1. Introduction
The etiopathogenesis of neurodegenerative disorders including Parkinson’s disease (PD) is not clear but interactions between genes and environmental pathogens seem to play a major role in PD [
1]. Multiple genes have been discovered that contribute to the development of PD but still the majority of the sporadic PD patients do not carry a specific identified gene [
2,
3]. On the other hand, multiple environmental factors have been suggested to produce oxidative stress, inflammation, and cell loss in localized brain regions, leading to PD [
4]. Interestingly, the majority of our population is still exposed to these suggested environmental factors such as pesticides, heavy metals, trauma, agent orange, etc., but never develop PD [
5]. Thus, gene–environment interactions seem to play a major role in the initiation of the PD disease process. Another possibility would be that there exist certain ubiquitous environmental factors that can cause PD in all people, but that the majority of the population carries one, or most likely multiple, unknown genes that protect against the development of the disease [
5].
Epigenetics, particularly DNA methylation, is a potential mechanism whereby the environment affects the genome [
6]. There is a growing body of evidence demonstrating the associations between chronic progressive diseases and aberrant DNA methylation modifications [
7]. Epigenetic mechanisms are essential for development and differentiation and allowing an organism to respond to the environment through changes in gene expression patterns. We believe there would be great value in identifying epigenetic alternations and the patterns that predispose or protect against PD. These changes accumulate over time in response to lifestyle and environmental exposures and subsequently affect gene regulation and disease susceptibility.
Altered DNA methylation in the brain has been observed in many neurodegenerative and neurological disorders [
7]. Several studies have found aberrant methylation of several genes related to the pathogenesis of PD [
8]. These studies showed that decreased methylation of CpG islands at intron 1 of the SNCA gene (synuclein, α) resulted in increased α-synuclein gene expression [
9]. A genome-wide DNA methylation study also revealed global hypomethylation in the brains of PD patients [
10]. Similarly, a recent study investigating the genome-wide DNA methylation profiles of brain and blood samples of PD patients and control subjects revealed a set of genes with differential methylation (DM) in brain and blood, with most of these loci previously reported in pathogenesis of PD [
11]. In this study, the authors identified 2908 CpG with DM in the brain and 3897 CpG in the blood of PD cases. Using DM analysis, they further identified 317 probes in brain and 476 in blood with increased methylation, and 2591 probes in brain and 3421 in blood with decreased methylation in patients with PD. Subsequently, after refining the analysis to the fraction of autosomal probes, they showed differential methylation in a total of 174 genes in the brain (84 with increased and 90 with decreased methylation in PD) and 233 genes in blood (127 with increased and 106 with decreased methylation in PD). Finally, using the unsupervised hierarchical clustering of the individual methylation profiles detected from blood, they demonstrated a clear separation between control and PD cases [
11].
Considering the possible interaction between environment and organism, we conducted a genome-wide DNA methylation analysis in brain tissue from 24 brain autopsies to provide preliminary information regarding the methylation pattern of patients with PD. In this study, we compared 12 PD subjects’ DNA methylome to that of 12 sex- and age-matched control subjects, using Illumina Infinium HumanMethylation 450 BeadChip which interrogates more than 450,000 methylation sites across the human genome.
2. Methods
Sample collections: The frozen tissue samples of the human cerebral cortex (superior frontal gyrus at the level of genu of the corpus callosum) from 12 PD patients and 12 subjects without PD pathology or other neurodegenerative disorders (age- and sex-similar matched controls) were obtained from the Banner Sun Health Research Institute Brain and Body Donation Program [
12].
DNA methylome analysis: A genome-wide DNA methylation profiling was performed using the Illumina Infinium HumanMethylation 450 BeadChip Kit at the University of California, Los Angeles Neuroscience Genomics Core. The DNA microarray was carried using 500 ng genomics DNA which was treated with the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s protocol. Methylation fraction values for the autosomal chromosomes were performed using the Infinium HumanMethylation 450 BeadChip assay [
13]. The intensity of the methylated probe relative to the total probe intensity for each site represented the fractional level of methylation at that site in the sample. These values were adjusted for internal controls with Illumina’s GenomeStudio software (Illumina, Incorporated, San Diego, CA, USA).
