
The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution

Abstract
: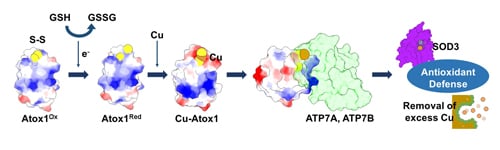
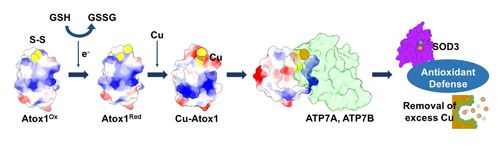{kind=link}
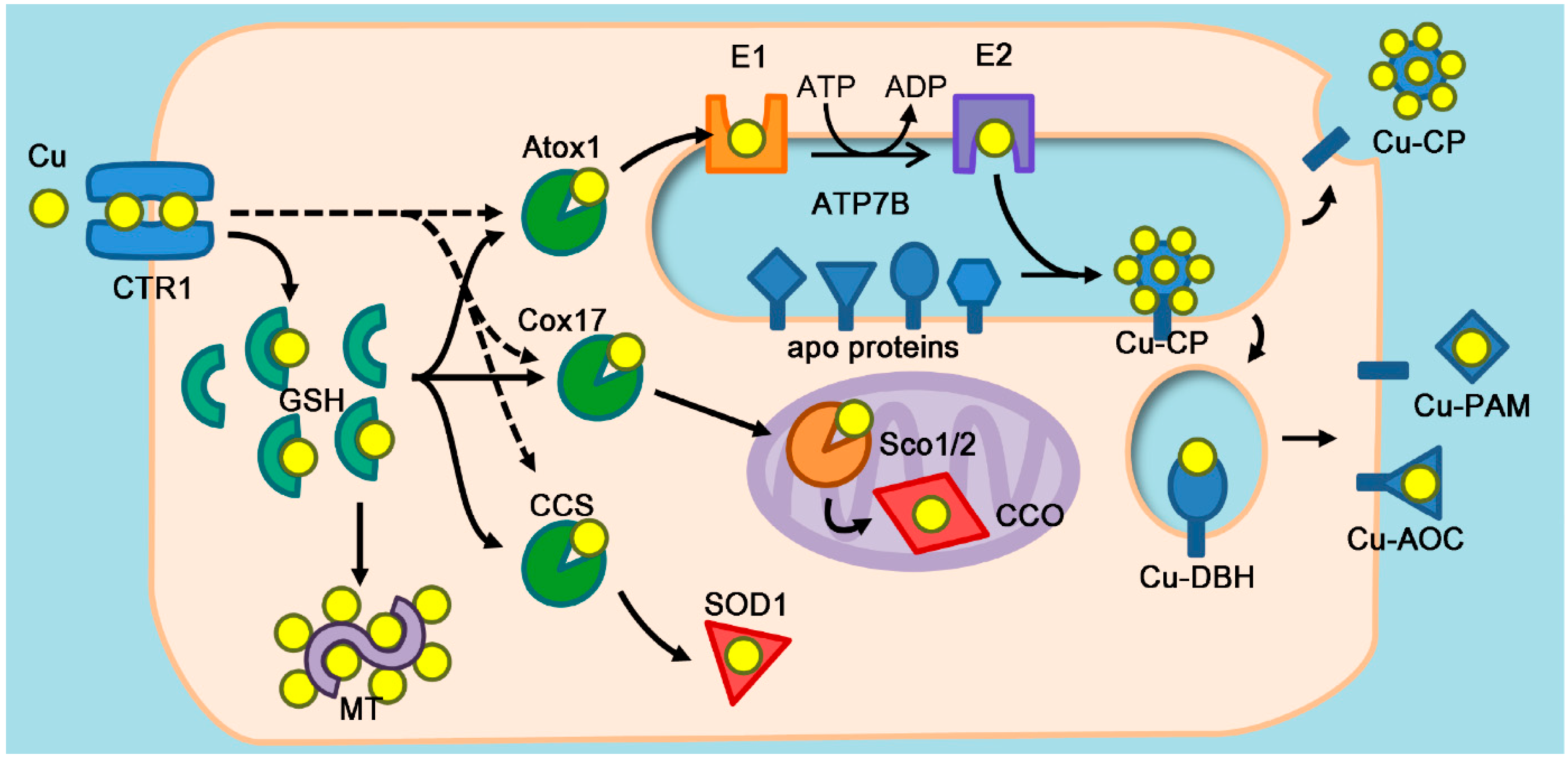{kind=link}
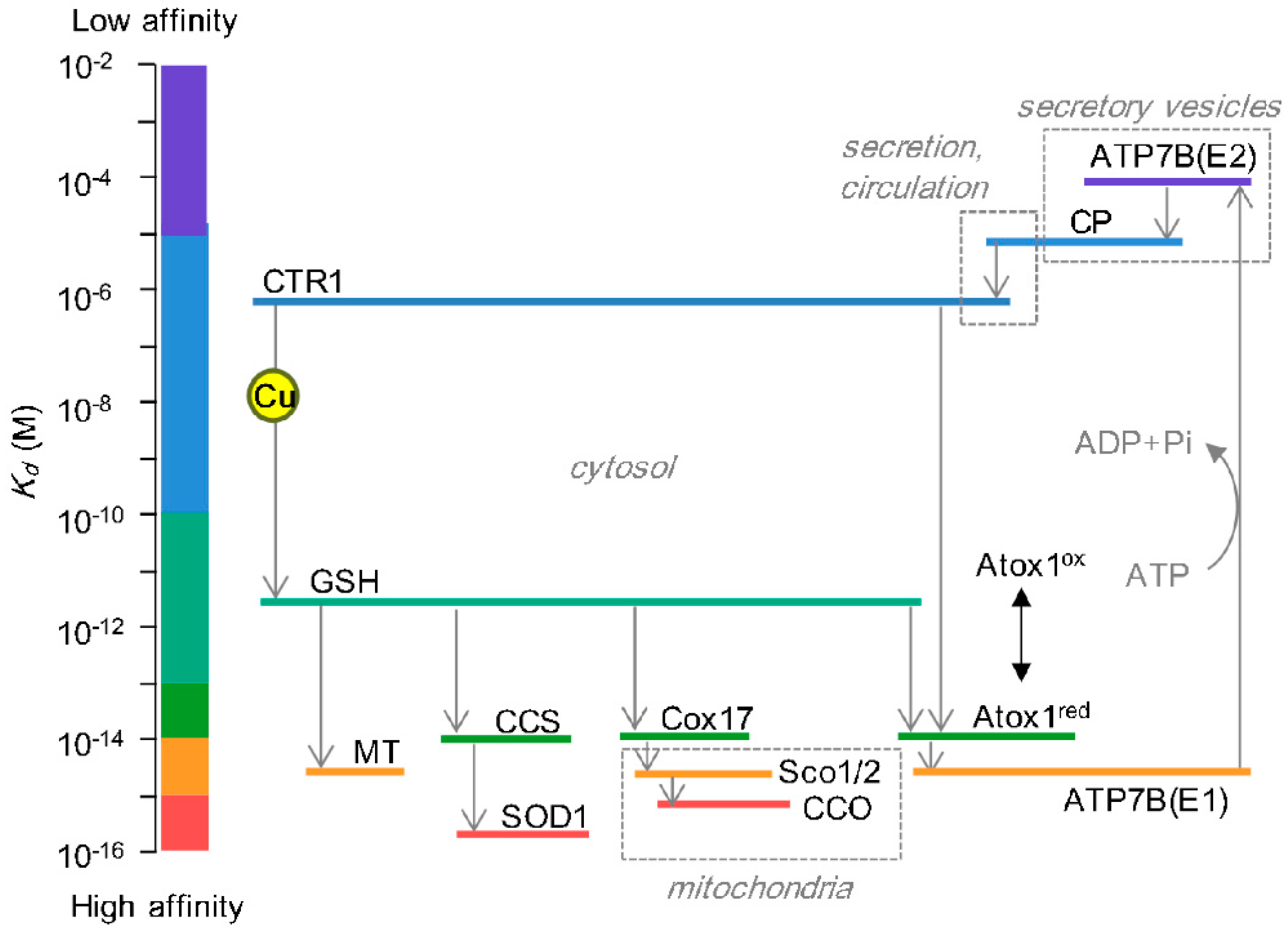{kind=link}
{kind=link}
{kind=link}
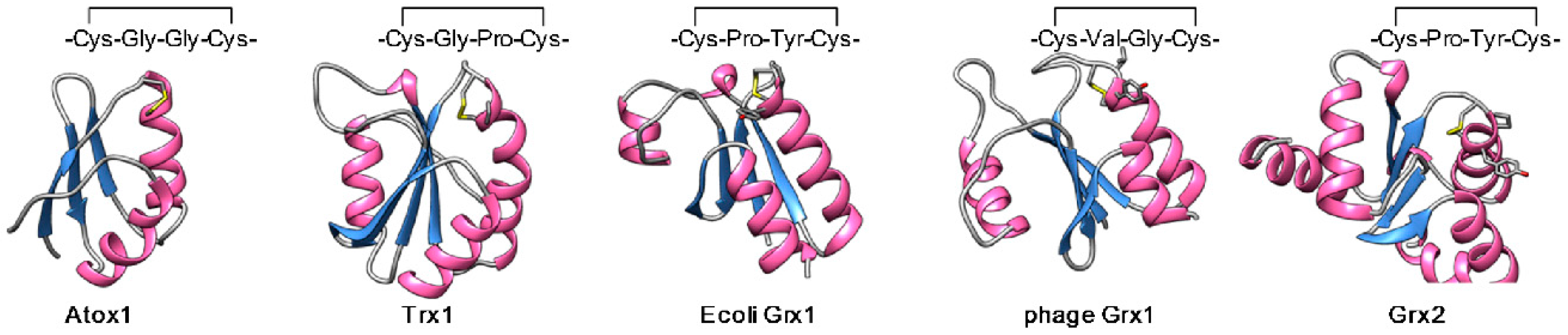{kind=link}
1. Introduction
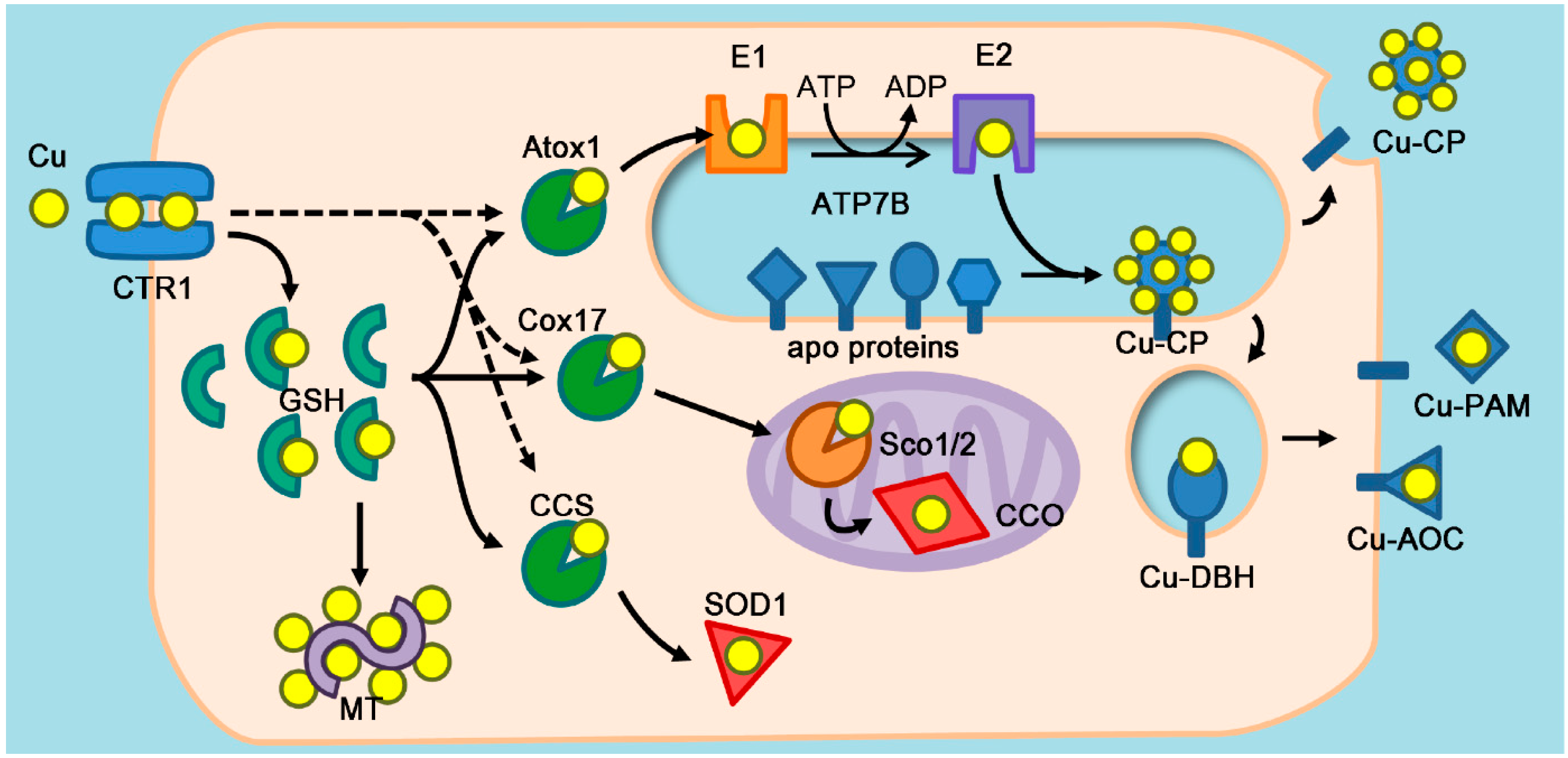
2. Atox1 as a Copper Chaperone
2.1. Copper Chaperones
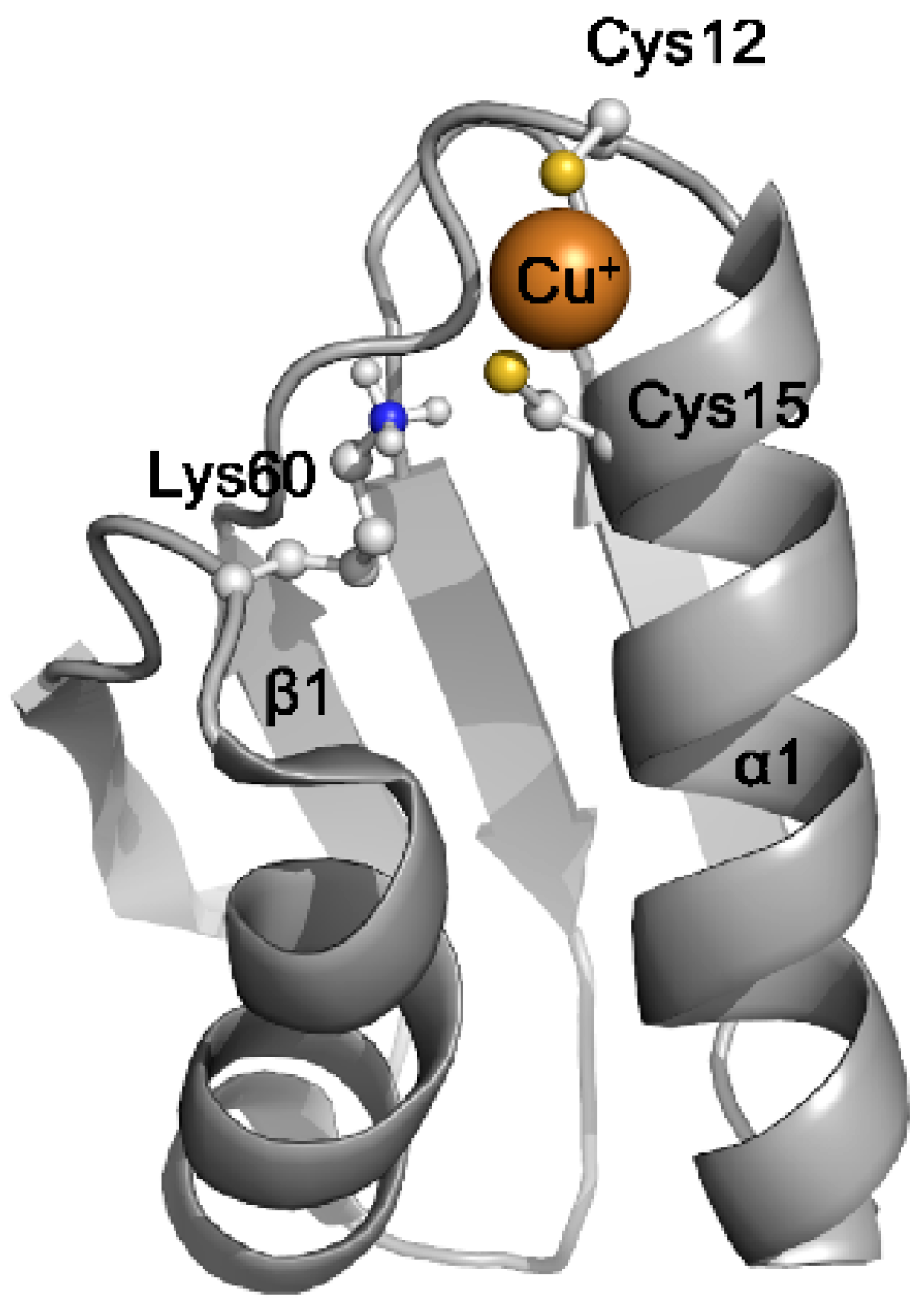
2.2. The Structural Basis of the Copper Chaperone Function of Atox1
2.3. Enzymes that Require Atox1 Function
2.4. Atox1 as a Calibrator of Cellular Copper Load
3. Antioxidant Role of Atox1
3.1. Atox1 Contributes to an Antioxidant Defense
3.2. The Proposed Mechanisms Underlying Antioxidant Role of Atox1
3.2.1. The SOD-like Activity
3.2.2. Transactivation of Antioxidant Genes
3.2.3. Coordination of Cellular Copper Distribution
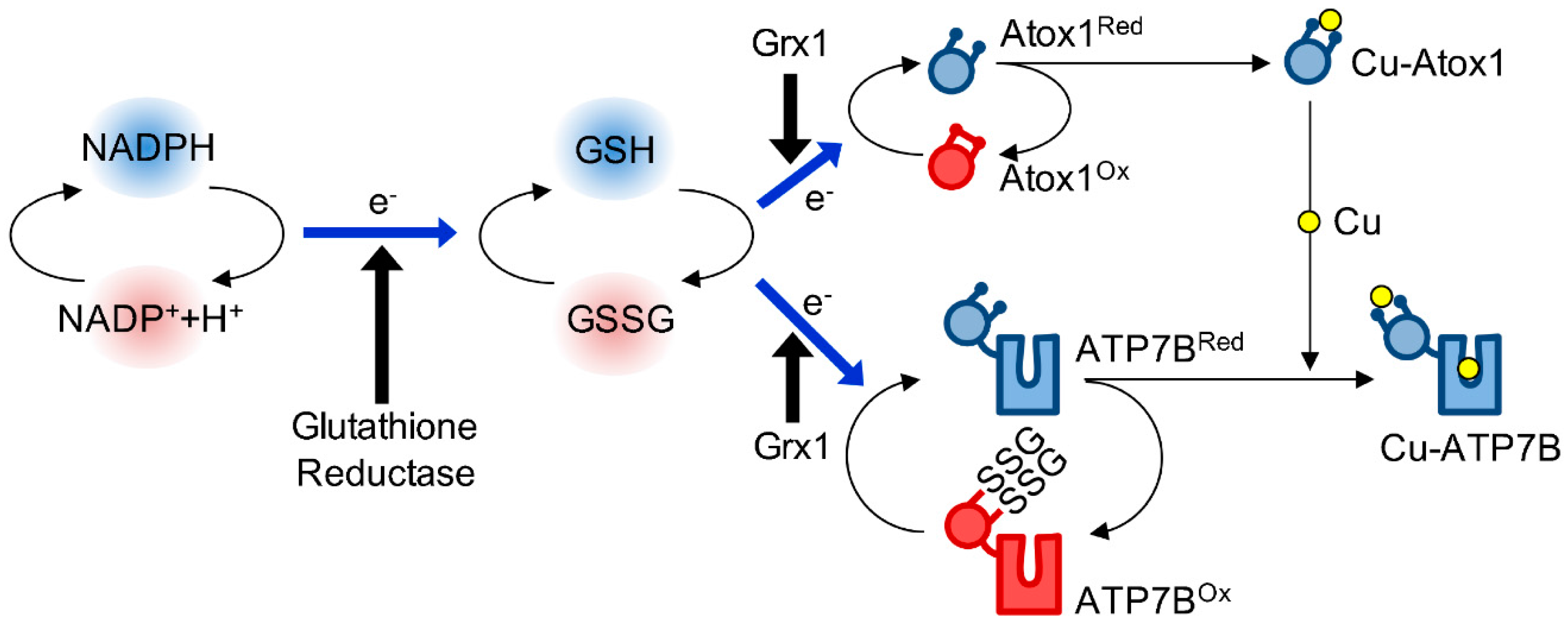
4. Redox Regulation of Atox1
4.1. The Biochemical Basis of Atox1 Redox Properties
4.2. The Redox Status of Atox1 in Cells
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kaler, S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Wooton-Kee, C.R.; Jain, A.K.; Wagner, M.; Grusak, M.A.; Finegold, M.J.; Lutsenko, S.; Moore, D.D. Elevated copper impairs hepatic nuclear receptor function in Wilson’s disease. J. Clin. Investig. 2015, 125, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Brose, J.; La Fontaine, S.; Wedd, A.G.; Xiao, Z. Redox sulfur chemistry of the copper chaperone Atox1 is regulated by the enzyme glutaredoxin 1, the reduction potential of the glutathione couple GSSG/2GSH and the availability of Cu(I). Metallomics 2014, 6, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Lutsenko, S. Human copper transporters: Mechanism, role in human diseases and therapeutic potential. Future Med. Chem. 2009, 1, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Culotta, V.C. The ATX1 gene of Saccharomyces cerevisiae encodes a small metal homeostasis factor that protects cells against reactive oxygen toxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Kelner, G.S.; Lee, M.; Clark, M.E.; Maciejewski, D.; McGrath, D.; Rabizadeh, S.; Lyons, T.; Bredesen, D.; Jenner, P.; Maki, R.A. The copper transport protein Atox1 promotes neuronal survival. J. Biol. Chem. 2000, 275, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Hatori, Y.; Clasen, S.; Hasan, N.M.; Barry, A.N.; Lutsenko, S. Functional partnership of the copper export machinery and glutathione balance in human cells. J. Biol. Chem. 2012, 287, 26678–26687. [Google Scholar] [CrossRef] [PubMed]
