
Immunogenic Subviral Particles Displaying Domain III of Dengue 2 Envelope Protein Vectored by Measles Virus

Abstract
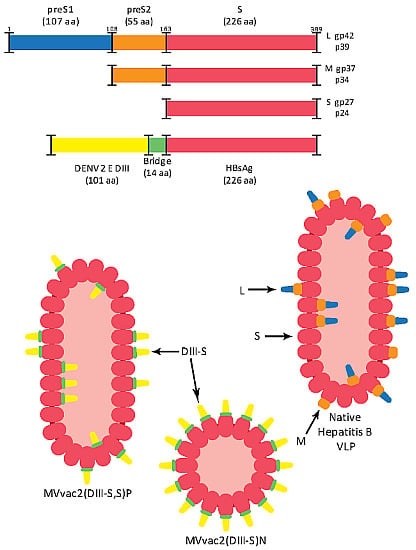
:
1. Introduction
2. Experimental Section
2.1. Cells and Viruses
2.2. Construction and Recovery of Recombinant MVs
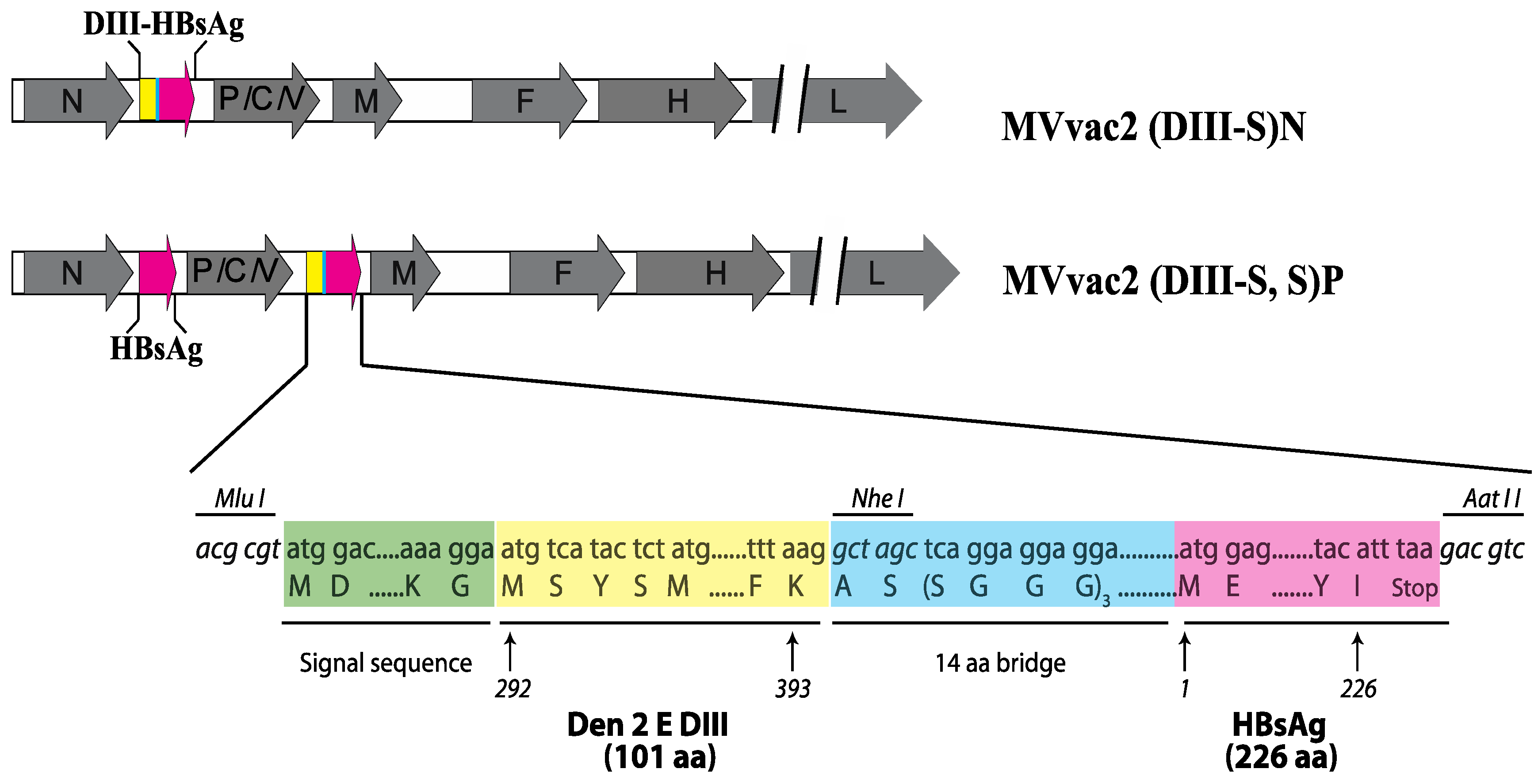
2.3. Protein Expression Analysis
2.4. Viral Particles Purification
2.5. Mouse Inoculations
2.6. Analysis of Immunogenicity
3. Results and Discussion
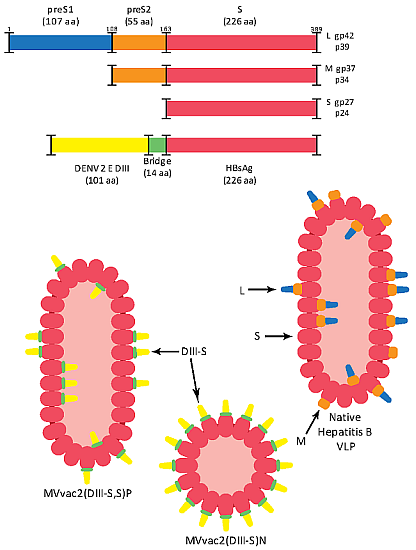
3.1. Design of Recombinant MVs Vectoring Hybrid s Secreting Hybrid DIII-HBsAg Forms
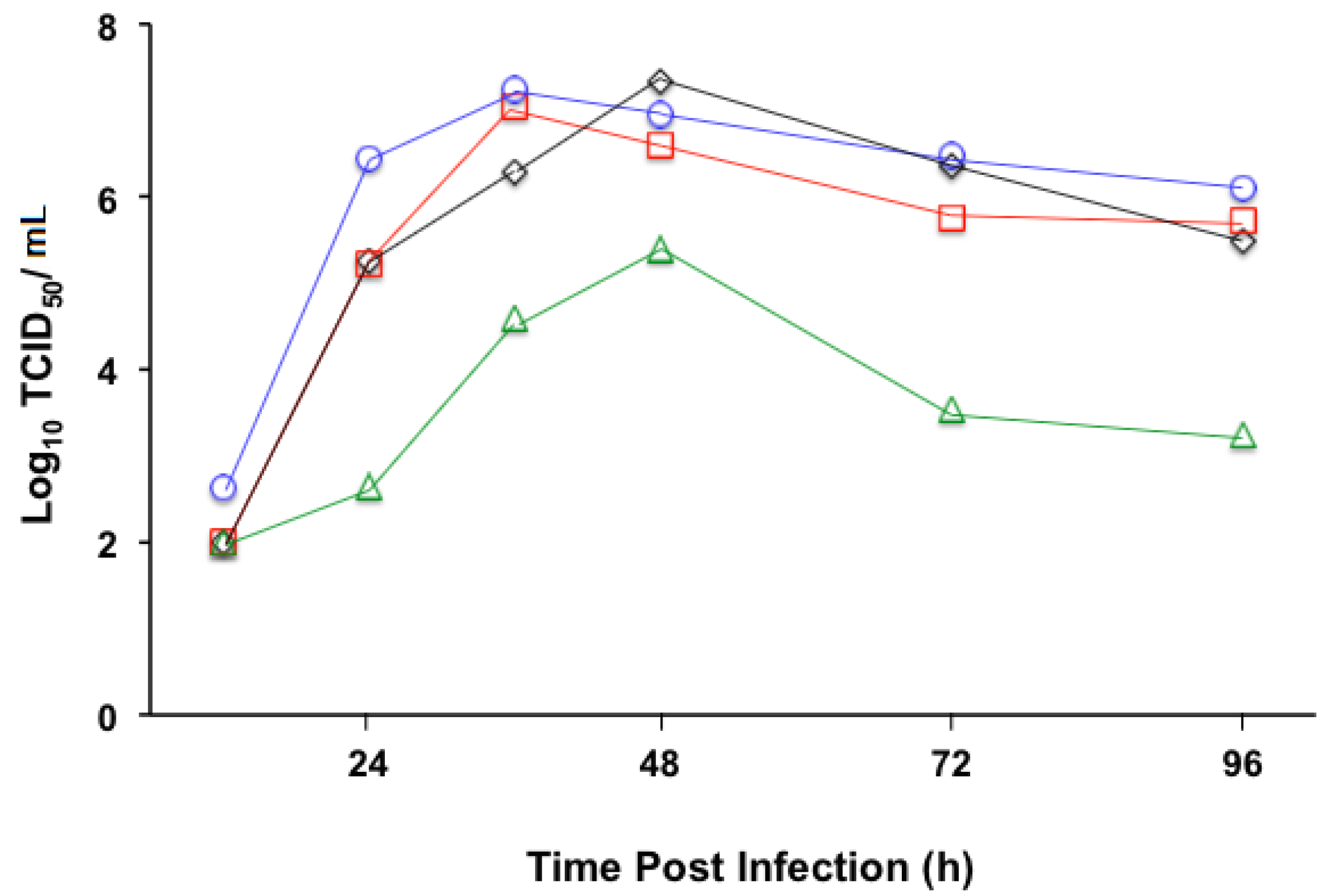
3.2. Replication Profile of Recombinant MV Vectors
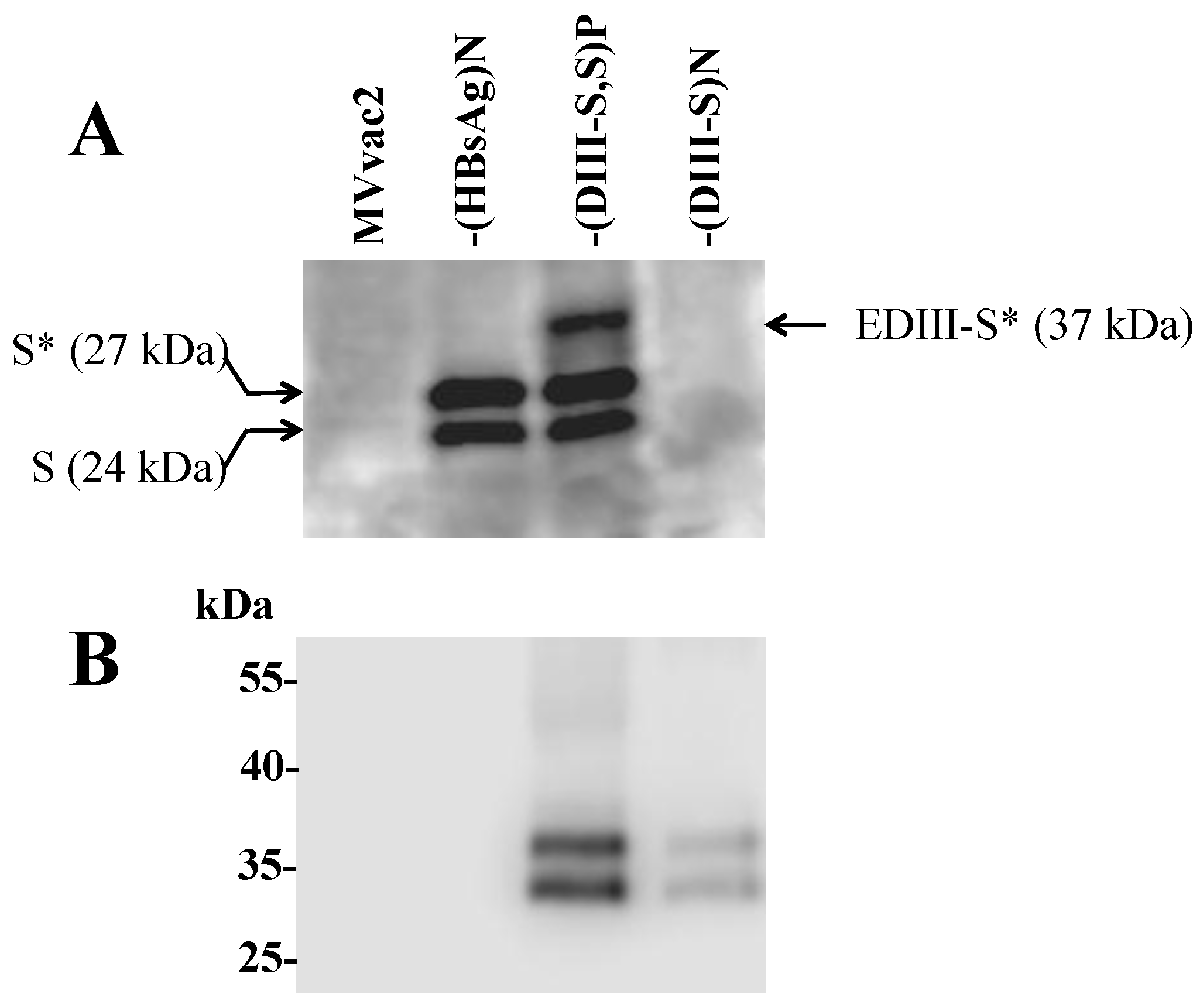
3.3. Expression of Hybrid DIII-HBsAg Antigens by MV Vectored Vaccines
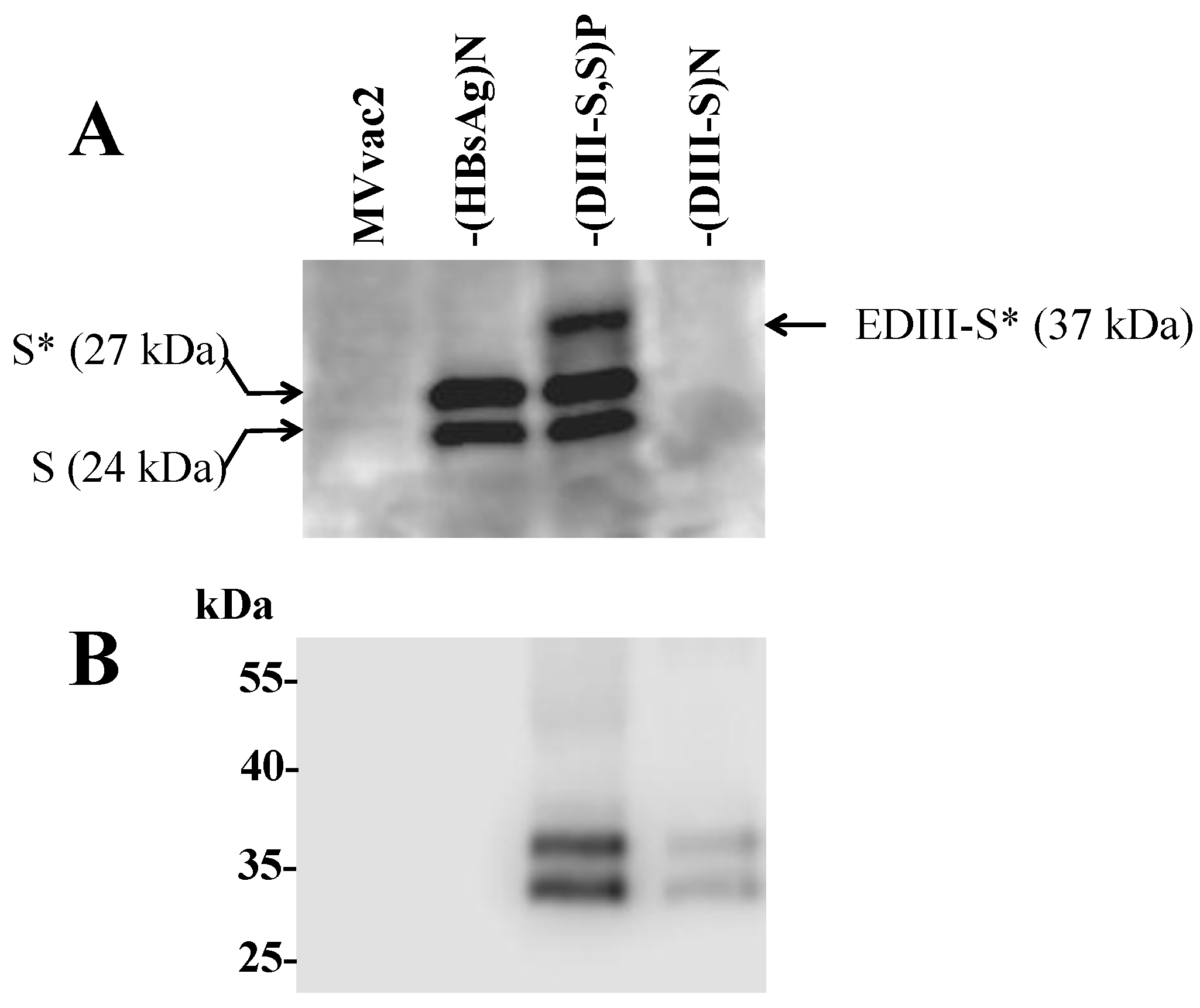
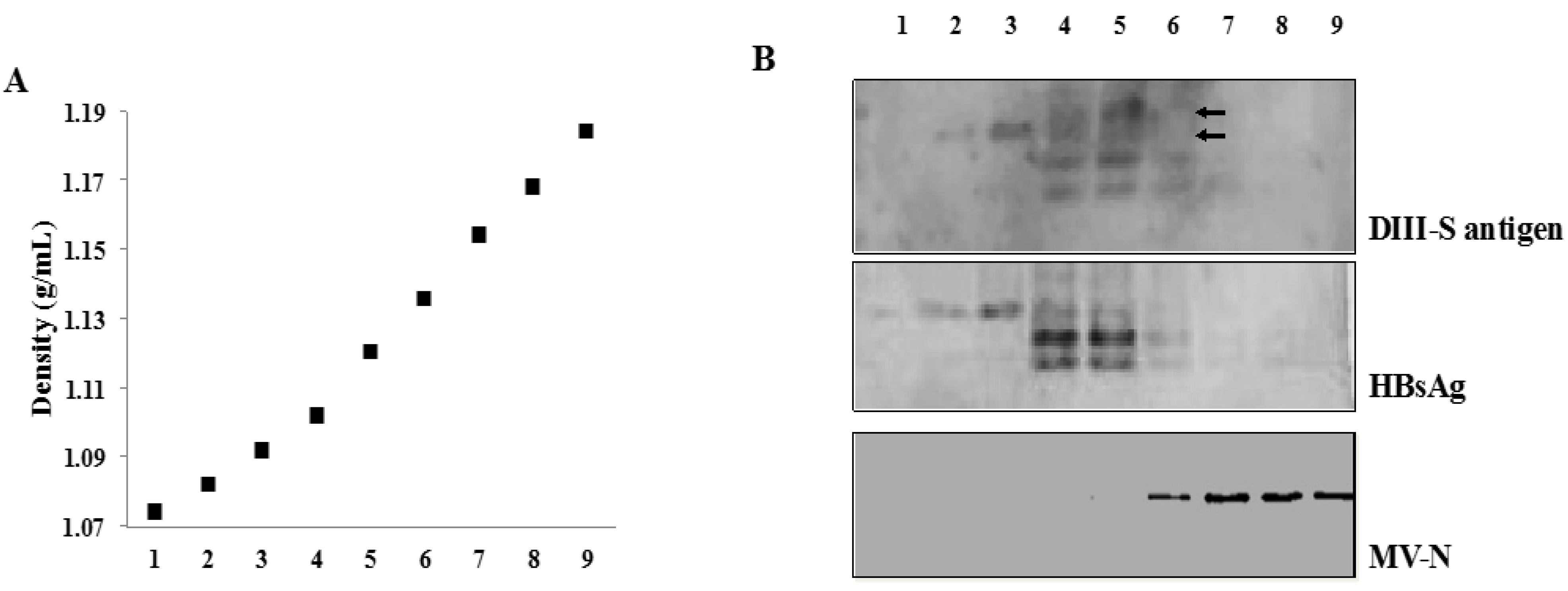
3.4. Secretion of Hybrid Virus-Like Particles with an HBsAg-Like Density
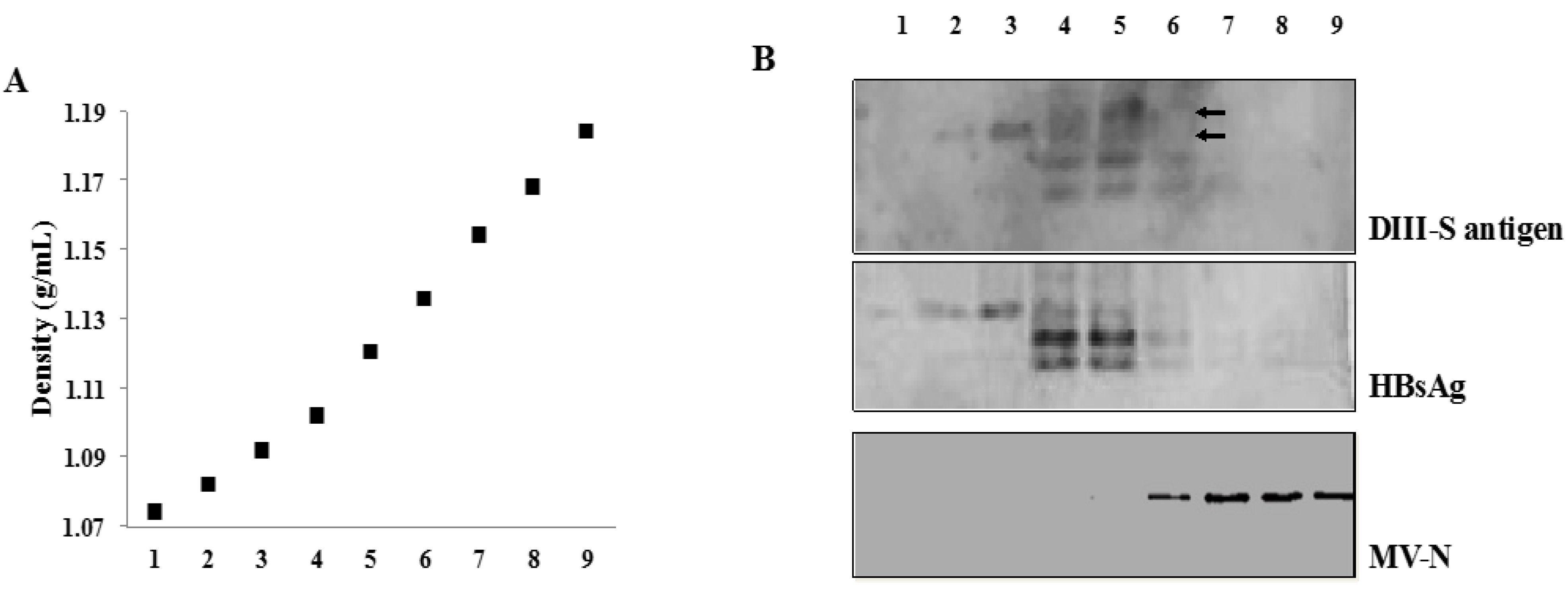
3.5. Immunogenicity of Recombinant MVs Vectoring Hybrid DIII-HBsAg Forms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MVvac2 X 1 a | -(HBsAg)N X 2 b | -(DIII-S, S)P X 1 a | -(DIII-S, S)P X 2 b | -(DIII-S)N X 2 b | |||||
|---|---|---|---|---|---|---|---|---|---|
| Pre | Post | Pre | Post | Pre | Post | Pre | Post | Pre | Post |
| <1:10 | 1:1280 | <1:10 | >1:2560 | <1:10 | 1:1066 | <1:10 | >1:2560 | <1:10 | >1:2560 |
| <1:10 | 1:1280 | <1:10 | >1:2560 | <1:10 | 1:640 | <1:10 | >1:2560 | <1:10 | 1:1280 |
| <1:10 | 1:1280 | <1:10 | >1:2560 | <1:10 | 1:320 | <1:10 | 1:1280 | <1:10 | 1:1066 |
