
Epigenetic Mechanisms in Developmental Alcohol-Induced Neurobehavioral Deficits


Abstract
:1. Introduction
2. Epigenetic Mechanisms
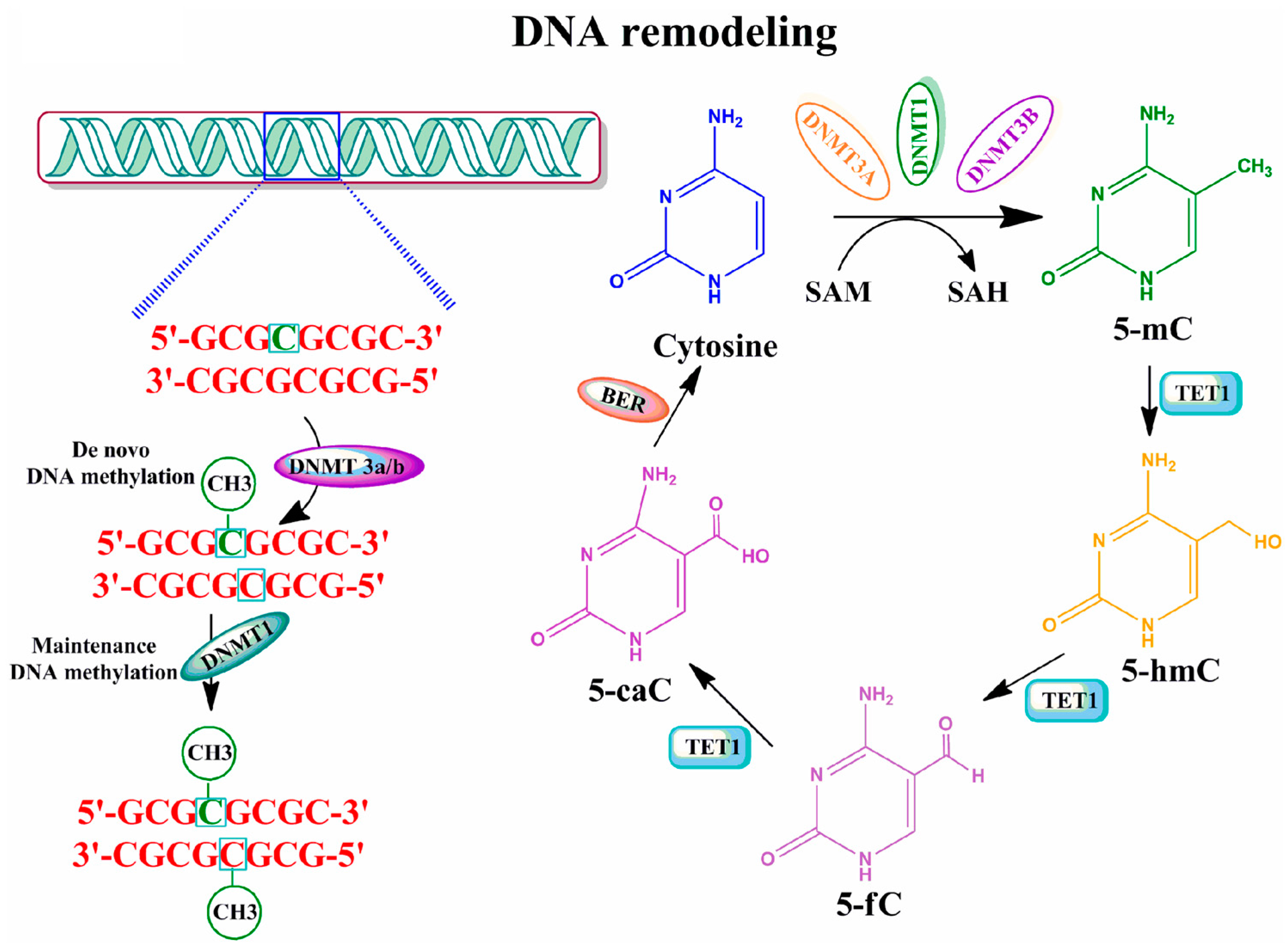
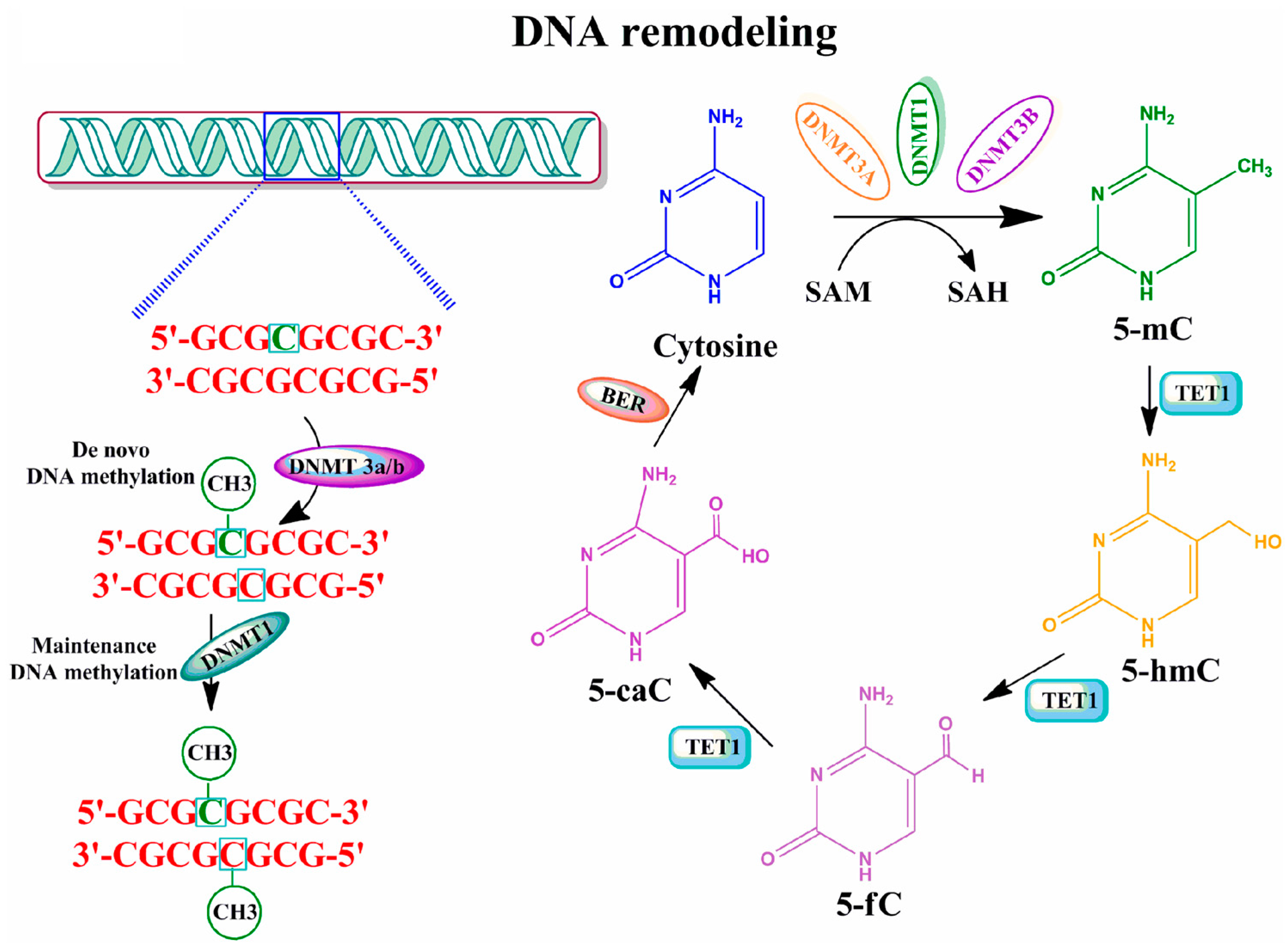
2.1. DNA Methylation