Bisulfite treatment, whole genome amplification, labeling, hybridization and scanning were performed following the manufacturer’s protocols for the EZ DNA Methylation-Gold and Infinium HumanMethylation 450 BeadChip kits. The methylation data generated by the array were analyzed using the Illumina GenomeStudio software package. The methylation status of a specific CpG site was expressed as β values, calculated as the ratio of the fluorescence intensity signals of the methylated (M) and unmethylated (U) alleles, β = Max(M,0)/[Max(M,0) + Max(U,0) + 100]. β values range between 0 (non-methylated) and 1 (completely methylated).
Bioinformatics data analysis: Bioinformatics data mining was carried out in the Loma Linda University Center for Genomics (Loma Linda University, Loma Linda, CA, USA). Illumina data array was processed using Partek Genomics Suite, under the assumption that β-values are normally distributed. In each individual, probes with a detection p value > 0.01 were removed from later analyses. We excluded CpG sites localized on sex chromosomes to prevent variability among samples within a tissue set related to gender. Differential methylation analysis between sets of samples representing two tissues was performed by using Bioconductor package minfi. Significant differentially methylated regions (DMRs) were selected with p-value < 0.01. DMRs where methylation values for PD were less than controls for the majority of individual CpGs were considered hypomethylated; otherwise, they were annotated as hypermethylated. Hierarchical clustering and principal component analysis were performed for a significant DMR list by using Partek Genomics Suite 6.0.
The autosomal probe sequences were annotated by using Illumina HumanMethylation 450 K BeadChip annotation file. We defined promoter regions as ±2 kb from transcriptional start sites (TSS), gene body regions as transcription start to transcription end after excluding the first 2 kb of the gene, and intergenic regions as those not annotated by the preceding categories. We also annotated individual CpG in context with CpG island (using Bioconductor package IlluminaHumanMethylation450k.db), CpG shore (±2 kb of island), CpG shelf (±2 kb of shore) and CpG sea (regions outside the previous three categories).
Gene ontology analysis and data set signature comparison: To identify enriched gene functions associated with the DMRs in PD brain tissue samples, we performed gene ontology (GO) analysis by using Partek Genomics Suite 6.0.