- Hatori, Y.; Yan, Y.; Schmidt, K.; Furukawa, E.; Hasan, N.M.; Yang, N.; Liu, C.-N.; Sockanathan, S.; Lutsenko, S. Neuronal differentiation is associated with a redox-regulated increase of copper flow to the secretory pathway. Nat. Commun. 2016, 7, 10640. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-F.; Sudhahar, V.; Youn, S.-W.; Das, A.; Cho, J.; Kamiya, T.; Urao, N.; McKinney, R.D.; Surenkhuu, B.; Hamakubo, T.; et al. Copper Transport Protein Antioxidant-1 Promotes Inflammatory Neovascularization via Chaperone and Transcription Factor Function. Sci. Rep. 2015, 5, 14780. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Thomassen, M.; Riley, C.H.; Kjær, L.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Skov, V. Whole blood transcriptional profiling reveals deregulation of oxidative and antioxidative defence genes in myelofibrosis and related neoplasms. Potential implications of downregulation of Nrf2 for genomic instability and disease progression. PLoS ONE 2014, 9, e112786. [Google Scholar] [CrossRef] [PubMed]
- Maryon, E.B.; Molloy, S.A.; Kaplan, J.H. Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am. J. Physiol. Cell Physiol. 2013, 304, C768–C779. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Kozyreva, T.; Zovo, K.; Palumaa, P. Affinity gradients drive copper to cellular destinations. Nature 2010, 465, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.C.; Girouard, J.B.; Ulloa, J.L.; Subramaniam, J.R.; Wong, P.C.; Valentine, J.S.; Culotta, V.C. Mechanisms for activating Cu- and Zn-containing superoxide dismutase in the absence of the CCS Cu chaperone. Proc. Natl. Acad. Sci. USA 2004, 101, 5964–5969. [Google Scholar] [CrossRef] [PubMed]
- Bamberg, E.; Schoner, W. The Sodium Pump: Structure Mechanism, Hormonal Control and Its Role in Disease; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Linse, S.; Helmersson, A.; Forsen, S. Calcium binding to calmodulin and its globular domains. J. Biol. Chem. 1991, 266, 8050–8054. [Google Scholar] [PubMed]
- Xiao, Z.; Brose, J.; Schimo, S.; Ackland, S.M.; La Fontaine, S.; Wedd, A.G. Unification of the copper(I) binding affinities of the metallo-chaperones Atx1, Atox1, and related proteins: Detection probes and affinity standards. J. Biol. Chem. 2011, 286, 11047–11055. [Google Scholar] [CrossRef] [PubMed]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Hasan, N.M.; Lutsenko, S. Regulation of copper transporters in human cells. Curr. Top. Membr. 2012, 69, 137–161. [Google Scholar] [PubMed]
- Flores, A.G.; Unger, V.M. Atox1 contains positive residues that mediate membrane association and aid subsequent copper loading. J. Membr. Biol. 2013, 246, 903–913. [Google Scholar] [CrossRef] [PubMed]
- De Feo, C.J.; Aller, S.G.; Siluvai, G.S.; Blackburn, N.J.; Unger, V.M. Three-dimensional structure of the human copper transporter hCTR1. Proc. Natl. Acad. Sci. USA 2009, 106, 4237–4242. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Hadjiloi, T.; Martinelli, M.; Palumaa, P. Mitochondrial copper(I) transfer from Cox17 to Sco1 is coupled to electron transfer. Proc. Natl. Acad. Sci. USA 2008, 105, 6803–6808. [Google Scholar] [CrossRef] [PubMed]
- Morgada, M.N.; Abriata, L.A.; Cefaro, C.; Gajda, K.; Banci, L.; Vila, A.J. Loop recognition and copper-mediated disulfide reduction underpin metal site assembly of CuA in human cytochrome oxidase. Proc. Natl. Acad. Sci. USA 2015, 112, 11771–11776. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Cantini, F.; Kozyreva, T.; Massagni, C.; Palumaa, P.; Rubino, J.T.; Zovo, K. Human superoxide dismutase 1 (hSOD1) maturation through interaction with human copper chaperone for SOD1 (hCCS). Proc. Natl. Acad. Sci. USA 2012, 109, 13555–13560. [Google Scholar] [CrossRef] [PubMed]
- Setty, S.R.G.; Tenza, D.; Sviderskaya, E.V.; Bennett, D.C.; Raposo, G.; Marks, M.S. Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes. Nature 2008, 454, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.W.; Peng, F.; Molloy, S.A.; Pendyala, V.S.; Muchenditsi, A.; Muzik, O.; Lee, J.; Kaplan, J.H.; Lutsenko, S. Urinary copper elevation in a mouse model of Wilson’s disease is a regulated process to specifically decrease the hepatic copper load. PLoS ONE 2012, 7, e38327. [Google Scholar] [CrossRef] [PubMed]
- Hatori, Y.; Majima, E.; Tsuda, T.; Toyoshima, C. Domain organization and movements in heavy metal ion pumps: Papain digestion of CopA, a Cu+-transporting ATPase. J. Biol. Chem. 2007, 282, 25213–25221. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W. Biology, structure and mechanism of P-type ATPases. Nat. Rev. Mol. Cell Biol. 2004, 5, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Kahra, D.; Kovermann, M.; Wittung-Stafshede, P. The C-Terminus of Human Copper Importer Ctr1 Acts as a Binding Site and Transfers Copper to Atox1. Biophys. J. 2016, 110, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zgirski, A.; Frieden, E. Binding of Cu(II) to non-prosthetic sites in ceruloplasmin and bovine serum albumin. J. Inorg. Biochem. 1990, 39, 137–148. [Google Scholar] [CrossRef]
- Rosenzweig, A.C.; Huffman, D.L.; Hou, M.Y.; Wernimont, A.K.; Pufahl, R.A.; O’Halloran, T. V Crystal structure of the Atx1 metallochaperone protein at 1.02 A resolution. Structure 1999, 7, 605–617. [Google Scholar] [CrossRef]
- Badarau, A.; Dennison, C. Copper trafficking mechanism of CXXC-containing domains: Insight from the pH-dependence of their Cu(I) affinities. J. Am. Chem. Soc. 2011, 133, 2983–2988. [Google Scholar] [CrossRef] [PubMed]
- Arnesano, F.; Banci, L.; Bertini, I.; Huffman, D.L.; O’Halloran, T. V Solution structure of the Cu(I) and apo forms of the yeast metallochaperone, Atx1. Biochemistry 2001, 40, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.H.; Casareno, R.L.; Labesse, G.; Mathews, F.S.; Gitlin, J.D. HAH1 is a copper-binding protein with distinct amino acid residues mediating copper homeostasis and antioxidant defense. J. Biol. Chem. 1998, 273, 1749–1754. [Google Scholar] [CrossRef] [PubMed]
- Hussain, F.; Olson, J.S.; Wittung-Stafshede, P. Conserved residues modulate copper release in human copper chaperone Atox1. Proc. Natl. Acad. Sci. USA 2008, 105, 11158–11163. [Google Scholar] [CrossRef] [PubMed]
- Anastassopoulou, I.; Banci, L.; Bertini, I.; Cantini, F.; Katsari, E.; Rosato, A. Solution structure of the apo and copper(I)-loaded human metallochaperone HAH1. Biochemistry 2004, 43, 13046–13053. [Google Scholar] [CrossRef] [PubMed]
- Hussain, F.; Rodriguez-Granillo, A.; Wittung-Stafshede, P. Lysine-60 in copper chaperone Atox1 plays an essential role in adduct formation with a target Wilson disease domain. J. Am. Chem. Soc. 2009, 131, 16371–16373. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Cantini, F.; Della-Malva, N.; Migliardi, M.; Rosato, A. The different intermolecular interactions of the soluble copper-binding domains of the menkes protein, ATP7A. J. Biol. Chem. 2007, 282, 23140–23146. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S.; Efremov, R.G.; Tsivkovskii, R.; Walker, J.M. Human copper-transporting ATPase ATP7B (the Wilson’s disease protein): Biochemical properties and regulation. J. Bioenerg. Biomembr. 2002, 34, 351–362. [Google Scholar] [CrossRef] [PubMed]
- González-Guerrero, M.; Argüello, J.M. Mechanism of Cu+-transporting ATPases: Soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites. Proc. Natl. Acad. Sci. USA 2008, 105, 5992–5997. [Google Scholar] [CrossRef] [PubMed]
- Gourdon, P.; Liu, X.-Y.; Skjørringe, T.; Morth, J.P.; Møller, L.B.; Pedersen, B.P.; Nissen, P. Crystal structure of a copper-transporting PIB-type ATPase. Nature 2011, 475, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Kwok, E.Y.; Severance, S.; Kosman, D.J. Evidence for iron channeling in the Fet3p-Ftr1p high-affinity iron uptake complex in the yeast plasma membrane. Biochemistry 2006, 45, 6317–6327. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.J.; Pufahl, R.; Dancis, A.; O’Halloran, T.V.; Culotta, V.C. A role for the Saccharomyces cerevisiae ATX1 gene in copper trafficking and iron transport. J. Biol. Chem. 1997, 272, 9215–9220. [Google Scholar] [PubMed]
- Bousquet-Moore, D.; Mains, R.E.; Eipper, B.A. Peptidylgycine α-amidating monooxygenase and copper: A gene-nutrient interaction critical to nervous system function. J. Neurosci. Res. 2010, 88, 2535–2545. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Urao, N.; Ashino, T.; Sudhahar, V.; McKinney, R.D.; Hamakubo, T.; Iwanari, H.; Ushio-Fukai, M.; Fukai, T. Novel role of copper transport protein antioxidant-1 in neointimal formation after vascular injury. Arterioscler Thromb Vasc. Biol. 2013, 33, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Ozumi, K.; Sudhahar, V.; Kim, H.W.; Chen, G.-F.; Kohno, T.; Finney, L.; Vogt, S.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Role of copper transport protein antioxidant 1 in angiotensin II-induced hypertension: A key regulator of extracellular superoxide dismutase. Hypertension 2012, 60, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S. Human copper homeostasis: A network of interconnected pathways. Curr. Opin. Chem. Biol. 2010, 14, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Hamza, I.; Faisst, A.; Prohaska, J.; Chen, J.; Gruss, P.; Gitlin, J.D. The metallochaperone Atox1 plays a critical role in perinatal copper homeostasis. Proc. Natl. Acad. Sci. USA 2001, 98, 6848–6852. [Google Scholar] [CrossRef] [PubMed]
- Hamza, I.; Prohaska, J.; Gitlin, J.D. Essential role for Atox1 in the copper-mediated intracellular trafficking of the Menkes ATPase. Proc. Natl. Acad. Sci. USA 2003, 100, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Miyayama, T.; Suzuki, K.T.; Ogra, Y. Copper accumulation and compartmentalization in mouse fibroblast lacking metallothionein and copper chaperone, Atox1. Toxicol. Appl. Pharmacol. 2009, 237, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Ralle, M.; Huster, D.; Vogt, S.; Schirrmeister, W.; Burkhead, J.L.; Capps, T.R.; Gray, L.; Lai, B.; Maryon, E.; Lutsenko, S. Wilson disease at a single cell level: Intracellular copper trafficking activates compartment-specific responses in hepatocytes. J. Biol. Chem. 2010, 285, 30875–30883. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Kim, H.W.; Nakagawa, O.; Ozumi, K.; Lessner, S.M.; Aoki, H.; Akram, K.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J. Biol. Chem. 2008, 283, 9157–9167. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Kambe, T.; Fulcher, Y.G.; Sachdev, S.W.; Bush, A.I.; Fritsche, K.; Lee, J.; Quinn, T.P.; Petris, M.J. Copper transport into the secretory pathway is regulated by oxygen in macrophages. J. Cell Sci. 2009, 122, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Cobine, P.A.; Nishimura, T.; Verdijk, R.M.; de Krijger, R.; de Coo, R.; Tarnopolsky, M.A.; Winge, D.R.; Shoubridge, E.A. COX19 mediates the transduction of a mitochondrial redox signal from SCO1 that regulates ATP7A-mediated cellular copper efflux. Mol. Biol. Cell 2013, 24, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.A.; Liou, L.-L.; Martins, J.; Clement, M.H.S.; Bailey, S.; Longo, V.D.; Valentine, J.S.; Gralla, E.B. Superoxide inhibits 4Fe-4S cluster enzymes involved in amino acid biosynthesis. Cross-compartment protection by CuZn-superoxide dismutase. J. Biol. Chem. 2004, 279, 32055–32062. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, C.; Shan, C.; You, Q.; Lu, J.; Elf, S.; Zhou, Y.; Wen, Y.; Vinkenborg, J.L.; Fan, J.; et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat. Chem. 2015, 7, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Peng, F. Knockdown of copper chaperone antioxidant-1 by RNA interference inhibits copper-stimulated proliferation of non-small cell lung carcinoma cells. Oncol. Rep. 2013, 30, 269–275. [Google Scholar] [PubMed]
- Liu, Y.-Y.; Nagpure, B.V.; Wong, P.T.-H.; Bian, J.-S. Hydrogen sulfide protects SH-SY5Y neuronal cells against d-galactose induced cell injury by suppression of advanced glycation end products formation and oxidative stress. Neurochem. Int. 2013, 62, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Hwang, I.K.; Yoo, D.Y.; Eum, W.S.; Kim, D.W.; Shin, M.J.; Ahn, E.H.; Jo, H.S.; Ryu, E.J.; Yong, J.I.; et al. Tat-antioxidant 1 protects against stress-induced hippocampal HT-22 cells death and attenuate ischaemic insult in animal model. J. Cell. Mol. Med. 2015, 19, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Sanokawa-Akakura, R.; Cao, W.; Allan, K.; Patel, K.; Ganesh, A.; Heiman, G.; Burke, R.; Kemp, F.W.; Bogden, J.D.; Camakaris, J.; et al. Control of Alzheimer’s amyloid beta toxicity by the high molecular weight immunophilin FKBP52 and copper homeostasis in Drosophila. PLoS ONE 2010, 5, e8626. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Caragounis, A.; Parker, S.J.; Meyerowitz, J.; La Fontaine, S.; Kanninen, K.M.; Perreau, V.M.; Crouch, P.J.; White, A.R. A potential copper-regulatory role for cytosolic expression of the DNA repair protein XRCC5. Free Radic. Biol. Med. 2011, 51, 2060–2072. [Google Scholar] [CrossRef] [PubMed]
