
Impact of Increased Astrocyte Expression of IL-6, CCL2 or CXCL10 in Transgenic Mice on Hippocampal Synaptic Function

Abstract
:1. Introduction
2. Astrocytes Are a Primary Source of Neuroimmune Factors in the CNS
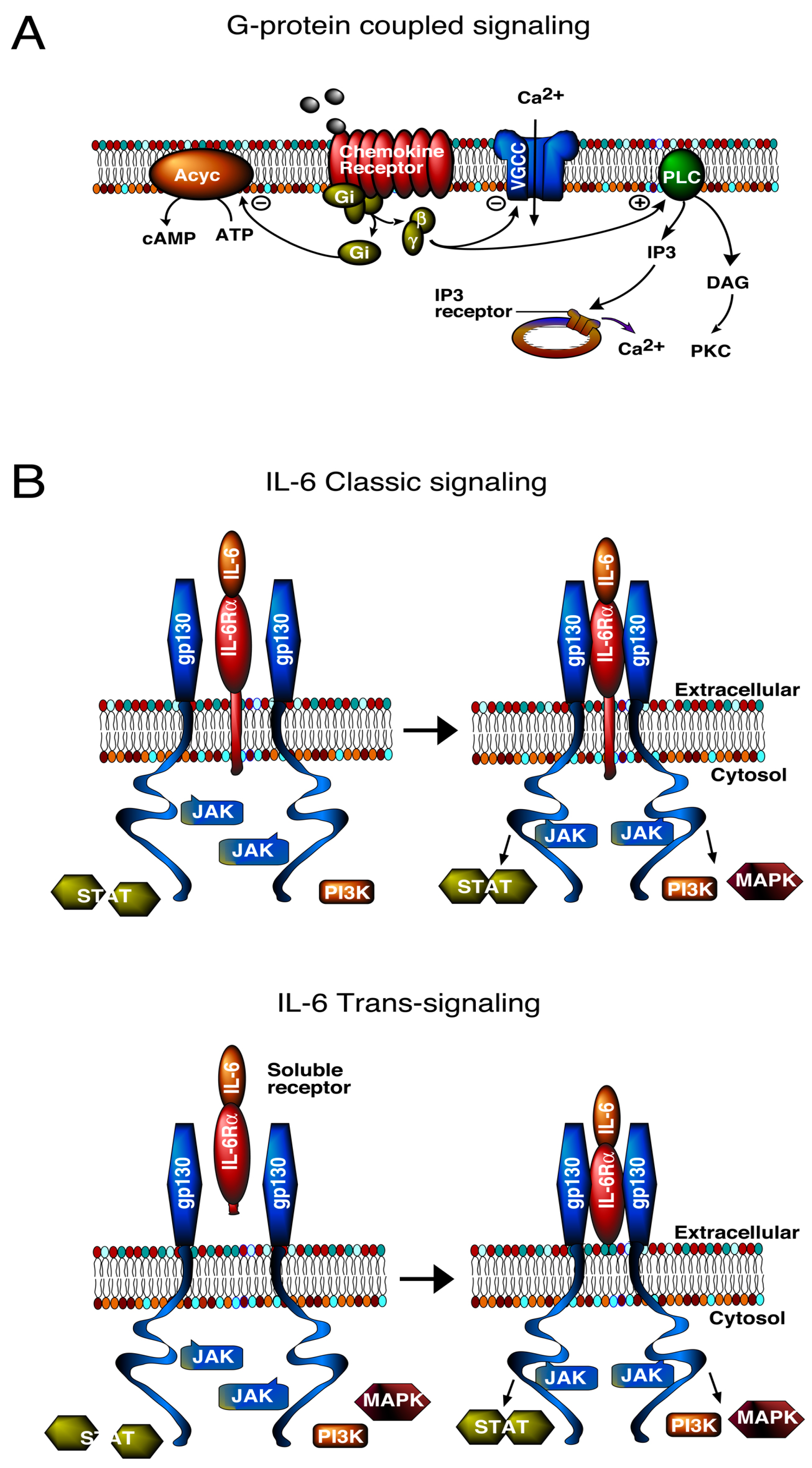
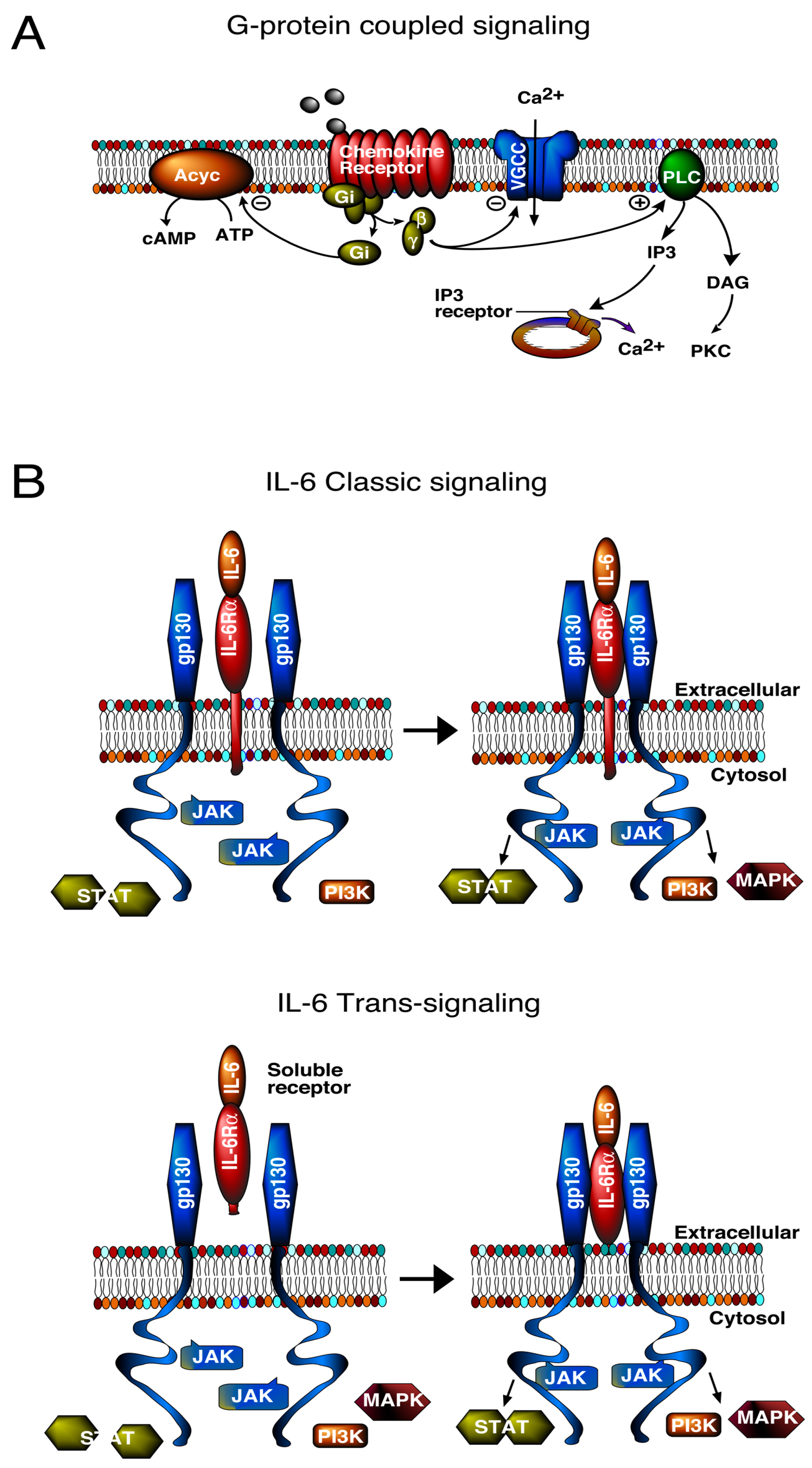
3. Signal Transduction Pathways
4. IL-6, CCL2, or CXCL10 Transgenic Mice
4.1. Expression of IL-6, CCL2, or CXCL10 in the Transgenic Mice
4.2. Neuropathology
5. Synaptic Function in the Hippocampus from IL-6, CCL2, and CXCL10 Transgenic Mice
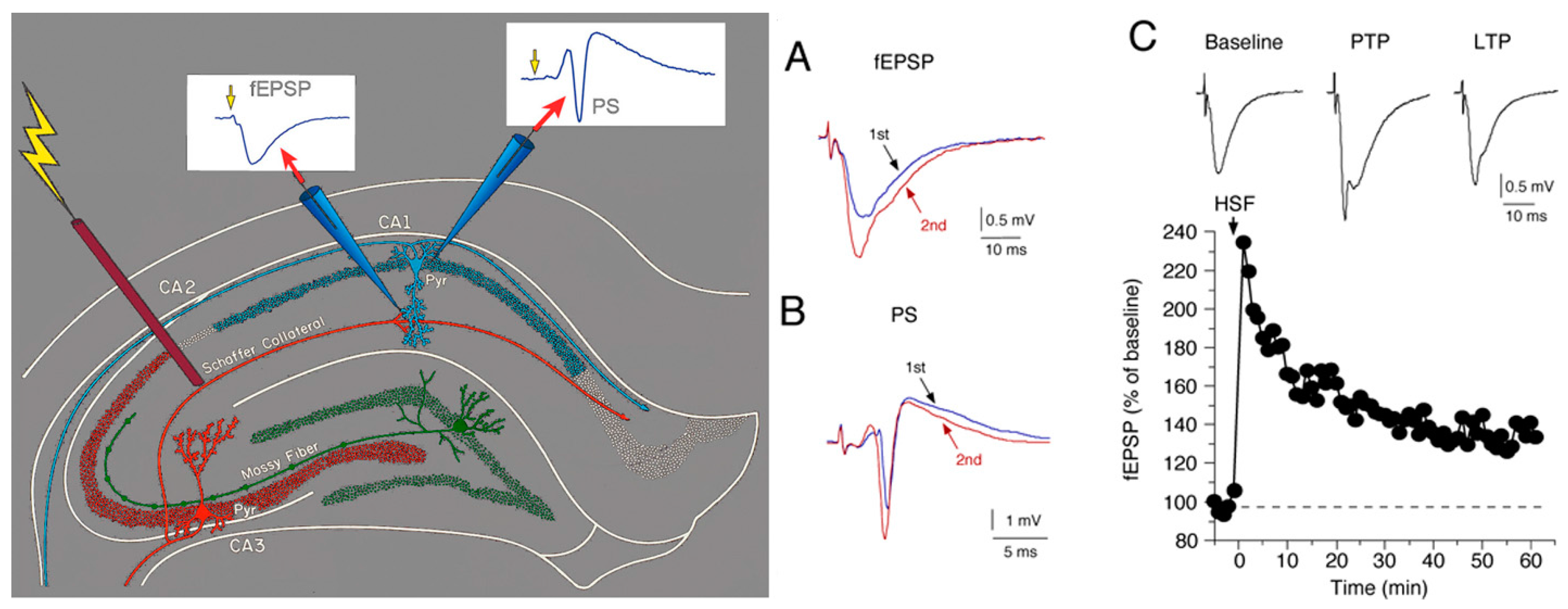
5.1. Synaptic Transmission
5.2. Synaptic Plasticity in IL-6 tg, CCL2-tg and CXCL-10 tg Mice
5.2.1. Short-Term Synaptic Plasticity
5.2.2. Long-Term Synaptic Plasticity
5.3. Effect of Acute Application of Neuroimmune Factors on Synaptic Function
6. Protein Levels in Hippocampus
7. Behavioral Studies
8. Covert Neuroadaptive Changes
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Nicolas, C.S.; Peineau, S.; Amici, M.; Csaba, Z.; Fafouri, A.; Javalet, C.; Collett, V.J.; Hildebrandt, L.; Seaton, G.; Choi, S.L.; et al. The JAK/STAT pathway is involved in synaptic plasticity. Neuron 2012, 73, 374–390. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhou, X.W.; Wang, J.Z. The dual roles of cytokines in Alzheimer’s disease: Update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 2016. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.E.; van den Munckhof, B.; Braun, K.P.; van Royen-Kerkhof, A.; de Jager, W.; Jansen, F.E. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2016, 63, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. Control of autoimmune CNS inflammation by astrocytes. Semin. Immunopathol. 2015, 37, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Vetreno, R.P. Neuroimmune basis of alcoholic brain damage. Int. Rev. Neurobiol. 2014, 118, 315–357. [Google Scholar] [PubMed]
- Drew, P.D.; Kane, C.J. Fetal alcohol spectrum disorders and neuroimmune changes. Int. Rev. Neurobiol. 2014, 118, 41–80. [Google Scholar] [PubMed]
- Chastain, L.G.; Sarkar, D.K. Role of microglia in regulation of ethanol neurotoxic action. Int. Rev. Neurobiol. 2014, 118, 81–103. [Google Scholar] [PubMed]
- Tomasik, J.; Rahmoune, H.; Guest, P.C.; Bahn, S. Neuroimmune biomarkers in schizophrenia. Schizophr. Res. 2014, in press. [Google Scholar]
- Shie, F.S.; Chen, Y.H.; Chen, C.H.; Ho, I.K. Neuroimmune pharmacology of neurodegenerative and mental diseases. J. Neuroimmune Pharmacol. 2011, 6, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, C.; Bambini-Junior, V.; Francis, F.; Riesgo, R.; Savino, W. The impact of neuroimmune alterations in autism spectrum disorder. Front. Psychiatry 2015. [Google Scholar] [CrossRef] [PubMed]
