryotic pathogens using subtractive reverse vaccinology. BMC Bioinform. 2017, 18, 106. [Google Scholar] [CrossRef]
- Oldstone, M.B.A. Molecular mimicry, microbial infection, and autoimmune disease: Evolution of the concept. Curr. Top. Microbiol. Immunol. 2005, 296, 1–17. [Google Scholar]
- Gardy, J.L.; Laird, M.R.; Chen, F.; Rey, S.; Walsh, C.J.; Ester, M.; Brinkman, F.S.L. PSORTb v.2.0: Expanded prediction of bacterial protein subcellular localization and insights gained from comparative proteome analysis. Bioinformatics 2005, 21, 617–623. [Google Scholar] [CrossRef]
- Naz, A.; Awan, F.M.; Obaid, A.; Muhammad, S.A.; Paracha, R.Z.; Ahmad, J.; Ali, A. Identification of putative vaccine candidates against Helicobacter pylori exploiting exoproteome and secretome: A reverse vaccinology based approach. Infect. Genet. Evol. 2015, 32, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Lin, Y.; Gao, F.; Zhang, C.-T.; Zhang, R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014, 42, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Grazziotin, A.L.; Vidal, N.M.; Venancio, T.M. Uncovering major genomic features of essential genes in Bacteria and a methanogenic Archaea. FEBS J. 2015, 282, 3395–3411. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.E.; Smith, J.; Lam, M.; Zemla, A.; Dyer, M.D.; Slezak, T. MvirDB—A microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res. 2007, 35, D391–D394. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]
- Tusnády, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Naz, A.; Obaid, A.; Paracha, R.Z.; Naz, K.; Awan, F.M.; Muhmmad, S.A.; Janjua, H.A.; Ahmad, J.; Ali, A. Pangenome and immuno-proteomics analysis of Acinetobacter baumannii strains revealed the core peptide vaccine targets. BMC Genom. 2016, 17, 732. [Google Scholar] [CrossRef]
- Walker, J.M. The Proteomics Protocols Handbook; Springer: Berlin, Germany, 2005. [Google Scholar]
- Baseer, S.; Ahmad, S.; Ranaghan, K.E.; Azam, S.S. Towards a peptide-based vaccine against Shigella sonnei: A subtractive reverse vaccinology based approach. Biologicals 2017, 50, 87–99. [Google Scholar] [CrossRef]