At the end, the methylation regions with an absolute delta β value ≥ 0.20 were selected for further gene function studies.
3. Results
3.1. Demographics
Twenty-four samples from the cortex of subjects were compared from two groups of PD and non-PD subjects.
Table 1 shows the demographics for each group. Mean age of each group was 78.9 and 79.4 years old for PD and control respectively. PD patients were all male however there were three females in the control group. Unified Parkinson disease rating scale (UPDRS) was measured during ON or OFF condition or both. UPDRS was available for eleven off of twelve PD subjects. Using the Unified Staging System for Lewy Body Disorders, seven patients were stage III and five were stage IV (
Table 1).
3.2. DNA Methylation Analysis
We identified 2794 differentially methylated CpG sites in the frontal cortex of PD cases with a detection
p-value of ≤ 0.01 and 328 differentially methylated CpG sites with a detection
p-value of ≤ 0.001. The majority of differentially methylated regions were hypomethylated, suggesting overall hypomethylation in the brain of PD cases compared to controls (
Table 2). The observed differences in DNA methylation between groups were small but widespread. A pattern of robust hypermethylation of synphilin-1, α-synuclein-interacting protein (SNCAIP) gene was found in the brain of PD cases (
p = 4.93 × 10
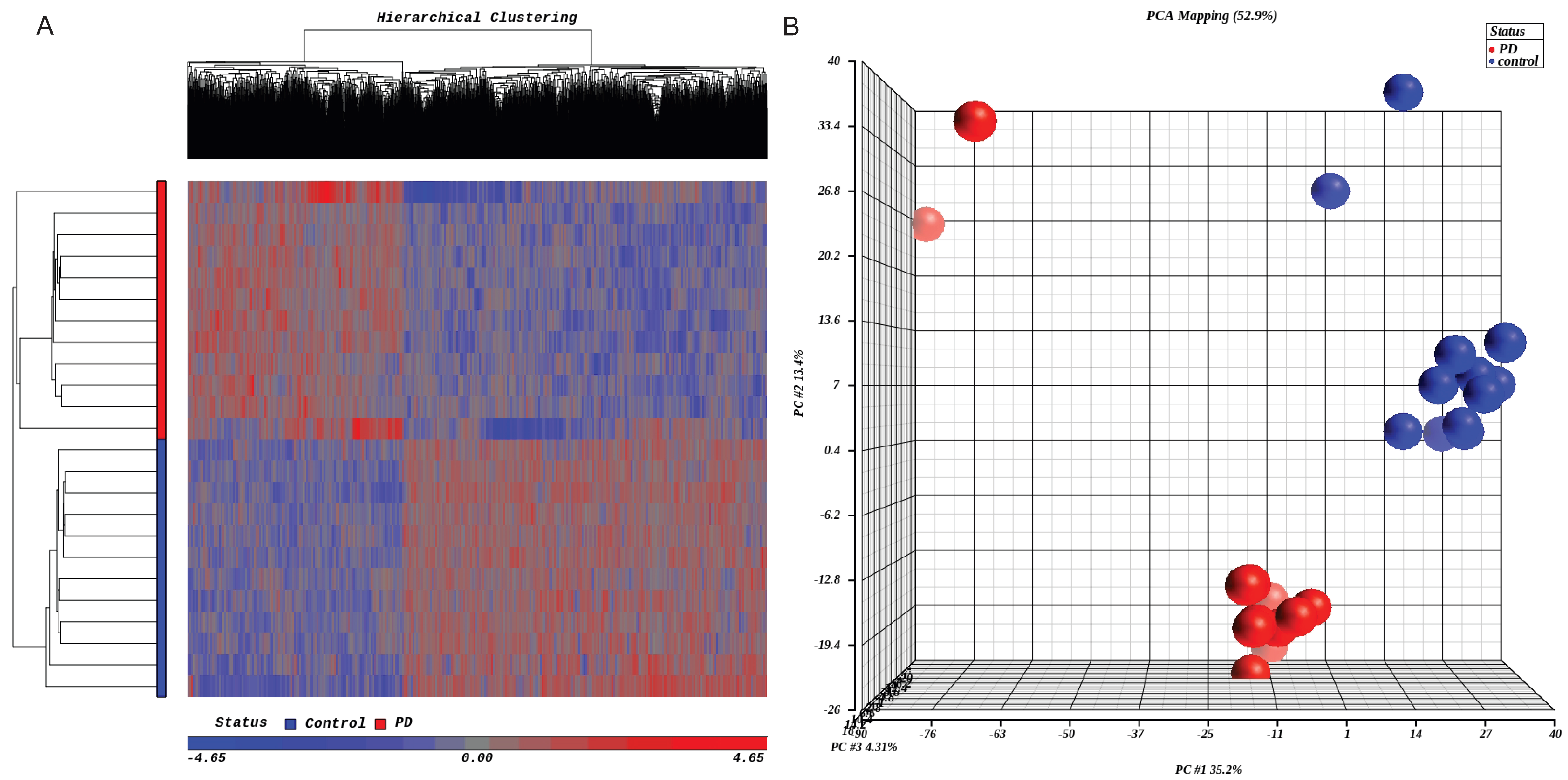
−7 and delta β = 0.60). Unsupervised hierarchical clustering of samples based on genome-wide DNA methylation profiles clearly delineated PD cases from controls (
Figure 1).
4. Discussion
PD is a synucleinopathy caused by an accumulation of misfolded α-synuclein. Lewy bodies, which are the pathological hallmark of PD, contain an abundant amount of this protein. While α-synuclein plays a role in the pathogenesis of Parkinson’s and other neurological diseases, it also has important physiological functions in normal neural tissue.
4.1. Physiological Role of α-Synuclein
α-synuclein (encoded by the gene SNCA) is a natively unfolded protein comprised of 140 amino acids, predominantly localized in the presynaptic terminals of neurons. α-synuclein is abundantly expressed in the human brain, constituting as much as 1% of protein content in the cytosol and is present at high levels in the neocortex, hippocampus, substantia nigra, thalamus and cerebellum [
14]. Considered an unstructured soluble protein, un-mutated α-synuclein may occur physiologically as a monomer or a helically folded tetramer that resists aggregation [
15].