| <1:10 | 1:640 | <1:10 | 1:240 | <1:10 | 1:1280 | ||||
| <1:10 | 1:640 | <1:10 | 1:640 | ||||||
| <1:10 | 1:640 | ||||||||
| <1:10 | 1:640 | ||||||||
| (DIII-S, S)P X 1 | (DIII-S, S)P X 2 | (DIII-S)N X 2 | |||
|---|---|---|---|---|---|
| LNI a | PRNT50 b | LNI | PRNT50 | LNI | PRNT50 |
| 0.64 | 1:10 | 1.41 | 1:160 | 0.21 | 1:20 |
| 0.81 | 1.51 | 0.33 | |||
| 0.64 | 1.51 | 0.21 | |||
| 0.51 | 1.81 | ||||
| 1.81 | |||||
| 0.51 | |||||
| 0.41 | |||||
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Messina, J.P.; Brady, O.J.; Scott, T.W.; Zou, C.; Pigott, D.M.; Duda, K.A.; Bhatt, S.; Katzelnick, L.; Howes, R.E.; Battle, K.E.; et al. Global spread of dengue virus types: Mapping the 70 year history. Trends Microbiol. 2014, 22, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Guy, B.; Guirakhoo, F.; Barban, V.; Higgs, S.; Monath, T.P.; Lang, J. Preclinical and clinical development of yfv 17d-based chimeric vaccines against dengue, west nile and japanese encephalitis viruses. Vaccine 2010, 28, 632–649. [Google Scholar] [CrossRef] [PubMed]
- Capeding, M.R.; Tran, N.H.; Hadinegoro, S.R.; Ismail, H.I.; Chotpitayasunondh, T.; Chua, M.N.; Luong, C.Q.; Rusmil, K.; Wirawan, D.N.; Nallusamy, R.; et al. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in asia: A phase 3, randomised, observer-masked, placebo-controlled trial. Lancet 2014, 384, 1358–1365. [Google Scholar] [CrossRef]