- Rashid, M.I.; Naz, A.; Ali, A.; Andleeb, S. Prediction of vaccine candidates against Pseudomonas aeruginosa: An integrated genomics and proteomics approach. Genomics 2017, 109, 274–283. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Singh, H.; Raghava, G.P. ProPred: Prediction of HLA-DR binding sites. Bioinformatics 2001, 17, 1236–1237. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Doytchinova, I.A.; Zygouri, C.; Flower, D.R. MHCPred: A server for quantitative prediction of peptide-MHC binding. Nucleic Acids Res. 2003, 31, 3621–3624. [Google Scholar] [CrossRef] [PubMed]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource in Epitope Discovery and Synthetic Vaccine Design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.; Gupta, D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Vir, P.; Raghava, G.P.S. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef]
- Singh, H.; Raghava, G.P.S. ProPred1: Prediction of promiscuous MHC Class-I binding sites. Bioinformatics 2003, 19, 1009–1014. [Google Scholar] [CrossRef]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef]
- Stratmann, T. Cholera Toxin Subunit B as Adjuvan—An Accelerator in Protective Immunity and a Break in Autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef]
- Magnan, C.N.; Zeller, M.; Kayala, M.A.; Vigil, A.; Randall, A.; Felgner, P.L.; Baldi, P. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics 2010, 26, 2936–2943. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity prediction by descriptor fingerprints. Bioinformatics 2013, 30, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed]
- Smialowski, P.; Doose, G.; Torkler, P.; Kaufmann, S.; Frishman, D. PROSO II—A new method for protein solubility prediction. FEBS J. 2012, 279, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H. Predicting Secretory Proteins with SignalP; Humana Press: New York, NY, USA, 2017; pp. 59–73. [Google Scholar]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T. ERRAT: An empirical atom-based method for validating protein structures. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Turner, P. XMGRACE, Version 5.1. 19; Center Coastal Land-Margin Reserach Oregon Graduate Insttitude Science Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Regueiro, V.; Moranta, D.; Campos, M.A.; Margareto, J.; Garmendia, J.; Bengoechea, J.A. Klebsiella pneumoniae increases the levels of Toll-like receptors 2 and 4 in human airway epithelial cells. Infect. Immun. 2009, 77, 714–724. [Google Scholar] [CrossRef] [PubMed]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- de Vries, S.J.; Bonvin, A.M.J.J. CPORT: A Consensus Interface Predictor and Its Performance in Prediction-Driven Docking with HADDOCK. PLoS ONE 2011, 6, e17695. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A. PDBsum: Summaries and analyses of PDB structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef] [PubMed]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahbar, M.R.; Ghasemi, Y. Designing an efficient multi-epitope oral vaccine against Helicobacter pylori using immunoinformatics and structural vaccinology approaches. Mol. Biosyst. 2017, 13, 699–713. [Google Scholar] [CrossRef]
- Grandi, G. Bacterial surface proteins and vaccines. F1000 Biol. Rep. 2010, 2, 36. [Google Scholar] [CrossRef]
- Handman, E. Leishmaniasis: Current status of vaccine development. Clin. Microbiol. Rev. 2001, 14, 229–243. [Google Scholar] [CrossRef]
- Minch, M.J. An Introduction to Hydrogen Bonding (Jeffrey, George A.). J. Chem. Educ. 1999, 76, 759. [Google Scholar] [CrossRef]
- Cryz, S.J.; Mortimer, P.M.; Mansfield, V.; Germanier, R.; Germanier, R. Seroepidemiology of Klebsiella bacteremic isolates and implications for vaccine development. J. Clin. Microbiol. 1986, 23, 687–690. [Google Scholar] [PubMed]
- Cryz, S.J.; Fürer, E.; Germanier, R. Safety and immunogenicity of Klebsiella pneumoniae K1 capsular polysaccharide vaccine in humans. J. Infect. Dis. 1985, 151, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.A.; El-Sayed, L.H.; Haroun, M.; Hussein, A.A.; El Ashry, E.S.H. Development of immunization trials against Klebsiella pneumoniae. Vaccine 2012, 30, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Huang, S.; Zhang, Q. Outer membrane proteins: Key players for bacterial adaptation in host niches. Microbes Infect. 2002, 4, 325–331. [Google Scholar] [CrossRef]
- Hellman, J.; Warren, H.S. Outer membrane protein A (OmpA), peptidoglycan-associated lipoprotein (PAL), and murein lipoprotein (MLP) are released in experimental Gram-negative sepsis. J. Endotoxin Res. 2001, 7, 69–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurupati, P.; Teh, B.K.; Kumarasinghe, G.; Poh, C.L. Identification of vaccine candidate antigens of an ESBL producingKlebsiella pneumoniae clinical strain by immunoproteome analysis. Proteomics 2006, 6, 836–844. [Google Scholar] [CrossRef]
- Kurupati, P.; Ramachandran, N.P.; Poh, C.L. Protective efficacy of DNA vaccines encoding outer membrane protein A and OmpK36 of Klebsiella pneumoniae in mice. Clin. Vaccine Immunol. 2011, 18, 82–88. [Google Scholar] [CrossRef]