- Portnoy, M.E.; Rosenzweig, A.C.; Rae, T.; Huffman, D.L.; O’Halloran, T.V.; Culotta, V.C. Structure-function analyses of the ATX1 metallochaperone. J. Biol. Chem. 1999, 274, 15041–15045. [Google Scholar] [CrossRef] [PubMed]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Jeney, V.; Itoh, S.; Wendt, M.; Gradek, Q.; Ushio-Fukai, M.; Harrison, D.G.; Fukai, T. Role of antioxidant-1 in extracellular superoxide dismutase function and expression. Circ. Res. 2005, 96, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Ozumi, K.; Kim, H.W.; Nakagawa, O.; McKinney, R.D.; Folz, R.J.; Zelko, I.N.; Ushio-Fukai, M.; Fukai, T. Novel mechanism for regulation of extracellular SOD transcription and activity by copper: Role of antioxidant-1. Free Radic. Biol. Med. 2009, 46, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-F.; Justice, A.E.; Melton, P.E. Longitudinal analytical approaches to genetic data. BMC Genet. 2016, 17 (Suppl. 2), 4. [Google Scholar] [CrossRef] [PubMed]
- Raps, S.P.; Lai, J.C.; Hertz, L.; Cooper, A.J. Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res. 1989, 493, 398–401. [Google Scholar] [CrossRef]
- Montes, S.; Rivera-Mancia, S.; Diaz-Ruiz, A.; Tristan-Lopez, L.; Rios, C. Copper and copper proteins in Parkinson’s disease. Oxid. Med. Cell. Longev. 2014, 2014, 147251. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Reyes, A.E.; Chacón, M.A.; Cerpa, W.; Villalón, A.; Montiel, J.; Merabachvili, G.; Aldunate, R.; Bozinovic, F.; Aboitiz, F. Human-like rodent amyloid-beta-peptide determines Alzheimer pathology in aged wild-type Octodon degu. Neurobiol. Aging 2005, 26, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, M.T. Neurologic Wilson’s disease. Ann. N. Y. Acad. Sci. 2010, 1184, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Fomenko, D.E.; Xing, W.; Adair, B.M.; Thomas, D.J.; Gladyshev, V.N. High-throughput identification of catalytic redox-active cysteine residues. Science 2007, 315, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Cater, M.A.; Materia, S.; Xiao, Z.; Wolyniec, K.; Ackland, S.M.; Yap, Y.W.; Cheung, N.S.; La Fontaine, S. Glutaredoxin1 protects neuronal cells from copper-induced toxicity. Biometals 2014, 27, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Singleton, W.C.J.; McInnes, K.T.; Cater, M.A.; Winnall, W.R.; McKirdy, R.; Yu, Y.; Taylor, P.E.; Ke, B.