- Hein, A.M.; O’Banion, M.K. Neuroinflammation and cognitive dysfunction in chronic disease and aging. J. Neuroimmune Pharmacol. 2012, 7, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.L. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1β, BDNF and synaptic plasticity. Neuropharmacology 2015, 96, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Eichenbaum, H. Hippocampus: Cognitive processes and neural representations that underlie declarative memory. Neuron 2004, 44, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Chiasserini, D.; Gardoni, F.; Viviani, B.; Tozzi, A.; Giampa, C.; Costa, C.; Tantucci, M.; Zianni, E.; Boraso, M.; et al. Effects of central and peripheral inflammation on hippocampal synaptic plasticity. Neurobiol. Dis. 2013, 52, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Zink, W.E.; Anderson, E.; Boyle, J.; Hock, L.; Rodriguez-Sierra, J.; Xiong, H.; Gendelman, H.E.; Persidsky, Y. Impaired spatial cognition and synaptic potentiation in a murine model of human immunodeficiency virus type 1 encephalitis. J. Neurosci. 2002, 22, 2096–2105. [Google Scholar] [PubMed]
- Savanthrapadian, S.; Wolff, A.R.; Logan, B.J.; Eckert, M.J.; Bilkey, D.K.; Abraham, W.C. Enhanced hippocampal neuronal excitability and LTP persistence associated with reduced behavioral flexibility in the maternal immune activation model of schizophrenia. Hippocampus 2013, 23, 1395–1409. [Google Scholar] [CrossRef] [PubMed]
- Mosayebi, G.; Soleyman, M.R.; Khalili, M.; Mosleh, M.; Palizvan, M.R. Changes in synaptic transmission and long-term potentiation induction as a possible mechanism for learning disability in an animal model of multiple sclerosis. Int. Neurourol. J. 2016, 20, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, D.; Dorner-Ciossek, C.; Kroker, K.S.; Rosenbrock, H. Age-related synaptic dysfunction in tg2576 mice starts as a failure in early long-term potentiation which develops into a full abolishment of late long-term potentiation. J. Neurosci. Res. 2016, 94, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Wang, H.; Matsumoto, N.; Muroya, T.; Shimazaki, J.; Ogura, H.; Shimazu, T. Interleukin-1β causes long-term potentiation deficiency in a mouse model of septic encephalopathy. Neuroscience 2011, 187, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Batti, L.; O’Connor, J.J. Tumor necrosis factor-α impairs the recovery of synaptic transmission from hypoxia in rat hippocampal slices. J. Neuroimmunol. 2010, 218, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Motta, C.; Studer, V.; Barbieri, F.; Buttari, F.; Bergami, A.; Sancesario, G.; Bernardini, S.; de Angelis, G.; Martino, G.; et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult. Scler. 2014, 20, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Bateup, H.S.; Johnson, C.A.; Denefrio, C.L.; Saulnier, J.L.; Kornacker, K.; Sabatini, B.L. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 2013, 78, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Sgobio, C.; Siliquini, S.; Tozzi, A.; Tantucci, M.; Ghiglieri, V.; di Filippo, M.; Pendolino, V.; de Iure, A.; Marti, M.; et al. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Br. J. Neurol. 2012, 135, 1884–1899. [Google Scholar] [CrossRef] [PubMed]