- Hsieh, P.-F.; Liu, J.-Y.; Pan, Y.-J.; Wu, M.-C.; Lin, T.-L.; Huang, Y.-T.; Wang, J.-T. Klebsiella pneumoniae Peptidoglycan-Associated Lipoprotein and Murein Lipoprotein Contribute to Serum Resistance, Antiphagocytosis, and Proinflammatory Cytokine Stimulation. J. Infect. Dis. 2013, 208, 1580–1589. [Google Scholar] [CrossRef] [Green Version]
- Tseng, I.-L.; Liu, Y.-M.; Wang, S.-J.; Yeh, H.-Y.; Hsieh, C.-L.; Lu, H.-L.; Tseng, Y.-C.; Mu, J.-J. Emergence of carbapenemase producing Klebsiella pneumonia and spread of KPC-2 and KPC-17 in Taiwan: A nationwide study from 2011 to 2013. PLoS ONE 2015, 10, e0138471. [Google Scholar] [CrossRef]
- Randall, C.P.; Gupta, A.; Jackson, N.; Busse, D.; O’neill, A.J. Silver resistance in Gram-negative bacteria: A dissection of endogenous and exogenous mechanisms. J. Antimicrob. Chemother. 2015, 70, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Bayan, U.; Saikia, K.K. In silico identification of vaccine candidates against Klebsiella oxytoca. Comput. Biol. Chem. 2017, 69, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Azam, S.S. A novel approach of virulome based reverse vaccinology for exploring and validating peptide-based vaccine candidates against the most troublesome nosocomial pathogen: Acinetobacter baumannii. J. Mol. Graph. Model. 2018, 83, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, I.; Alcalá-Franco, B.; Sacrselli, M.; Dalsass, M.; Buccato, S.; Masignani, V.; Bragonzi, A. Genome-based approach delivers vaccine candidates against Pseudomonas aeruginosa. Front. Immunol. 2018, 9, 3021. [Google Scholar] [CrossRef] [PubMed]
- Baarda, B.I.; Martinez, F.G.; Sikora, A.E. Proteomics, Bioinformatics and Structure-Function Antigen Mining For Gonorrhea Vaccines. Front. Immunol. 2018, 9, 2793. [Google Scholar] [CrossRef] [PubMed]
- Clemente, A.M.; Castronovo, G.; Antonelli, A.; D’Andrea, M.M.; Tanturli, M.; Perissi, E.; Paccosi, S.; Parenti, A.; Cozzolino, F.; Rossolini, G.M.; et al. Differential Th17 response induced by the two clades of the pandemic ST258 Klebsiella pneumoniae clonal lineages producing KPC-type carbapenemase. PLoS ONE 2017, 12, e0178847. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Lu, M.-C.; Lin, C.; Chiang, M.-K.; Jan, M.-S.; Tang, H.-L.; Liu, H.-C.; Lin, W.-L.; Huang, C.-Y.; Chen, C.