Several studies have suggested that α-synuclein regulates levels of phosphatidic acid in neurons. Specifically, α-synuclein is thought to inhibit the function of phospholipase D2, which synthesizes phosphatidic acid [
16]. Researchers believe that α-synuclein therefore participates in important feedback loops for phosphatidic acid synthesis, preventing its levels from becoming too high [
17]. Phosphatidic acid is widely recognized as an important molecule for the budding of intracellular vesicles [
18]. Thus, the physiological function of α-synuclein is thought to be the regulation of phosphatidic acid and vesicle budding, which is important in neuronal function.
α-synuclein has been shown to play an important role in the formation of SNARE complexes. These complexes are required for the fusion of synaptic vesicles with the presynaptic membrane, making α-synuclein doubly important in cell signaling. Specifically, Burré and colleagues [
19] infected neurons with a lentivirus that expressed α-synuclein. They found that the addition of this lentivirus linearly increased the formation of SNARE protein complexes in these neurons. This led researchers to conclude that α-synuclein plays the physiological role of supporting SNARE complex assembly in nerve cells.
Lastly, α-synuclein has been implicated in microtubule formation [
20]. Alim and colleagues [
20] found that, at various concentrations, a solution of tubulin polymerized effectively in the presence of α-synuclein, but not mutated α-synuclein. This led the researchers to conclude that α-synuclein is important for the polymerization of tubulin particles into microtubules. Microtubules have long been implicated in the vital process of axoplasmal transport [
21]. Thus, α-synuclein is likely to be very important in physiological nerve cell function.
4.2. Physiological Role of Synphilin-1
Another protein associated with the neurodegeneration of Parkinson’s disease is synphilin-1.
Synphilin-1 has been shown to interact with the protein tbp7, which is part of the cellular proteasome. Specifically, Marx and colleagues [
22] mixed a solution of tagged synphilin-1 with tbp7 protein (protein S6 ATPase) to observe any binding interaction between the proteins. They found that synphilin-1 precipitated from solution along with a subunit of the tbp7 protein, leading the researchers to conclude that synphillin-1 binds to the tbp7 protein in cells. Accordingly, this result indicates that synphilin-1 may play a key role in the cellular proteasome, which is important in ubiquitination and protein degradation [
23], a vital process in cell function.
Another study found that synphilin-1 is localized with lipid fractions in brain samples of rats [
24]. Specifically, Murray and colleagues [
24] tagged rat brain extracts with anti-synphilin-1 antibodies and conducted immunohistochemistry to locate synphilin-1 protein in lipid fractions. This finding led some researchers to conclude that synphilin-1 participates in synaptic vesicle formation and secretion [
25].
Furthermore, using immunocytochemistry and in silico rendering, other researchers have demonstrated that synphilin-1 also interacts with phosphoprotein phosphatase 1 [
26]. The authors hypothesize that synphilin-1 may localize phosphoprotein phosphatase 1 to the synaptic membrane and modulate vesicle release, although this is mostly speculation. Other researchers have described that this phosphatase plays important roles in maintaining long-term potentiation/signal strength or modulating axonal ion channel opening and closing [
27,
28].
4.3. Physiological Interaction between α-Synuclein and Synphilin-1
The interaction between these two proteins is primarily associated with the pathophysiology of Parkinson’s disease. However, these two proteins have been shown to interact under physiological conditions, not in the disease state. Specifically, Ribeiro and colleagues [
29] used immunoprecipitation and chromagen staining to find that α-synuclein and synphilin-1 localize together in presynaptic axon terminals. Researchers speculate that synphilin-1 promotes α-synuclein binding to synaptic vesicle membranes [
25]. In vitro studies showed that overexpression of Synphilin-1 inhibits the degradation of α-synuclein by the 20S proteasome, due in part to the interaction of the Synphilin-1 central region 331–355 with the N-terminal region 1–60 of α-synuclein [
30]. The overexpression of synphilin-1 by cell transfection increases the half-life of α-synuclein, resulting in an increased turnover [
30]. Furthermore, Synphilin-1 is present in Lewy bodies, interacting with α-synuclein in vivo and in vitro and promotes its sequestration into aggresomes [
31].
4.4. Hypermethylation of Synphilin-1
Our data shows a robust hypermethylation of the SNCAIP gene in the brain cortex of PD case, suggesting that this gene is down-regulated in PD subjects. It seems that this protein interacts with α-synuclein and our data supports the previous in vivo studies, suggesting the protective effect of SNCAIP. Accordingly, we hypothesize that SNCAIP might serve a protective role in the development of PD in normal patients without down-regulated SNCAIP.
The small sample size of this study is one of the limitations of our analysis. Looking at the methylome pattern of larger samples with minimizing environmental variables such as medical therapies is warranted. Furthermore, there was at high rate of co-occurrence of dementia with PD in our samples. Thus, given the nature of this study, it is difficult to comment on any possible interactive role that dementia may play in our findings. It is possible that the presence of dementia can have epigenetic effects; likewise, it is also possible that the epigenetic modifications present may be particularly related to PD with dementia, and not PD alone. While this is not a particularly critical distinction to make in this study, given that dementia is one of the sequelae of PD, this may be an important avenue for further research: it is possible that different epigenetic modification patterns tend to be present in patients who have certain motor or non-motor complications (i.e., freezing of gait, dyskinesia, depression, cognitive impairment, etc.) versus those who do not. Broadly, it may prove fruitful to examine the role that epigenetics may play in the appearance of specific components or subtypes of neurodegenerative disease
Acknowledgments and Funding
This study was funded by Department of Neurology at Loma Linda University, Loma Linda, CA and Johnston-Teal Parkinson Endowment and Kenneth Vincent Cope Parkinson Endowment. The Brain and Body Donation Program has been supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. The authors thank Camellia Kani and Ali Hakimi for research and editorial assistance. Publication Professionals’ “Good Publication Practice for Communicating Company-Sponsored Medical Research: The GPP3 Guidelines.”
Author Contributions
Research project Conception: K.D. and C.W.; Research project Organization: K.D., C.A., T.B., G.S. and C.W.; Research project Execution: K.D., S.T., T.B., C.A., G.S. and X.C. Statistical Analysis Design: K.D.; Statistical Analysis Execution: K.D. and X.C.; Statistical Analysis Review and Critique: K.D., X.C. and S.T. Manuscript Writing of the first draft: K.D. and A.T.; Manuscript Review and Critique: K.D., A.T., X.C., S.T., C.A., T.B., G.S. and C.W.
Conflicts of Interest
The authors declare no conflict of interest. K.D. has received compensation/honoraria for services as a consultant/advisory committee member/speaker from AbbVie, Acadia, Allergan, Cynapsus, Impax, Ipsen, Lundbeck, Merz, Teva, US World Meds (12 months).
References
- Wirdefeldt, K.; Adami, H.O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of Parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 2011, 26, S1–S58. [Google Scholar] [CrossRef] [PubMed]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M. Is the cause of Parkinson’s disease environmental or hereditary? Evidence from twin studies. Adv. Neurol. 2003, 91, 133–142. [Google Scholar] [PubMed]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Rev. 1997, 25, 335–358. [Google Scholar] [CrossRef]
- Dashtipour, K. Do genetic factors protect against Parkinson’s disease? What I can learn from my healthy grandma. Med. Hypotheses 2014, 83, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.M.; Johnson, A.; Tarapore, P.; Janakiram, V.; Zhang, X.; Leung, Y.K. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR J. 2012, 53, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Methylation and acetylation in nervous system development and neurodegenerative disorders. Ageing Res. Rev. 2003, 2, 329–342. [Google Scholar] [CrossRef]
- Matsumoto, L.; Takuma, H.; Tamaoka, A.; Kurisaki, H.; Date, H.; Tsuji, S.; Itawa, A. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS ONE 2010, 5, e15522. [Google Scholar] [CrossRef] [PubMed]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wullner, U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef] [PubMed]