2.2. Post-Translational Modification of DNA-Associated Histone Proteins
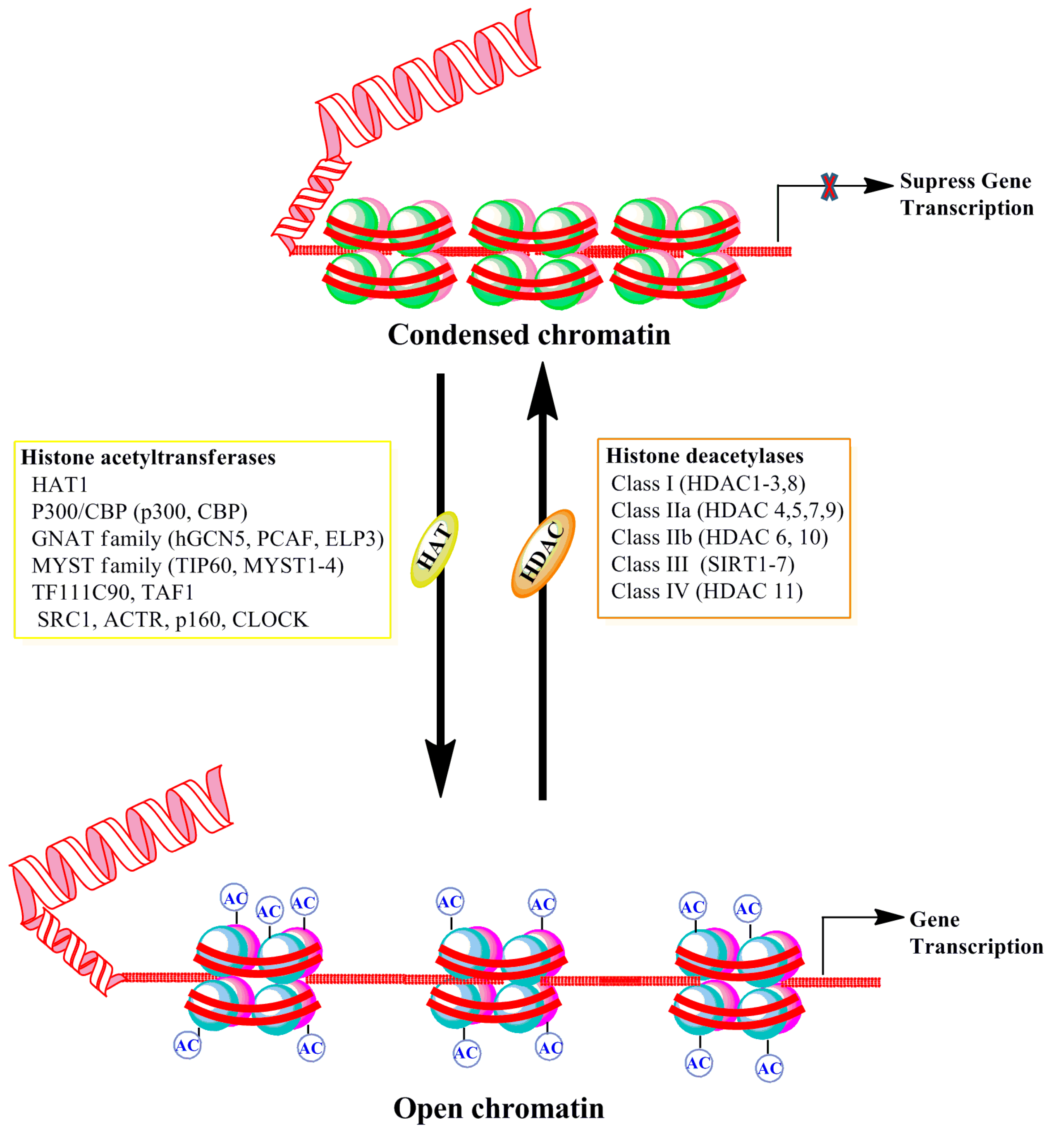
2.2.1. Histone Acetylation and Deacetylation
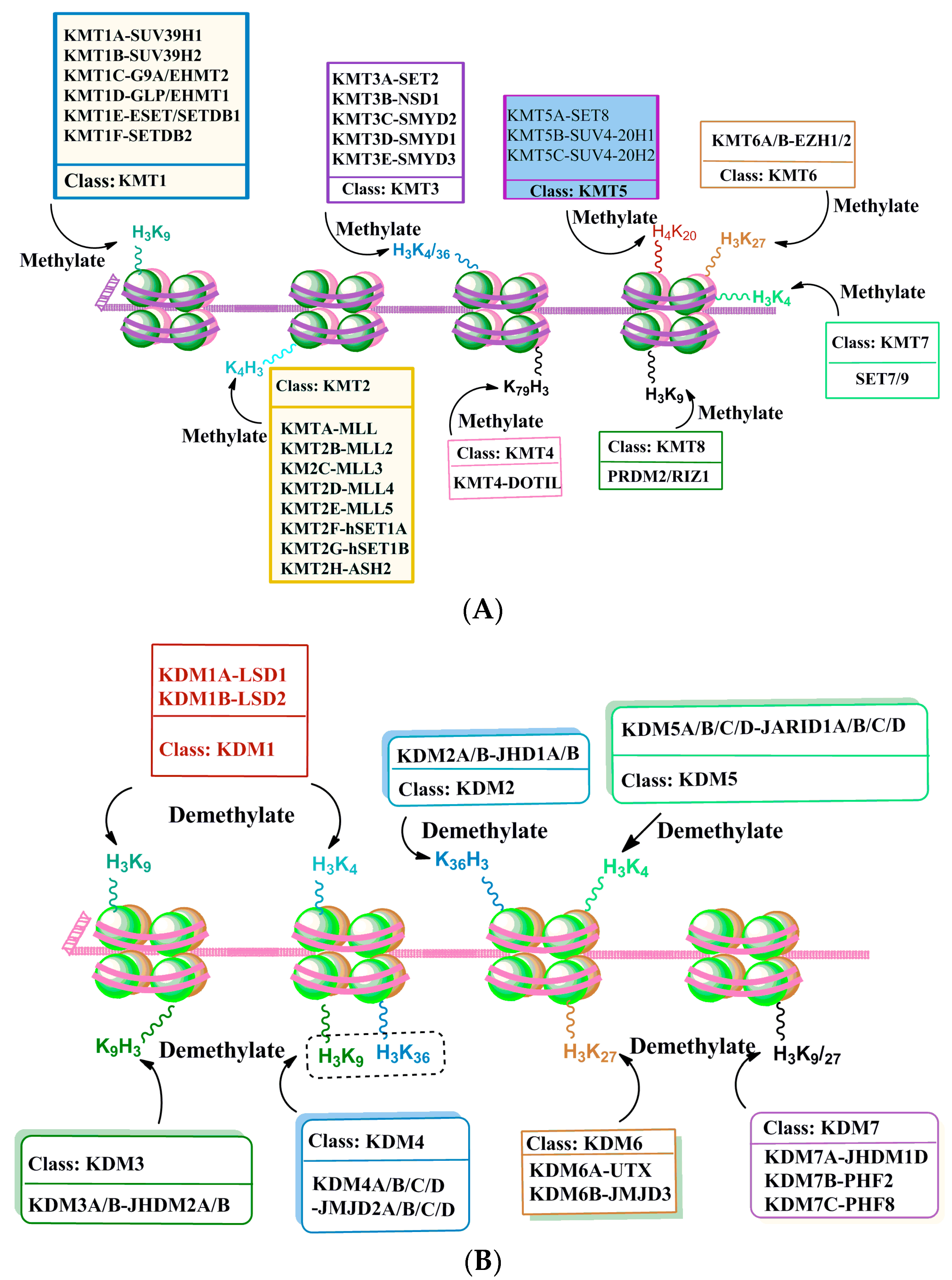
2.2.2. Histone Methylation and Demethylation