-M.; et al. Activation of IFN-γ/STAT/IRF-1 in Hepatic Responses to Klebsiella pneumoniae Infection. PLoS ONE 2013, 8, e79961. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.A.; Perry, M.L.; Getsoian, A.G.; Newstead, M.W.; Standiford, T.J. Divergent role of gamma interferon in a murine model of pulmonary versus systemic Klebsiella pneumoniae infection. Infect. Immun. 2002, 70, 6310–6318. [Google Scholar] [CrossRef]
- Amanna, I.J.; Slifka, M.K. Contributions of humoral and cellular immunity to vaccine-induced protection in humans. Virology 2011, 411, 206–215. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
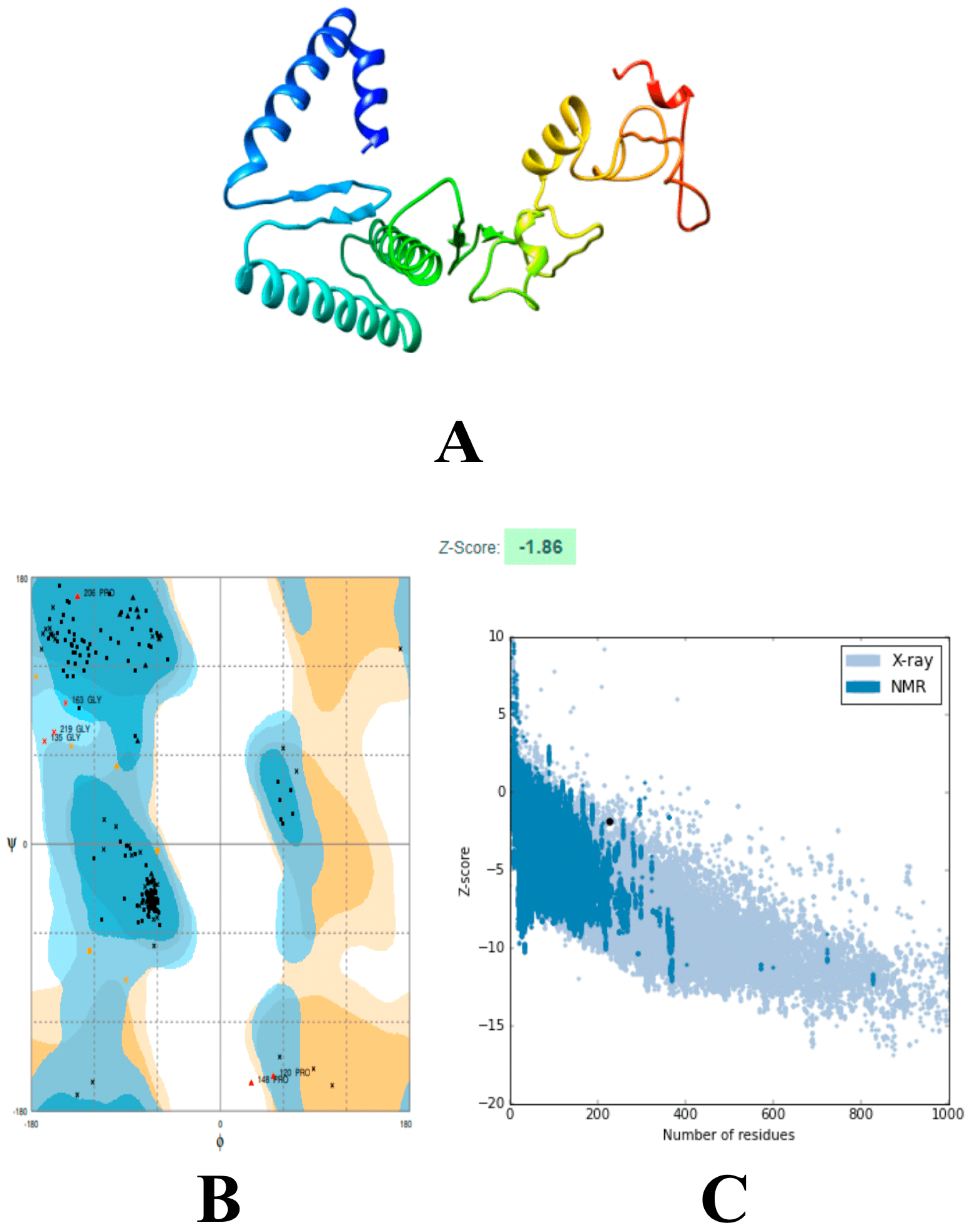{kind=link}
{kind=link}
{kind=link}
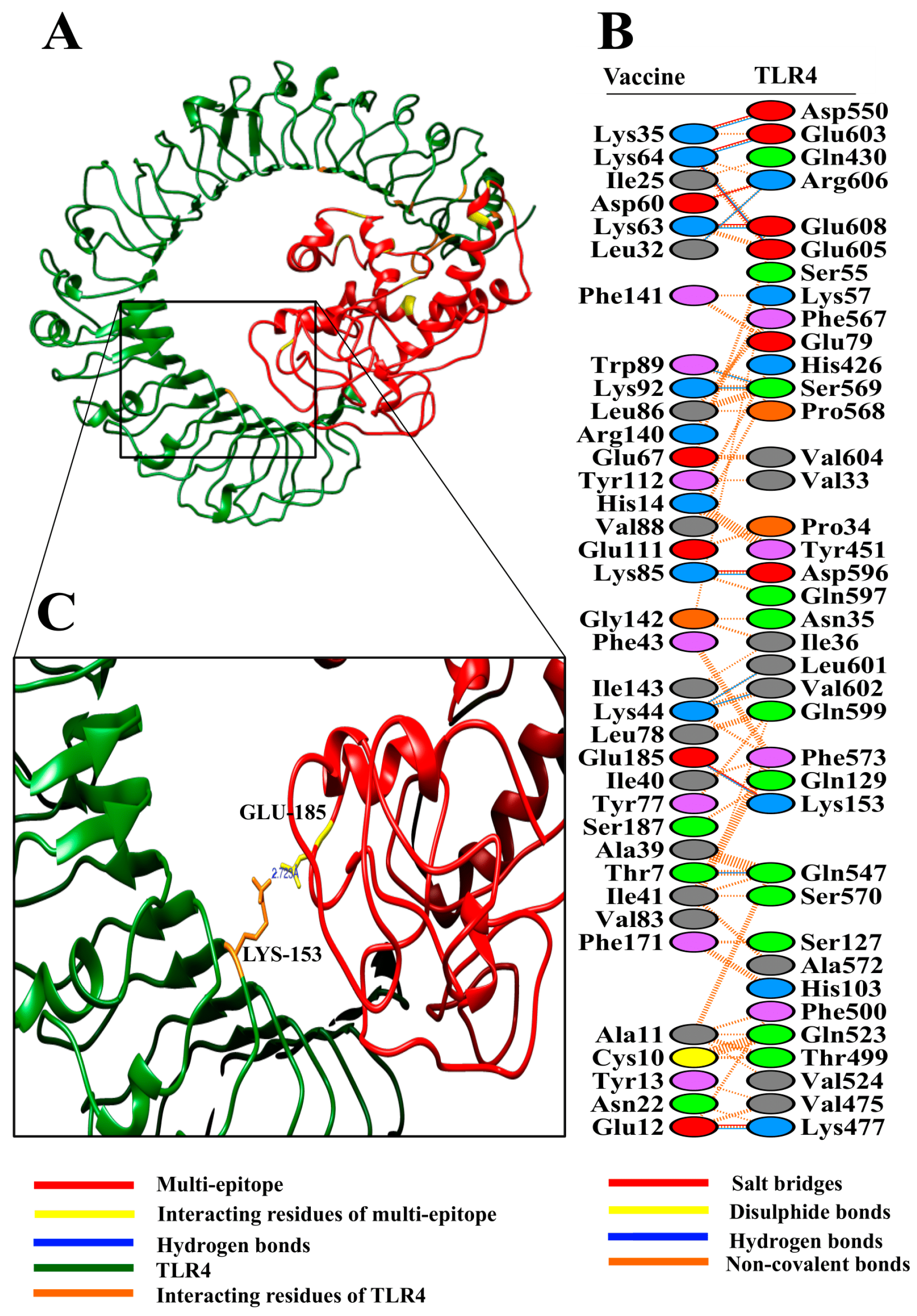{kind=link}
| Parameters | Value |
|---|---|
| HADDOCK score | −237.1 +/−3.3 |
| Cluster size | 20 |
| RMSD from the overall lowest-energy structure | 0.3 +/−0.2 |
| Van der Waals energy | −156.8 +/−1.9 |
| Electrostatic energy | −435.7 +/−19.0 |
| Desolvation energy | 6.8 +/−6.1 |
| Restraints violation energy | 0.0 +/−0.00 |
| Buried Surface Area | 4411.6 +/−24.1 |
| Z-Score | 0 |
| Parameters | Value |
|---|---|
| HADDOCK score | −235.6 +/−3.7 |
| Cluster size | 20 |
| RMSD from the overall lowest-energy structure | 0.3 +/−0.2 |
| Van der Waals energy | −121.2 +/−3.5 |
| Electrostatic energy | −517.3 +/−6.3 |
| Desolvation energy | −11.0 +/−4.1 |
| Restraints violation energy | 0.0 +/−0.00 |
| Buried Surface Area | 4009.3 +/−30.5 |
| Z-Score | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dar, H.A.; Zaheer, T.; Shehroz, M.; Ullah, N.; Naz, K.; Muhammad, S.A.; Zhang, T.; Ali, A. Immunoinformatics-Aided Design and Evaluation of a Potential Multi-Epitope Vaccine against Klebsiella Pneumoniae. Vaccines 2019, 7, 88. https://doi.org/10.3390/vaccines7030088
Dar HA, Zaheer T, Shehroz M, Ullah N, Naz K, Muhammad SA, Zhang T, Ali A. Immunoinformatics-Aided Design and Evaluation of a Potential Multi-Epitope Vaccine against Klebsiella Pneumoniae. Vaccines. 2019; 7(3):88. https://doi.org/10.3390/vaccines7030088
Chicago/Turabian StyleDar, Hamza Arshad, Tahreem Zaheer, Muhammad Shehroz, Nimat Ullah, Kanwal Naz, Syed Aun Muhammad, Tianyu Zhang, and Amjad Ali. 2019. "Immunoinformatics-Aided Design and Evaluation of a Potential Multi-Epitope Vaccine against Klebsiella Pneumoniae" Vaccines 7, no. 3: 88. https://doi.org/10.3390/vaccines7030088