- Sabchareon, A.; Wallace, D.; Sirivichayakul, C.; Limkittikul, K.; Chanthavanich, P.; Suvannadabba, S.; Jiwariyavej, V.; Dulyachai, W.; Pengsaa, K.; Wartel, T.A.; et al. Protective efficacy of the recombinant, live-attenuated, cyd tetravalent dengue vaccine in thai schoolchildren: A randomised, controlled phase 2b trial. Lancet 2012, 380, 1559–1567. [Google Scholar] [CrossRef]
- Villar, L.; Dayan, G.H.; Arredondo-Garcia, J.L.; Rivera, D.M.; Cunha, R.; Deseda, C.; Reynales, H.; Costa, M.S.; Morales-Ramirez, J.O.; Carrasquilla, G.; et al. Efficacy of a tetravalent dengue vaccine in children in Latin America. N. Engl. J. Med. 2015, 372, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Roehrig, J.T. Antigenic structure of flavivirus proteins. Adv. Virus Res. 2003, 59, 141–175. [Google Scholar] [PubMed]
- Del Angel, R.M.; Reyes-del Valle, J. Dengue vaccines: Strongly sought but not a reality just yet. PLoS Pathog. 2013, 9, e1003551. [Google Scholar] [CrossRef] [PubMed]
- Rouvinski, A.; Guardado-Calvo, P.; Barba-Spaeth, G.; Duquerroy, S.; Vaney, M.C.; Kikuti, C.M.; Navarro Sanchez, M.E.; Dejnirattisai, W.; Wongwiwat, W.; Haouz, A.; et al. Recognition determinants of broadly neutralizing human antibodies against dengue viruses. Nature 2015, 520, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.J.; Hsieh, M.T.; Young, M.J.; Kao, C.L.; King, C.C.; Chang, W. An external loop region of domain III of dengue virus type 2 envelope protein is involved in serotype-specific binding to mosquito but not mammalian cells. J. Virol. 