- Scott-McKean, J.J.; Costa, A.C. Exaggerated NMDA mediated LTD in a mouse model of down syndrome and pharmacological rescuing by memantine. Learn. Mem. 2011, 18, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Roberto, M.; Nelson, T.E.; Ur, C.L.; Gruol, D.L. Long-term potentiation in the rat hippocampus is reversibly depressed by chronic intermittent ethanol exposure. J. Neurophysiol. 2002, 87, 2385–2397. [Google Scholar] [PubMed]
- Biber, K.; Pinto-Duarte, A.; Wittendorp, M.C.; Dolga, A.M.; Fernandes, C.C.; von Frijtag Drabbe Kunzel, J.; Keijser, J.N.; de Vries, R.; Ijzerman, A.P.; Ribeiro, J.A.; et al. Interleukin-6 upregulates neuronal adenosine A1 receptors: Implications for neuromodulation and neuroprotection. Neuropsychopharmacology 2008, 33, 2237–2250. [Google Scholar] [CrossRef] [PubMed]
- Penkowa, M.; Giralt, M.; Lago, N.; Camats, J.; Carrasco, J.; Hernandez, J.; Molinero, A.; Campbell, I.L.; Hidalgo, J. Astrocyte-targeted expression of IL-6 protects the CNS against a focal brain injury. Exp. Neurol. 2003, 181, 130–148. [Google Scholar] [CrossRef]
- Millington, C.; Sonego, S.; Karunaweera, N.; Rangel, A.; Aldrich-Wright, J.R.; Campbell, I.L.; Gyengesi, E.; Munch, G. Chronic neuroinflammation in Alzheimer’s disease: New perspectives on animal models and promising candidate drugs. BioMed Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Boztug, K.; Carson, M.J.; Pham-Mitchell, N.; Asensio, V.C.; DeMartino, J.; Campbell, I.L. Leukocyte infiltration, but not neurodegeneration, in the CNS of transgenic mice with astrocyte production of the CXC chemokine ligand 10. J. Immunol. 2002, 169, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, T.; Yamamoto, M.; Xiong, H.; Lambert, M.P.; Klein, W.L.; Gendelman, H.E.; Ransohoff, R.M.; Ikezu, T. CCL2 accelerates microglia-mediated Aβ oligomer formation and progression of neurocognitive dysfunction. PLoS ONE 2009, 4, e6197. [Google Scholar] [CrossRef] [PubMed]
- Elhofy, A.; Wang, J.; Tani, M.; Fife, B.T.; Kennedy, K.J.; Bennett, J.; Huang, D.; Ransohoff, R.M.; Karpus, W.J. Transgenic expression of CCL2 in the central nervous system prevents experimental autoimmune encephalomyelitis. J. Leukoc. Biol. 2005, 77, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Wujek, J.; Kidd, G.; He, T.T.; Cardona, A.; Sasse, M.E.; Stein, E.J.; Kish, J.; Tani, M.; Charo, I.F.; et al. Chronic expression of monocyte chemoattractant protein-1 in the central nervous system causes delayed encephalopathy and impaired microglial function in mice. FASEB J. 2005, 19, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Almolda, B.; Villacampa, N.; Manders, P.; Hidalgo, J.; Campbell, I.L.; Gonzalez, B.; Castellano, B. Effects of astrocyte-targeted production of interleukin-6 in the mouse on the host response to nerve injury. Glia 2014, 62, 1142–1161. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Tani, M.; Wang, J.; Han, Y.; He, T.T.; Weaver, J.; Charo, I.F.; Tuohy, V.K.; Rollins, B.J.; Ransohoff, R.M. Pertussis toxin-induced reversible encephalopathy dependent on monocyte chemoattractant protein-1 overexpression in mice. J. Neurosci. 2002, 22, 10633–10642. [Google Scholar] [PubMed]
- Jensen, C.J.; Massie, A.; de Keyser, J. Immune players in the CNS: The astrocyte. J. Neuroimmune Pharmacol. 2013, 8, 824–839. [Google Scholar] [CrossRef] [PubMed]
- Ransom, B.R.; Ransom, C.B. Astrocytes: Multitalented stars of the central nervous system. Methods Mol. Biol. 2012, 814, 3–7. [Google Scholar] [PubMed]
- Finsterwald, C.; Magistretti, P.J.; Lengacher, S. Astrocytes: New targets for the treatment of neurodegenerative diseases. Curr. Pharma. Des. 2015, 21, 3570–3581. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Zanetti, A.T.; Hallock, R.M. The role of astrocytes in the regulation of synaptic plasticity and memory formation. Neural Plast. 2013. [Google Scholar] [CrossRef] [PubMed]
- Gruart, A.; Delgado-Garcia, J.M. Activity-dependent changes of the hippocampal CA3-CA1 synapse during the acquisition of associative learning in conscious mice. Genes Brain Behav. 2007, 6, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Gruol, D.L. IL-6 regulation of synaptic function in the CNS. Neuropharmacology 2015. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Erta, M.; Lim, S.L.; Frausto, R.; May, U.; Rose-John, S.; Scheller, J.; Hidalgo, J. Trans-signaling is a dominant mechanism for the pathogenic actions of interleukin-6 in the brain. J. Neurosci. 2014, 34, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Vollenweider, F.; Herrmann, M.; Otten, U.; Nitsch, C. Interleukin-6 receptor expression and localization after transient global ischemia in gerbil hippocampus. Neurosci. Lett. 2003, 341, 49–52. [Google Scholar] [CrossRef]
- Chang, G.Q.; Karatayev, O.; Leibowitz, S.F. Prenatal exposure to ethanol stimulates hypothalamic CCR2 chemokine receptor system: Possible relation to increased density of orexigenic peptide neurons and ethanol drinking in adolescent offspring. Neuroscience 2015, 310, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Van der Meer, P.; Ulrich, A.M.; Gonzalez-Scarano, F.; Lavi, E. Immunohistochemical analysis of CCR2, CCR3, CCR5, and CXCR4 in the human brain: Potential mechanisms for HIV dementia. Exp. Mol. Pathol. 2000, 69, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Banisadr, G.; Gosselin, R.D.; Mechighel, P.; Rostene, W.; Kitabgi, P.; Parsadaniantz, S.M. Constitutive neuronal expression of CCR2 chemokine receptor and its colocalization with neurotransmitters in normal rat brain: Functional effect of MCP-1/CCL2 on calcium mobilization in primary cultured neurons. J. Comp. Neurol. 2005, 492, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Ragozzino, D. CXC chemokine receptors in the central nervous system: Role in cerebellar neuromodulation and development. J. Neurovirol. 2002, 8, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.Q.; Bacskai, B.J.; Knowles, R.B.; Qin, S.X.; Hyman, B.T. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: In vitro ERK1/2 activation and role in Alzheimer’s disease. J. Neuroimmunol. 2000, 108, 227–235. [Google Scholar] [CrossRef]
- Campbell, I.L.; Abraham, C.R.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef] [PubMed]
- Brenner, M.; Messing, A. GFAP transgenic mice. Methods 1996, 10, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Hu, H.; Lee, Y.; D’Azzo, A.; Messing, A.; Brenner, M. Expression specificity of GFAP transgenes. Neurochem. Res. 2004, 29, 2075–2093. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.S.; Stalder, A.; Samimi, A.; Campbell, I.L. Reactive gliosis as a consequence of interleukin-6 expression in the brain: Studies in transgenic mice. Dev. Neurosci. 1994, 16, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, J.; Kim, Y.; Yang, M.; Jang, H.; Kang, S.; Kim, J.C.; Kim, S.H.; Shin, T.; Moon, C. Differential patterns of nestin and glial fibrillary acidic protein expression in mouse hippocampus during postnatal development. J. Vet. Sci. 2011, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Heyser, C.J.; Masliah, E.; Samimi, A.; Campbell, I.L.; Gold, L.H. Progressive decline in avoidance learning paralleled by inflammatory neurodegeneration in transgenic mice expressing interleukin 6 in the brain. Proc. Natl. Acad. Sci. USA 1997, 94, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Vallieres, L.; Campbell, I.L.; Gage, F.H.; Sawchenko, P.E. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J. Neurosci. 2002, 22, 486–492. [Google Scholar] [PubMed]
- Gruol, D.L.; Vo, K.; Bray, J.G.; Roberts, A.J. CCL2-ethanol interactions and hippocampal synaptic protein expression in a transgenic mouse model. Front. Integr. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.; Erta, M.; Ferrer, B.; Comes, G.; Giralt, M.; Hidalgo, J. Astrocyte-specific deficiency of interleukin-6 and its receptor reveal specific roles in survival, body weight and behavior. Brain Behav. Immun. 2013, 27, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Gruol, D.L.; Vo, K.; Bray, J.G. Increased astrocyte expression of IL-6 or CCL2 in transgenic mice alters levels of hippocampal and cerebellar proteins. Front. Cell Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Sanz, E.; Hofer, M.J.; Unzeta, M.; Campbell, I.L. Minimal role for stat1 in interleukin-6 signaling and actions in the murine brain. Glia 2008, 56, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.E.; Engberink, A.O.; Hernandez, R.; Puro, A.; Huitron-Resendiz, S.; Hao, C.; de Graan, P.N.; Gruol, D.L. Altered synaptic transmission in the hippocampus of transgenic mice with enhanced central nervous systems expression of interleukin-6. Brain Behav. Immun. 2012, 26, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Nelson, T.E.; Hao, C.; Manos, J.; Ransohoff, R.M.; Gruol, D.L. Altered hippocampal synaptic transmission in transgenic mice with astrocyte-targeted enhanced CCL2 expression. Brain Behav. Immun. 2011, 25 (Suppl. S1), S106–S119. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L. Structural and functional impact of the transgenic expression of cytokines in the CNS. Annu. N. Y. Acad. Sci. 1998, 840, 83–96. [Google Scholar] [CrossRef]
- Brett, F.M.; Mizisin, A.P.; Powell, H.C.; Campbell, I.L. Evolution of neuropathologic abnormalities associated with blood-brain barrier breakdown in transgenic mice expressing interleukin-6 in astrocytes. J. Neuropathol. Exp. Neurol. 1995, 54, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, S.C.; Campbell, I.L.; Henriksen, S.J. Site-specific hippocampal pathophysiology due to cerebral overexpression of interleukin-6 in transgenic mice. Brain Res. 1994, 652, 149–153. [Google Scholar] [CrossRef]
- Bellinger, F.P.; Madamba, S.G.; Campbell, I.L.; Siggins, G.R. Reduced long-term potentiation in the dentate gyrus of transgenic mice with cerebral overexpression of interleukin-6. Neurosci. Lett. 1995, 198, 95–98. [Google Scholar] [CrossRef]
- Nelson, T.E.; Campbell, I.L.; Gruol, D.L. Altered physiology of purkinje neurons in cerebellar slices from transgenic mice with chronic central nervous system expression of interleukin-6. Neuroscience 1999, 89, 127–136. [Google Scholar] [CrossRef]
- Bray, J.G.; Reyes, K.C.; Roberts, A.J.; Ransohoff, R.M.; Gruol, D.L. Synaptic plasticity in the hippocampus shows resistance to acute ethanol exposure in transgenic mice with astrocyte-targeted enhanced CCL2 expression. Neuropharmacology 2013, 67, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Vlkolinsky, R.; Siggins, G.R.; Campbell, I.L.; Krucker, T. Acute exposure to cxc chemokine ligand 10, but not its chronic astroglial production, alters synaptic plasticity in mouse hippocampal slices. J. Neuroimmunol. 2004, 150, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Samland, H.; Huitron-Resendiz, S.; Masliah, E.; Criado, J.; Henriksen, S.J.; Campbell, I.L. Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J. Neurosci. Res. 2003, 73, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, R.V.; Puro, A.C.; Manos, J.C.; Huitron-Resendiz, S.; Reyes, K.C.; Liu, K.; Vo, K.; Roberts, A.J.; Gruol, D.L. Transgenic mice with increased astrocyte expression of IL-6 show altered effects of acute ethanol on synaptic function. Neuropharmacology 2015, 103, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.A. Long-term potentiation and memory. Physiol. Rev. 2004, 84, 87–136. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.G.; Saggau, P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area ca1 of hippocampus. J. Neurosci. 1994, 14, 645–654. [Google Scholar] [PubMed]
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Annu. Rev Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [PubMed]
- Jackman, S.L.; Turecek, J.; Belinsky, J.E.; Regehr, W.G. The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 2016, 529, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tang, H.; Liu, J.; Dong, J.; Xiong, H. Chemokine CCL2 modulation of neuronal excitability and synaptic transmission in rat hippocampal slices. J. Neurochem. 2011, 116, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tang, H.; Xiong, H. Chemokine CCL2 enhances nmda receptor-mediated excitatory postsynaptic current in rat hippocampal slices-a potential mechanism for HIV-1-associated neuropathy? J. Neuroimmune Pharmacol. 2016, 11, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Tancredi, V.; D’Antuono, M.; Cafe, C.; Giovedi, S.; Bue, M.C.; D’Arcangelo, G.; Onofri, F.; Benfenati, F. The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase erk. J. Neurochem. 2000, 75, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Li, A.J.; Katafuchi, T.; Oda, S.; Hori, T.; Oomura, Y. Interleukin-6 inhibits long-term potentiation in rat hippocampal slices. Brain Res. 1997, 748, 30–38. [Google Scholar] [CrossRef]
- Tancredi, V.; D’Arcangelo, G.; Grassi, F.; Tarroni, P.; Palmieri, G.; Santoni, A.; Eusebi, F. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci. Lett. 1992, 146, 176–178. [Google Scholar] [CrossRef]
- Rose, C.F.; Verkhratsky, A.; Parpura, V. Astrocyte glutamine synthetase: Pivotal in health and disease. Biochem. Soc. Trans. 2013, 41, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.; Jurado-Coronel, J.C.; Avila, M.F.; Sabogal, A.; Capani, F.; Barreto, G.E. NMDARs in neurological diseases: A potential therapeutic target. Int. J. Neurosci. 2015, 125, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Sheng, M. NMDA receptors in nervous system diseases. Neuropharmacology 2013, 74, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.B.; Pal, N. Alcohol dependence produced in mice by inhalation of ethanol: Grading the withdrawal reaction. Science 1971, 172, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Metten, P.; Crabbe, J.C. Alcohol withdrawal severity in inbred mouse (mus musculus) strains. Behav. Neurosci. 2005, 119, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Raber, J.; O’Shea, R.D.; Bloom, F.E.; Campbell, I.L. Modulation of hypothalamic-pituitary-adrenal function by transgenic expression of interleukin-6 in the CNS of mice. J. Neurosci. 1997, 17, 9473–9480. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
| Measurement | IL-6 tg vs. Non-tg | CCL2-tg vs. Non-tg | CCL2-tg SJL vs. Non-tg | CXCL10-tg vs. Non-tg | |
|---|---|---|---|---|---|
| Age (months) | 1–2 | 3–6 | 2–3 | 7–12 | 5–6 |
| Synaptic transmission | |||||
| -fEPSP | ↑ | ↑ | no Δ | ↓ | no Δ |
| -PS | ↑ | ↑ | ↑ | ↓ | no Δ |
| P-P synaptic plasticity | |||||
| -fEPSP (PPF) | no Δ | no Δ | no Δ | ↑ | no Δ |
| -PS (PPR) | no Δ | no Δ | no Δ | ↑ | no Δ |
| Long-term synaptic plasticity | |||||
| -PTP | ↓ | no Δ | no Δ | ↑ | no Δ |
| -LTP | no Δ | no Δ | no Δ | no Δ | no Δ |
| Reference | [60] | [67] | [61] | [68] | |
| Measurement | Neuroimmune Factor | ||||
|---|---|---|---|---|---|
| IL-6 | CCL2 | CXCL10 non-tg | CXCL10 tg | ||
| species | rat | rat | rat | mouse | mouse |
| Age (months) or weight (gm) | 2–3 months | 200–250 gm | 0.5–1 month | 5–6 months | 5–6 months |
| Concentration | 1, 5, 50 ng/mL | 50–2000 U/mL | 2.3 nM | 10 ng/mL | 10 ng/mL |
| Synaptic transmission | |||||
| -fEPSP or EPSC | nd | no Δ | ↑ | no Δ | nd |
| -Population spike | no Δ | nd | nd | nd | nd |
| Short-term synaptic plasticity | |||||
| -fEPSP (PPF) | no Δ | nd | nd | ↑ | no Δ |
| -Population spike (PPR) | nd | nd | nd | nd | nd |
| Long-term synaptic plasticity | |||||
| -PTP | ↓ | ↓ | nd | ↓ | no Δ |
| -LTP | ↓ | ↓ | nd | ↓ | ↓ |
| Reference | [79] | [78] | [75] | [68] | |
| Measurement | IL-6 tg vs. Non-tg | CCL2-tg vs. Non-tg | CCL2-tg SJL vs. Non-tg | |||
|---|---|---|---|---|---|---|
| Age (months) | 1–2 | 3–5 | 1–3 | 3–5 | 3–4 | 7–9 |
| Housekeeping proteins | ||||||
| -β-actin | no Δ | no Δ | no Δ | no Δ | no Δ | no Δ |
| Astrocyte proteins | ||||||
| -GFAP | ↑ | ↑ | no Δ | no Δ | no Δ | ↑ |
| -Glutamine synthetase | no Δ | no Δ | no Δ | no Δ | nd | nd |
| Microglial protein | ||||||
| -CD11b | nd | no Δ | no Δ | nd | ↑ | no Δ |
| Neuronal proteins | ||||||
| -Enolase | no Δ | no Δ | no Δ | no Δ | no Δ | no Δ |
| -GAD65/67 | no Δ | ↓ | no Δ | no Δ | no Δ | no Δ |
| Synaptic proteins | ||||||
| -Synapsin 1 | no Δ | no Δ | no Δ | ↑ | no Δ | no Δ |
| -VGLUT1 | nd | no Δ | no Δ | nd | nd | nd |
| -GluA1 | no Δ | no Δ | no Δ | no Δ | no Δ | no Δ |
| -GluN1 | no Δ | no Δ | ↑ | ↑ | no Δ | no Δ |
| Signal transduction | ||||||
| -STAT3 | ↑ | ↑ | no Δ | nd | nd | nd |
| -p42/44 MAPK | no Δ | no Δ | no Δ | no Δ | nd | nd |
| Reference | [58,60] | [58,67] | [61] | |||
| Measurement | 60 mM Alcohol vs. Baseline | |||
|---|---|---|---|---|
| Non-tg | IL-6 tg | Non-tg | CCL2-tg | |
| Synaptic transmission | ||||
| -fEPSP | ↓ | ↑ | ↓ | ↓ |
| -PS | ↓ | ↑ | ↓ | ↓ |
| P-P synaptic plasticity | ||||
| -fEPSP (PPF) | no Δ | no Δ | no Δ | no Δ |
| -PS (PPR) | ↑ | no Δ | ↑ | no Δ |
| Long-term synaptic plasticity | ||||
| -PTP | ↓ | no Δ | ↓ | no Δ |
| -LTP | ↓ | no Δ | ↓ | no Δ |
| Reference | [70] | [67] | ||
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruol, D.L. Impact of Increased Astrocyte Expression of IL-6, CCL2 or CXCL10 in Transgenic Mice on Hippocampal Synaptic Function. Brain Sci. 2016, 6, 19. https://doi.org/10.3390/brainsci6020019
Gruol DL. Impact of Increased Astrocyte Expression of IL-6, CCL2 or CXCL10 in Transgenic Mice on Hippocampal Synaptic Function. Brain Sciences. 2016; 6(2):19. https://doi.org/10.3390/brainsci6020019
Chicago/Turabian StyleGruol, Donna L. 2016. "Impact of Increased Astrocyte Expression of IL-6, CCL2 or CXCL10 in Transgenic Mice on Hippocampal Synaptic Function" Brain Sciences 6, no. 2: 19. https://doi.org/10.3390/brainsci6020019