- Kaut, O.; Schmitt, I.; Wullner, U. Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics 2012, 13, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Dumaop, W.; Galasko, D.; Desplats, P. Distinctive patterns of DNA methylation associated with Parkinson disease: Identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 2013, 8, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Serrano, G.; Shill, H.A.; Walker, D.G.; Lue, L.; Roher, A.E.; Dugger, B.N.; Maarouf, C.; et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 2015, 35, 354–389. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.L.; Dozmorov, M.G.; Wolen, A.R.; Jackson-Cook, C.; Starkweather, A.R.; Lyon, D.E.; York, T.P. Establishing an analytic pipeline for genome-wide DNA methylation. Clin. Epigenet. 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Kagedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. Alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Exton, J.H. Phospholipase D: Enzymology, mechanisms of regulation, and function. Physiol. Rev. 1997, 77, 303–320. [Google Scholar] [PubMed]
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef]
- Siddhanta, A.; Shields, D. Secretory vesicle budding from the trans-Golgi network is mediated by phosphatidic acid levels. J. Biol. Chem. 1998, 273, 17995–17998. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Alim, M.A.; Ma, Q.L.; Takeda, K.; Aizawa, T.; Matsubara, M.; Asada, A.; Saito, T.; Kaji, M.; Yoshii, M.; Hisanaga, S.; et al. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J. Alzheimers Dis. 2004, 6, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.C.; McClure, W.O. Microtubules and axoplasmic transport. Brain Res. 1974, 73, 333–337. [Google Scholar] [CrossRef]
- Marx, F.P.; Soehn, A.S.; Berg, D.; Melle, C.; Schliesling, C.; Lang, M.; Kautzmann, S.; Strauss, K.M.; Franck, T.; Engelender, S.; et al. The proteasomal subunit S6 ATPase is a novel synphilin-1 interacting protein—Implications for Parkinson’s disease. FASEB J. 2007, 21, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef]
- Murray, I.J.; Medford, M.A.; Guan, H.P.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M. Synphilin in normal human brains and in synucleinopathies: Studies with new antibodies. Acta Neuropathol. 2003, 105, 177–184. [Google Scholar] [PubMed]
- Kruger, R. The role of synphilin-1 in synaptic function and protein degradation. Cell Tissue Res. 2004, 318, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Fernandes, E.; Esteves, S.L.; Korrodi-Gregorio, L.; Luers, G.; Afreixo, V.; Fardilha, M.; da Cruz e Silva, O.A. Synphilin-1A is a phosphoprotein phosphatase 1-interacting protein and affects PPP1 sorting to subcellular compartments. J. Mol. Neurosci. 2015, 55, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Allen, P.B.; Hvalby, O.; Jensen, V.; Errington, M.L.; Ramsay, M.; Chaudhry, F.A.; Bliss, T.V.; Storm-Mathisen, J.; Morris, R.G.; Andersen, P.; et al. Protein phosphatase-1 regulation in the induction of long-term potentiation: Heterogeneous molecular mechanisms. J. Neurosci. 2000, 20, 3537–3543. [Google Scholar] [PubMed]
- Levitan, I.B. Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu. Rev. Physiol. 1994, 56, 193–212. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.S.; Carneiro, K.; Ross, C.A.; Menezes, J.R.; Engelender, S. Synphilin-1 is developmentally localized to synaptic terminals, and its association with synaptic vesicles is modulated by alpha-synuclein. J. Biol. Chem. 2002, 277, 23927–23933. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Castelao, B.; Castano, J.G. Synphilin-1 inhibits alpha-synuclein degradation by the proteasome. Cell. Mol. Life Sci. 2011, 68, 2643–2654. [Google Scholar] [CrossRef] [PubMed]
- Casadei, N.; Pöhler, A.M.; Tomás-Zapico, C.; Torres-Peraza, J.; Schwedhelm, I.; Witz, A.; Zamolo, I.; DeHeer, R.; Sprujit, B.; Noldus, L.P.; et al. Overexpression of synphilin-1 promotes clearance of soluble and misfolded alpha-synuclein without restoring the motor phenotype in aged A30P transgenic mice. Hum. Mol. Genet. 2014, 23, 767–781. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



{kind=link}