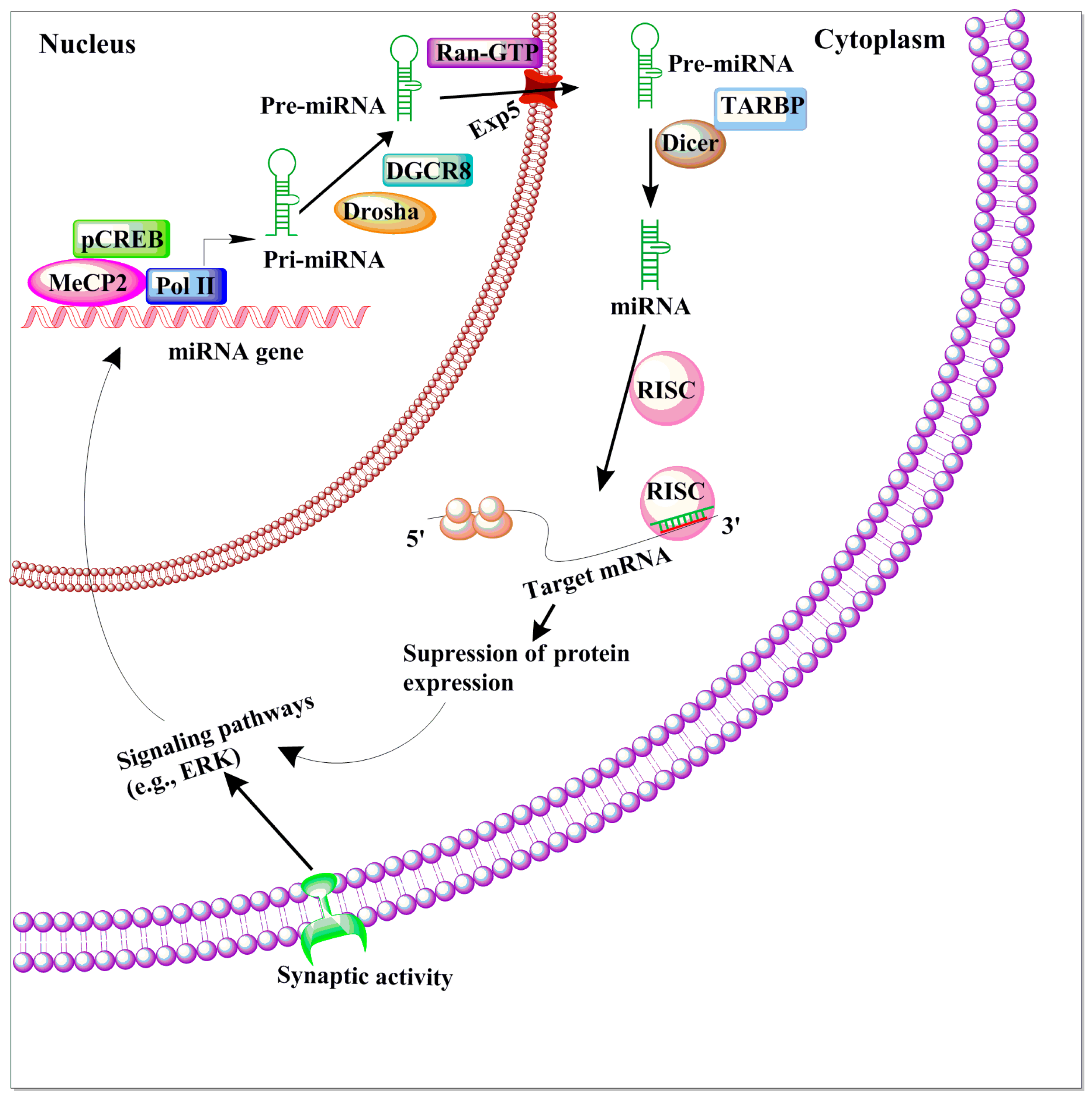
2.3. MicroRNAs
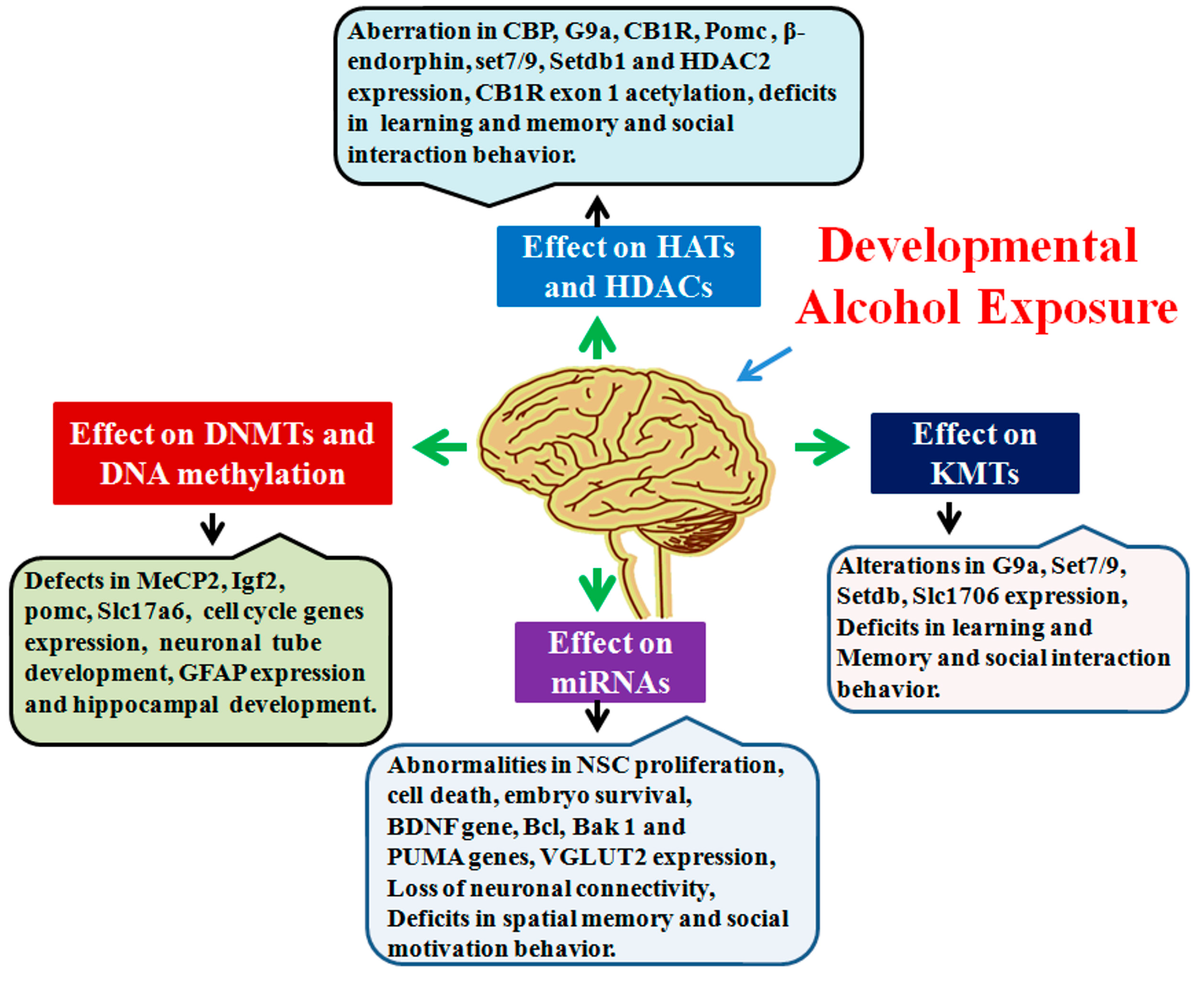
3. Alcohol’s Influence on Epigenetic Mechanisms in the Developing Brain
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
Abbreviations
References
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Choi, K.H.; Renthal, W.; Tsankova, N.M.; Theobald, D.E.; Truong, H.T.; Russo, S.J.; Laplant, Q.; Sasaki, T.S.; Whistler, K.N.; et al. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 2005, 48, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Madrigano, J.; Baccarelli, A.; Mittleman, M.A.; Wright, R.O.; Sparrow, D.; Vokonas, P.S.; Tarantini, L.; Schwartz, J. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ. Health Perspect. 2011, 119, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Nagaraja, N.N.; Umapathy, N.S.; Pace, B.S.; Basavarajappa, B.S. Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int. J. Neuropsychopharmacol. 2015. [Google Scholar] [CrossRef]
- