Patient-Derived Stem Cell Models in SPAST HSP: Disease Modelling and Drug Discovery


Abstract
:1. Genetics of Hereditary Spastic Paraplegia (HSP)
2. Radiology of HSP
3. Histological Findings in HSP Patients and Animal Models
3.1. HSP Patients
3.2. Mouse Models
3.3. Other Animal Models
4. Patient-Derived Stem Cell Models in HSP
4.1. Induced Pluripotent Stem Cells
4.2. Adult Olfactory Stem Cells
4.3. Two Stem Cell Models Combined
4.4. Tubulin-Binding Drugs as Therapeutic Candidates
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Salinas, S.; Proukakis, C.; Crosby, A.; Warner, T. Hereditary spastic paraplegia: Clinical features and pathogenetic mechanisms. Lancet Neurol. 2008, 7, 1127–1138. [Google Scholar] [CrossRef]
- Hedera, P.; Eldevik, O.; Maly, P.; Rainier, S.; Fink, J. Spinal cord magnetic resonance imaging in autosomal dominant hereditary spastic paraplegia. Neuroradiology 2005, 47, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Tesson, C.; Koht, J.; Stevanin, G. Delving into the complexity of hereditary spastic paraplegias: How unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet. 2015, 134, 511–538. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.E. Classification of the hereditary ataxias and paraplegias. Lancet 1983, 321, 1151–1155. [Google Scholar] [CrossRef]
- Fink, J.K. The hereditary spastic paraplegias: Nine genes and counting. Arch. Neurol. 2003, 60, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Vandebona, H.; Kerr, N.; Liang, C.; Sue, C. Spast mutations in Australian patients with hereditary spastic paraplegia. Internal Med. J. 2012, 42, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, N.T.; Sun, Q.; Xue, M.; Zhang, B.; Zinn, K. Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2004, 2, e429. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, K.-U. An AAA family tree. J. Cell Sci. 2001, 114, 1601–1602. [Google Scholar] [PubMed]
- Errico, A.; Ballabio, A.; Rugarli, E. Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum. Mol. Genet. 2002, 11, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Zhu, P.-P.; Parker, R.L.; Blackstone, C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Investig. 2010, 120, 1097–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, C.; Orso, G.; Mancuso, G.; Herholz, M.; Gumeni, S.; Tadepalle, N.; Jüngst, C.; Tzschichholz, A.; Schauss, A.; Höning, S.; et al. Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet. 2015, 11, e1005149. [Google Scholar] [CrossRef] [PubMed]
- Basser, P.J.; Pierpaoli, C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J. Magn. Reson. 2011, 213, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Le Bihan, D.; Mangin, J.F.; Poupon, C.; Clark, CA.; Pappata, S.; Molko, N.; Chabriat, H. Diffusion tensor imaging: Concepts and applications. J. Magn. Reson. Imaging 2001, 13, 534–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, B. The basis of anisotropic water diffusion in the nervous system—A technical review. NMR Biomed. 2002, 15, 435–455. [Google Scholar]
- Rezende, T.J.R.; de Albuquerque, M.; Lamas, G.M.; Martinez, A.R.M.; Campos, B.M.; Casseb, R.F.; Silva, C.B.; Branco, L.M.T.; D’Abreu, A.; Lopes-Cendes, I.; et al. Multimodal MRI-based study in patients with spg4 mutations. PLoS ONE 2015, 10, e0117666. [Google Scholar] [CrossRef] [PubMed]
- Agosta, F.; Scarlato, M.; Spinelli, E.G.; Canu, E.; Benedetti, S.; Bassi, M.; Casali, C.; Sessa, M.; Copetti, M.; Pagani, E.; et al. Hereditary spastic paraplegia: Beyond clinical phenotypes toward a unified pattern of central nervous system damage. Radiology 2015, 276, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Aghakhanyan, G.; Martinuzzi, A.; Frijia, F.; Vavla, M.; Hlavata, H.; Baratto, A.; Martino, N.; Paparella, G.; Montanaro, D. Brain white matter involvement in hereditary spastic paraplegias: Analysis with multiple diffusion tensor indices. Am. J. Neuroradiol. 2014, 35, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Lindig, T.; Bender, B.; Hauser, T.-K.; Mang, S.; Schweikardt, D.; Klose, U.; Karle, K.N.; Schüle, R.; Schöls, L.; Rattay, T.W. Gray and white matter alterations in hereditary spastic paraplegia type spg4 and clinical correlations. J. Neurol. 2015, 262, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Oğuz, K.; Sanverdi, E.; Has, A.; Temuçin, Ç.; Türk, S.; Doerschner, K. Tract-based spatial statistics of diffusion tensor imaging in hereditary spastic paraplegia with thin corpus callosum reveals widespread white matter changes. Diagn. Interv. Radiol. 2013, 19, 181–186. [Google Scholar] [PubMed]
- Garaci, F.; Toschi, N.; Lanzafame, S.; Meschini, A.; Bertini, E.; Simonetti, G.; Santorelli, F.; Guerrisi, M.; Floris, R. Diffusion tensor imaging in SPG11- and SPG4-linked hereditary spastic paraplegia. Int. J. Neurosci. 2013, 124, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Unrath, A.; Müller, H.P.; Riecker, A.; Ludolph, A.C.; Sperfeld, A.D.; Kassubek, J. Whole brain-based analysis of regional white matter tract alterations in rare motor neuron diseases by diffusion tensor imaging. Hum. Brain Mapp. 2010, 31, 1727–1740. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, T.; Duning, T.; Schwan, A.; Lohmann, H.; Epplen, J.T.; Young, P. A novel form of autosomal recessive hereditary spastic paraplegia caused by a new SPG7 mutation. Neurology 2007, 69, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Duning, T.; Warnecke, T.; Schirmacher, A.; Schiffbauer, H.; Lohmann, H.; Mohammadi, S.; Young, P.; Deppe, M. Specific pattern of early white-matter changes in pure hereditary spastic paraplegia. Mov. Disord. 2010, 25, 1986–1992. [Google Scholar] [CrossRef] [PubMed]
- França, M.C.; Yasuda, C.L.; Pereira, F.R.S.; D’Abreu, A.; Lopes-Ramos, C.M.; Rosa, M.V.; Cendes, F.; Lopes-Cendes, I. White and grey matter abnormalities in patients with SPG11 mutations. J. Neurol. Neurosurg. Psychiatry 2012, 83, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Assaf, Y.; Pasternak, O. Diffusion tensor imaging (DTI)-based white matter mapping in brain research: A review. J. Mol. Neurosci. 2008, 34, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Fink, J. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [PubMed]
- Deluca, G.; Ebers, G.; Esiri, M. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol. Appl. Neurobiol. 2004, 30, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Denton, K.R.; Lei, L.; Grenier, J.; Rodionov, V.; Blackstone, C.; Li, X.-J. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells 2014, 32, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Havlicek, S.; Kohl, Z.; Mishra, H.; Prots, I.; Eberhardt, E.; Denguir, N.; Wend, H.; Plötz, S.; Boyer, L.; Marchetto, M.; et al. Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum. Mol. Genet. 2014, 23, 2527–2541. [Google Scholar] [CrossRef] [PubMed]
- McDermott, C.J.; Grierson, A.J.; Wood, J.D.; Bingley, M.; Wharton, S.B.; Bushby, K.M.D.; Shaw, P.J. Hereditary spastic paraparesis: Disrupted intracellular transport associated with spastin mutation. Ann. Neurol. 2003, 54, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Kasher, P.; De Vos, K.; Wharton, S.; Manser, C.; Bennett, E.; Bingley, M.; Wood, J.; Milner, R.; McDermott, C.; Miller, C.; et al. Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J. Neurochem. 2009, 110, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarrade, A.; Fassier, C.; Courageot, S.; Charvin, D.; Vitte, J.; Peris, L.; Thorel, A.; Mouisel, E.; Fonknechten, N.; Roblot, N.; et al. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum. Mol. Genet. 2006, 15, 3544–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orso, G.; Martinuzzi, A.; Rossetto, M.; Sartori, E.; Feany, M.; Daga, A. Disease-related phenotypes in a drosophila model of hereditary spastic paraplegia are ameliorated by treatment with vinblastine. J. Clin. Investig. 2005, 115, 3026–3034. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.D.; Landers, J.A.; Bingley, M.; McDermott, C.J.; Thomas-McArthur, V.; Gleadall, L.J.; Shaw, P.J.; Cunliffe, V.T. The microtubule-severing protein spastin is essential for axon outgrowth in the zebrafish embryo. Hum. Mol. Genet. 2006, 15, 2763–2771. [Google Scholar] [CrossRef] [PubMed]
- Trotta, N.; Orso, G.; Rossetto, M.; Daga, A.; Broadie, K. The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr. Biol. 2004, 14, 1135–1147. [Google Scholar] [CrossRef] [PubMed]
- Munos, B. Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discov. 2009, 8, 959. [Google Scholar] [CrossRef] [PubMed]
- Mackay-Sim, A. Concise review: Patient-derived olfactory stem cells: New models for brain diseases. Stem Cells 2012, 30, 2361–2365. [Google Scholar] [CrossRef] [PubMed]
- Mackay-Sim, A. Patient-derived stem cells: Pathways to drug discovery for brain diseases. Front. Cell. Neurosci. 2013, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Matigian, N.A.; McCurdy, R.D.; Féron, F.; Perry, C.; Smith, H.; Filippich, C.; McLean, D.; McGrath, J.; Mackay-Sim, A.; Mowry, B.; et al. Fibroblast and lymphoblast gene expression profiles in schizophrenia: Are non-neural cells informative? PLoS ONE 2008, 3, e2412. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2008, 457, 277. [Google Scholar] [CrossRef] [PubMed]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling familial alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef] [PubMed]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell. Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kirwan, P.; Smith, J.; Robinson, H.P.C.; Livesey, F.J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 2012, 15. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Jaenisch, R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell 2009, 5, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.-L.; Young, J.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assou, S.; Bouckenheimer, J.; Vos, J. Concise review: Assessing the genome integrity of human induced pluripotent stem cells: What quality control metrics? Stem Cells 2018, 36, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, V.; Orlando, L.; Giachino, C. Induced pluripotent stem cells: Advances in the quest for genetic stability during reprogramming process. Int. J. Mol. Sci. 2017, 18, 1952. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.-Y.; Weick, J.P.; Yu, J.; Ma, L.-X.; Zhang, X.-Q.; Thomson, J.A.; Zhang, S.-C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgmann-Winter, K.; Willard, S.L.; Sinclair, D.; Mirza, N.; Turetsky, B.; Berretta, S.; Hahn, C.G. Translational potential of olfactory mucosa for the study of neuropsychiatric illness. Transl. Psychiatry 2015, 5, e527. [Google Scholar] [CrossRef] [PubMed]
- Féron, F.; Perry, C.; McGrath, J.J.; Mackay-Sim, A. New techniques for biopsy and culture of human olfactory epithelial neurons. Arch. Otolaryngol.–Head Neck Surg. 1998, 124, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Murrell, W.; Féron, F.; Wetzig, A.; Cameron, N.; Splatt, K.; Bellette, B.; Bianco, J.; Perry, C.; Lee, G.; Mackay-Sim, A. Multipotent stem cells from adult olfactory mucosa. Dev. Dyn. 2005, 233, 496–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Féron, F.; Perry, C.; Girard, S.D.; Mackay-Sim, A. Isolation of adult stem cells from the human olfactory mucosa. In Neural Progenitor Cells: Methods and Protocols; Reynolds, A.B., Deleyrolle, P.L., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 107–114. [Google Scholar]
- Matigian, N.; Abrahamsen, G.; Sutharsan, R.; Cook, A.L.; Vitale, A.M.; Nouwens, A.; Bellette, B.; An, J.; Anderson, M.; Beckhouse, A.G.; et al. Disease-specific, neurosphere-derived cells as models for brain disorders. Dis. Models Mech. 2010, 3, 785–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Wali, G.; Sutharsan, R.; Bellette, B.; Crane, D.I.; Sue, C.M.; Mackay-Sim, A. Low dose tubulin-binding drugs rescue peroxisome trafficking deficit in patient-derived stem cells in hereditary spastic paraplegia. Biol. Open 2014, 3, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human atp13a2 (park9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Kozlov, S.; Matigan, N.; Wali, G.; Gatei, M.; Sutharsan, R.; Bellette, B.; Wraith-Kijas, A.; Cochrane, J.; Coulthard, M.; et al. A patient-derived olfactory stem cell disease model for ataxia-telangiectasia. Hum. Mol. Genet. 2013, 22, 2495–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahamsen, G.; Fan, Y.; Matigian, N.; Wali, G.; Bellette, B.; Sutharsan, R.; Raju, J.; Wood, S.; Veivers, D.; Sue, C.; et al. A patient-derived stem cell model of hereditary spastic paraplegia with spast mutations. Dis. Models Mech. 2013, 6, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Wali, G.; Sutharsan, R.; Fan, Y.; Stewart, R.; Tello Velasquez, J.; Sue, C.M.; Crane, D.I.; Mackay-Sim, A. Mechanism of impaired microtubule-dependent peroxisome trafficking and oxidative stress in spast-mutated cells from patients with hereditary spastic paraplegia. Sci. Rep. 2016, 6, 27004. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.S.; Lepleux, M.; Makhlouf, M.; Martin, C.; Fregeac, J.; Siquier-Pernet, K.; Philippe, A.; Feron, F.; Gepner, B.; Rougeulle, C.; et al. Profiling olfactory stem cells from living patients identifies miRNAs relevant for autism pathophysiology. Mol. Autism 2016, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boone, N.; Loriod, B.; Bergon, A.; Sbai, O.; Formisano-Tréziny, C.; Gabert, J.; Khrestchatisky, M.; Nguyen, C.; Féron, F.; Axelrod, F.B.; et al. Olfactory stem cells, a new cellular model for studying molecular mechanisms underlying familial dysautonomia. PLoS ONE 2010, 5, e15590. [Google Scholar] [CrossRef] [PubMed]
- Guzik, B.; Goldstein, L. Microtubule-dependent transport in neurons: Steps towards an understanding of regulation, function and dysfunction. Curr. Opin. Cell Biol. 2004, 16, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Wiemer, E.A.C.; Wenzel, T.; Deerinck, T.J.; Ellisman, M.H.; Subramani, S. Visualization of the peroxisomal compartment in living mammalian cells: Dynamic behavior and association with microtubules. J. Cell Biol. 1997, 136, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.; Grunau, S.; Ohlmeier, S.; Hiltunen, J. Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 2010, 13, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Yoon, Y. Mitochondria and peroxisomes: Are the ‘big brother’ and the ‘little sister’ closer than assumed? Bioessays 2007, 29, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Molon, A.; Di Giovanni, S.; Chen, Y.W.; Clarkson, P.M.; Angelini, C.; Pegoraro, E.; Hoffman, E.P. Large-scale disruption of microtubule pathways in morphologically normal human spastin muscle. Neurology 2004, 62, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Denis, I.C. Revisiting the neuropathogenesis of zellweger syndrome. Neurochem. Int. 2014, 69, 18. [Google Scholar]
- Barry, D.; O’Keeffe, G. Peroxisomes: The neuropathological consequences of peroxisomal dysfunction in the developing brain. Int. J. Biochem. Cell Biol. 2013, 45, 2012–2015. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.J.; Dodt, G.; Raymond, G.V.; Braverman, N.E.; Moser, A.B.; Moser, H.W. Peroxisome biogenesis disorders. Biochim. Biophys. Acta 2006, 1763, 1733–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.R.; Wali, G.; Davis, R.L.; Mallawaarachchi, A.C.; Palmer, E.E.; Gayeskiy, V.; Minoche, A.; Vievers, D.; Mackay-Sim, A.; Cowley, M.; et al. Expanding the spectrum of PEX16 mutations and novel insights into disease mechanisms. Mol. Genet. Metab. Rep. 2018, 16, 46–51. [Google Scholar] [CrossRef]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atorino, L.; Silvestri, L.; Koppen, M.; Cassina, L.; Ballabio, A.; Marconi, R.; Langer, T.; Casari, G. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J. Cell Biol. 2003, 163, 777–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Kevin, J.B.; Colin, L.M.; Ashley, I.B. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar]
- Mertsch, K.; Blasig, I.; Grune, T. 4-hydroxynonenal impairs the permeability of an in vitro rat blood–brain barrier. Neurosci. Lett. 2001, 314, 135–138. [Google Scholar] [CrossRef]
- Wali, G.; Recasens, A.; Blair, N.F.; Sutharsan, R.; Park, J.-S.; Mackay-Sim, A.; Sue, C.M. Enhanced axon degeneration in Hereditary Spastic Paraplegia patient-derived neurons is reversed by tubulin-binding drugs. under review.
- Saxton, W.M.; Hollenbeck, P.J. The axonal transport of mitochondria. J. Cell Sci. 2012, 125, 2095–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, S.; Weber, K. Post-translational modifications regulate microtubule function. Nat. Rev. Mol. Cell Biol. 2003, 4, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Owens, G.C.; Makarenkova, H.; Edelman, D.B. Hdac6 regulates mitochondrial transport in hippocampal neurons. PLoS ONE 2010, 5, e10848. [Google Scholar] [CrossRef] [PubMed]
- Checchi, P.M.; Nettles, J.H.; Zhou, J.; Snyder, J.P.; Joshi, H.C. Microtubule-interacting drugs for cancer treatment. Trends Pharmacol. Sci. 2003, 24, 361–365. [Google Scholar] [CrossRef]
- Amos, L.A. What tubulin drugs tell us about microtubule structure and dynamics. Semin. Cell Dev. Biol. 2011, 22, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Gigant, B.; Wang, C.; Ravelli, R.B.G.; Roussi, F.; Steinmetz, M.O.; Curmi, P.A.; Sobel, A.; Knossow, M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gupta, K.; Aggarwal, S.; Aneja, R.; Chandra, R.; Panda, D.; Joshi, H.C. Brominated derivatives of noscapine are potent microtubule-interfering agents that perturb mitosis and inhibit cell proliferation. Mol. Pharmacol. 2003, 63, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Fassier, C.; Tarrade, A.; Peris, L.; Courageot, S.; Mailly, P.; Dalard, C.; Delga, S.; Roblot, N.; Lefèvre, J.; Job, D.; et al. Microtubule-targeting drugs rescue axonal swellings in cortical neurons from spastin knockout mice. Dis. Models Mech. 2013, 6, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Millecamps, S.; Julien, J.-P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Chevalier-Larsen, E.; Holzbaur, E. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 2006, 1762, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.; Grierson, A.; Ackerley, S.; Miller, C. Role of axonal transport in neurodegenerative diseases. Ann. Rev. Neurosci. 2008, 31, 151–173. [Google Scholar] [CrossRef] [PubMed]
- Nordgren, M.; Fransen, M. Peroxisomal metabolism and oxidative stress. Biochimie 2013, 98, 56–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ros/rns-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Axiak-Bechtel, S.; Kumar, S.; Dank, K.; Clarkson, N.; Selting, K.; Bryan, J.; Rosol, T.; Espinosa, J.; Decedue, C. Nanoparticulate paclitaxel demonstrates antitumor activity in PC3 and Ace-1 aggressive prostate cancer cell lines. Investig. New Drugs 2013, 31, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Zhang, B.; Carroll, J.; Yao, Y.; Potuzak, J.S.; Hogan, A.-M.L.; Iba, M.; James, M.J.; Xie, S.X.; Ballatore, C.; et al. Epothilone d improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 2010, 30, 13861–13866. [Google Scholar] [CrossRef] [PubMed]
- Aneja, R.; Dhiman, N.; Idnani, J.; Awasthi, A.; Arora, S.; Chandra, R.; Joshi, H. Preclinical pharmacokinetics and bioavailability of noscapine, a tubulin-binding anticancer agent. Cancer Chemother. Pharmacol. 2007, 60, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.-H.; Wartmann, M.; O’Reilly, T. Epothilones and related structures—A new class of microtubule inhibitors with potent in vivo antitumor activity. Biochim. Biophys. Acta 2000, 1470, M79–M91. [Google Scholar] [CrossRef]
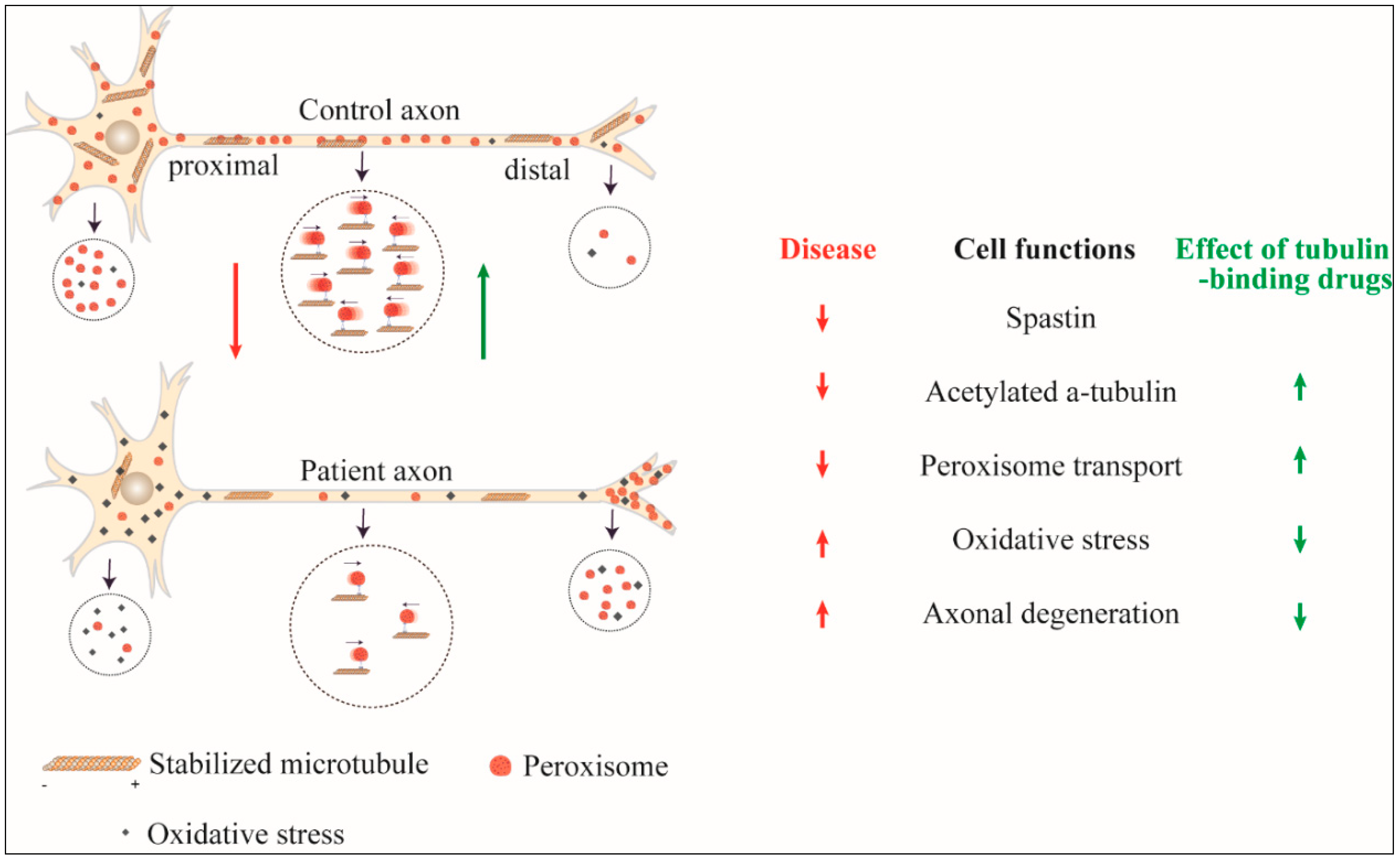

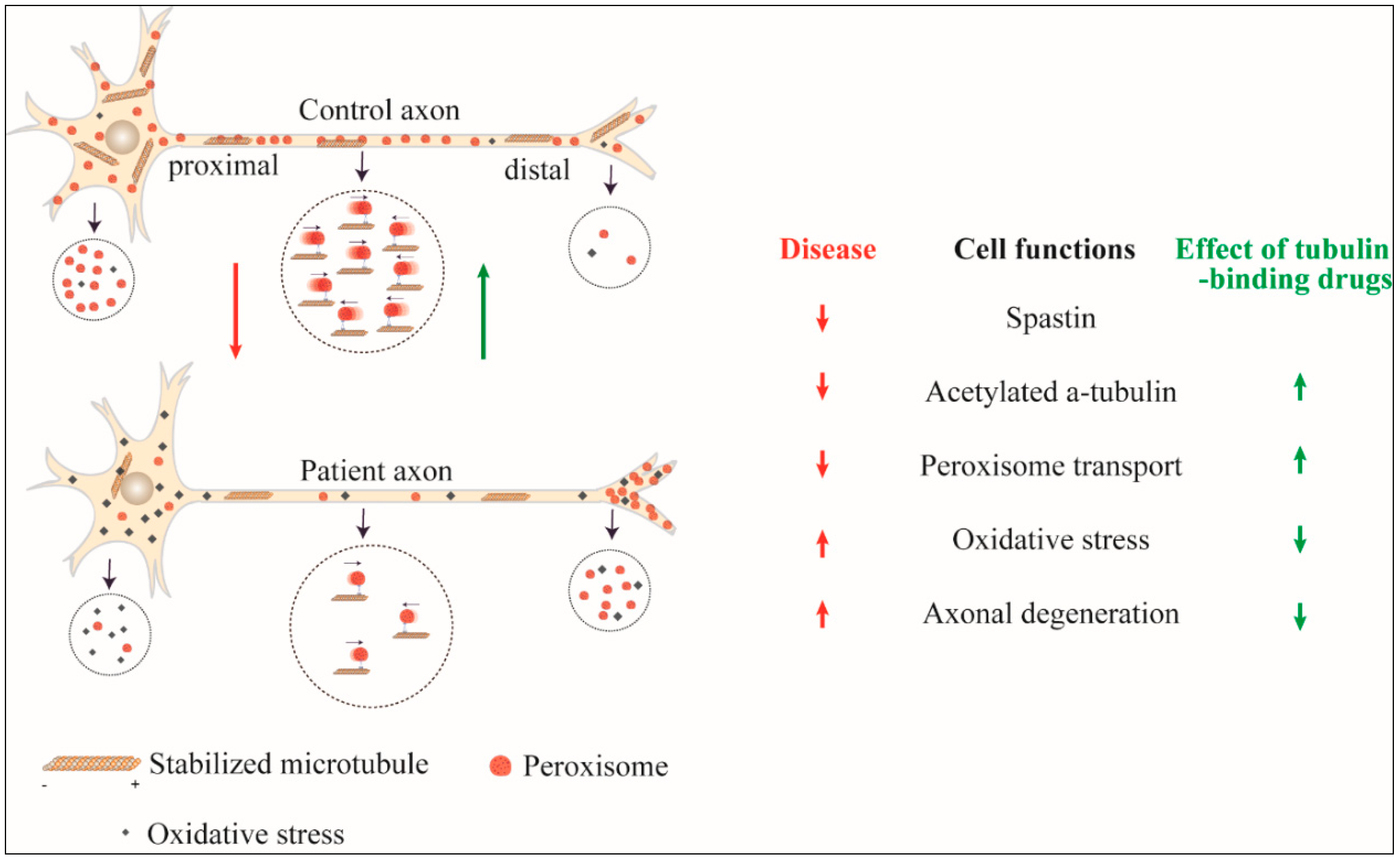{kind=link}
| Source of Patient-Derived Cells | Cell Model | Number of Cell Lines Used | Patient Cell Findings | Effects of Tubulin-Binding Drugs |
|---|---|---|---|---|
| Skin fibroblast cells [30] | iPS-derived glutamatergic neurons | 2 patients/2 controls | Reduced spastin | - |
| Increased p60 katanin | ||||
| Reduced axon number, length and branching | ||||
| Increased axonal swellings | ||||
| Reduced mitochondria retrograde transport | ||||
| Skin fibroblast cells [29] | iPS-derived glutamatergic neurons | 1 patient/1 control | Reduced spastin | Vinblastine reduced axonal swellings |
| Increased stabilised microtubules | ||||
| Increased axonal swellings | ||||
| Reduced mitochondria retrograde transport | ||||
| Olfactory mucosa cells [61] | ONS | 9 patients/10 controls | 57% genes dysregulated | Taxol and vinblastine restored stabilised microtubules and cell size |
| Reduced spastin | ||||
| Reduced stabilised microtubules | ||||
| Altered mitochondria and peroxisome distribution | ||||
| Reduced peroxisome transport speed | ||||
| Reduced cell size | ||||
| Olfactory mucosa cells [58] | ONS | 9 patients/8 controls | Reduced stabilised microtubules | Taxol, vinblastine, noscapine and epothilone D restored stabilised microtubules and rescued peroxisome transport |
| Reduced peroxisome transport speed | ||||
| Olfactory mucosa cells [62] | ONS and ONS-derived neuron-like cells | 5 patients/5 controls | Altered peroxisome distribution | Epothilone D rescued increased vulnerability to oxidative stress |
| Reduced microtubule-dependent peroxisome transport | ||||
| Reduced retrograde peroxisome transport | ||||
| Increased oxidative stress | ||||
| Increased vulnerability to induced oxidative stress |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wali, G.; Sue, C.M.; Mackay-Sim, A. Patient-Derived Stem Cell Models in SPAST HSP: Disease Modelling and Drug Discovery. Brain Sci. 2018, 8, 142. https://doi.org/10.3390/brainsci8080142
Wali G, Sue CM, Mackay-Sim A. Patient-Derived Stem Cell Models in SPAST HSP: Disease Modelling and Drug Discovery. Brain Sciences. 2018; 8(8):142. https://doi.org/10.3390/brainsci8080142
Chicago/Turabian StyleWali, Gautam, Carolyn M. Sue, and Alan Mackay-Sim. 2018. "Patient-Derived Stem Cell Models in SPAST HSP: Disease Modelling and Drug Discovery" Brain Sciences 8, no. 8: 142. https://doi.org/10.3390/brainsci8080142