2004, 78, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Zidane, N.; Dussart, P.; Bremand, L.; Villani, M.E.; Bedouelle, H. Thermodynamic stability of domain iii from the envelope protein of flaviviruses and its improvement by molecular design. Protein Eng. Des. Sel. 2013, 26, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Crill, W.D.; Roehrig, J.T. Monoclonal antibodies that bind to domain III of dengue virus E glycoprotein are the most efficient blockers of virus adsorption to vero cells. J. Virol. 2001, 75, 7769–7773. [Google Scholar] [CrossRef] [PubMed]
- Sukupolvi-Petty, S.; Austin, S.K.; Purtha, W.E.; Oliphant, T.; Nybakken, G.E.; Schlesinger, J.J.; Roehrig, J.T.; Gromowski, G.D.; Barrett, A.D.; Fremont, D.H.; et al. Type- and subcomplex-specific neutralizing antibodies against domain iii of dengue virus type 2 envelope protein recognize adjacent epitopes. J. Virol. 2007, 81, 12816–12826. [Google Scholar] [CrossRef] [PubMed]
- Roehrig, J.T.; Bolin, R.A.; Kelly, R.G. Monoclonal antibody mapping of the envelope glycoprotein of the dengue 2 virus, jamaica. Virology 1998, 246, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Leggatt, G.R.; Frazer, I.H. Hpv vaccines: The beginning of the end for cervical cancer. Curr. Opin. Immunol. 2007, 19, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M.; Noble, S. Recombinant hepatitis B vaccine (engerix-b): A review of its immunogenicity and protective efficacy against hepatitis b. Drugs 2003, 63, 1021–1051. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.M.; McGovern, T.W.; Krzych, U.; Cohen, J.C.; Schneider, I.; LaChance, R.; Heppner, D.G.; Yuan, G.; Hollingdale, M.; Slaoui, M.; et al. Safety, immunogenicity, and efficacy of a recombinantly produced plasmodium falciparum