-X.; Richardson, D.R.; Mercer, J.F.B.; et al. Role of glutaredoxin1 and glutathione in regulating the activity of the copper-transporting P-type ATPases, ATP7A and ATP7B. J. Biol. Chem. 2010, 285, 27111–27121. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.M.; Cater, M.A.; Mercer, J.F.B.; La Fontaine, S. Copper-dependent interaction of glutaredoxin with the N termini of the copper-ATPases (ATP7A and ATP7B) defective in Menkes and Wilson diseases. Biochem. Biophys. Res. Commun. 2006, 348, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Bouldin, S.D.; Darch, M.A.; Hart, P.J.; Outten, C.E. Redox properties of the disulfide bond of human Cu,Zn superoxide dismutase and the effects of human glutaredoxin 1. Biochem. J. 2012, 446, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, E.; Barbieri, L.; Luchinat, E.; Banci, L. Direct structural evidence of protein redox regulation obtained by in-cell NMR. Biochim. Biophys. Acta 2016, 1863, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.-M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Kirlin, W.G.; Cai, J.; Thompson, S.A.; Diaz, D.; Kavanagh, T.J.; Jones, D.P. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic. Biol. Med. 1999, 27, 1208–1218. [Google Scholar] [CrossRef]
- Nkabyo, Y.S.; Ziegler, T.R.; Gu, L.H.; Watson, W.H.; Jones, D.P. Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G1352–G1359. [Google Scholar] [CrossRef] [PubMed]
- Yanes, O.; Clark, J.; Wong, D.M.; Patti, G.J.; Sánchez-Ruiz, A.; Benton, H.P.; Trauger, S.A.; Desponts, C.; Ding, S.; Siuzdak, G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 2010, 6, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Sakai, N.; Enokido, Y.; Uchiyama, Y.; Hatanaka, H. Survival factor-insensitive generation of reactive oxygen species induced by serum deprivation in neuronal cells. Brain Res. 1996, 733, 9–14. [Google Scholar] [CrossRef]
- Filomeni, G.; Ciriolo, M.R. Redox control of apoptosis: An update. Antioxid. Redox Signal. 2006, 8, 2187–2192. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatori, Y.; Lutsenko, S. The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution. Antioxidants 2016, 5, 25. https://doi.org/10.3390/antiox5030025
Hatori Y, Lutsenko S. The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution. Antioxidants. 2016; 5(3):25. https://doi.org/10.3390/antiox5030025
Chicago/Turabian StyleHatori, Yuta, and Svetlana Lutsenko. 2016. "The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution" Antioxidants 5, no. 3: 25. https://doi.org/10.3390/antiox5030025