circumsporozoite protein-hepatitis b surface antigen subunit vaccine. J. Infect. Dis. 1995, 171, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Scott Thomson, O.H. Allan Gould and Robert tindle Genetically modified hepatitis b surface antigen: A powerful vaccine technology for the delivery of disease-associated foreign antigens. Curr. Drug Ther. 2008, 3, 226–234. [Google Scholar] [CrossRef]
- Bisht, H.; Chugh, D.A.; Raje, M.; Swaminathan, S.S.; Khanna, N. Recombinant dengue virus type 2 envelope/hepatitis b surface antigen hybrid protein expressed in pichia pastoris can function as a bivalent immunogen. J. Biotechnol. 2002, 99, 97–110. [Google Scholar] [CrossRef]
- Puaux, A.L.; Marsac, D.; Prost, S.; Singh, M.K.; Earl, P.; Moss, B.; le Grand, R.; Riviere, Y.; Michel, M.L. Efficient priming of simian/human immunodeficiency virus (shiv)-specific t-cell responses with DNA encoding hybrid shiv/hepatitis b surface antigen particles. Vaccine 2004, 22, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Marth, K.; Breyer, I.; Focke-Tejkl, M.; Blatt, K.; Shamji, M.H.; Layhadi, J.; Gieras, A.; Swoboda, I.; Zafred, D.; Keller, W.; et al. A nonallergenic birch pollen allergy vaccine consisting of hepatitis pres-fused bet v 1 peptides focuses blocking igg toward ige epitopes and shifts immune responses to a tolerogenic and th1 phenotype. J. Immunol. 2013, 190, 3068–3078. [Google Scholar] [CrossRef] [PubMed]
- Niespodziana, K.; Focke-Tejkl, M.; Linhart, B.; Civaj, V.; Blatt, K.; Valent, P.; van Hage, M.; Gronlund, H.; Valenta, R. A hypoallergenic cat vaccine based on Fel d 1-derived peptides fused to hepatitis B Pres. J. Allergy Clin. Immunol. 2011, 127, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- RTS,S Clinical Trials Partnership. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in africa: Final results of a phase 3, individually randomised, controlled trial. The Lancet 2015. [Google Scholar] [CrossRef]
- Del Valle, J.R.; Devaux, P.; Hodge, G.; Wegner, N.J.; McChesney, M.B.; Cattaneo, R. A vectored measles virus induces hepatitis b surface antigen antibodies while protecting macaques against measles virus challenge. J. Virol. 2007, 81, 10597–10605. [Google Scholar] [CrossRef] [PubMed]
- Reyes-del Valle, J.; Hodge, G.; McChesney, M.B.; Cattaneo, R. Protective anti-hepatitis B virus responses in rhesus monkeys primed with a vectored measles virus and boosted with a single dose of hepatitis B surface antigen. J. Virol. 2009, 83, 9013–9017. [Google Scholar] [CrossRef] [PubMed]
- Mrkic, B.; Pavlovic, J.; Rulicke, T.; Volpe, P.; Buchholz, C.J.; Hourcade, D.; Atkinson, J.P.; Aguzzi, A.; Cattaneo, R. Measles virus spread and pathogenesis in genetically modified mice. J. Virol. 1998, 72, 7420–7427. [Google Scholar] [PubMed]
- Roscic-Mrkic, B.; Schwendener, R.A.; Odermatt, B.; Zuniga, A.; Pavlovic, J.; Billeter, M.A.; Cattaneo, R. Roles of macrophages in measles virus infection of genetically modified mice. J. Virol. 2001, 75, 3343–3351. [Google Scholar] [CrossRef] [PubMed]
- Ono, N.; Tatsuo, H.; Hidaka, Y.; Aoki, T.; Minagawa, H.; Yanagi, Y. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J. Virol. 2001, 75, 4399–4401. [Google Scholar] [CrossRef] [PubMed]
- Radecke, F.; Spielhofer, P.; Schneider, H.; Kaelin, K.; Huber, M.; Dotsch, C.; Christiansen, G.; Billeter, M.A. Rescue of measles viruses from cloned DNA. EMBO J. 1995, 14, 5773–5784. [Google Scholar] [PubMed]
- Parks, C.L.; Lerch, R.A.; Walpita, P.; Wang, H.P.; Sidhu, M.S.; Udem, S.A. Analysis of the noncoding regions of measles virus strains in the edmonston vaccine lineage. J. Virol. 2001, 75, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar]
- Calain, P.; Roux, L. The rule of six, a basic feature for efficient replication of sendai virus defective interfering RNA. J. Virol. 1993, 67, 4822–4830. [Google Scholar] [PubMed]
- Billeter, M.A.; Naim, H.Y.; Udem, S.A. Reverse genetics of measles virus and resulting multivalent recombinant vaccines: Applications of recombinant measles viruses. Curr. Top. Microbiol. Immunol. 2009, 329, 129–162. [Google Scholar] [PubMed]
- Bukreyev, A.; Skiadopoulos, M.H.; Murphy, B.R.; Collins, P.L. Nonsegmented negative-strand viruses as vaccine vectors. J. Virol. 2006, 80, 10293–10306. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Cattaneo, R.; Billeter, M.A. A recombinant measles virus expressing hepatitis b virus surface antigen induces humoral immune responses in genetically modified mice. J. Virol. 1999, 73, 4823–4828. [Google Scholar] [PubMed]
- Gerin, J.L.; Purcell, R.H.; Hoggan, M.D.; Holland, P.V.; Chanock, R.M. Biophysical properties of australia antigen. J. Virol. 1969, 4, 763–768. [Google Scholar] [PubMed]
- Mason, R.A.; Tauraso, N.M.; Spertzel, R.O.; Ginn, R.K. Yellow fever vaccine: Direct challenge of monkeys given graded doses of 17d vaccine. Appl. Microbiol. 1973, 25, 538–544. [Google Scholar]
- Brandler, S.; Lucas-Hourani, M.; Moris, A.; Frenkiel, M.-P.; Combredet, C.; Février, M.; Bedouelle, H.; Schwartz, O.; Desprès, P.; Tangy, F. Pediatric measles vaccine expressing a dengue antigen induces durable serotype-specific neutralizing antibodies to dengue virus. PLoS Negl. Trop. Dis. 2007. [Google Scholar] [CrossRef] [PubMed]
- Guirakhoo, F.; Weltzin, R.; Chambers, T.J.; Zhang, Z.X.; Soike, K.; Ratterree, M.; Arroyo, J.; Georgakopoulos, K.; Catalan, J.; Monath, T.P. Recombinant chimeric yellow fever-dengue type 2 virus is immunogenic and protective in nonhuman primates. J. Virol. 2000, 74, 5477–5485. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harahap-Carrillo, I.S.; Ceballos-Olvera, I.; Valle, J.R.-d. Immunogenic Subviral Particles Displaying Domain III of Dengue 2 Envelope Protein Vectored by Measles Virus. Vaccines 2015, 3, 503-518. https://doi.org/10.3390/vaccines3030503
Harahap-Carrillo IS, Ceballos-Olvera I, Valle JR-d. Immunogenic Subviral Particles Displaying Domain III of Dengue 2 Envelope Protein Vectored by Measles Virus. Vaccines. 2015; 3(3):503-518. https://doi.org/10.3390/vaccines3030503
Chicago/Turabian StyleHarahap-Carrillo, Indira S., Ivonne Ceballos-Olvera, and Jorge Reyes-del Valle. 2015. "Immunogenic Subviral Particles Displaying Domain III of Dengue 2 Envelope Protein Vectored by Measles Virus" Vaccines 3, no. 3: 503-518. https://doi.org/10.3390/vaccines3030503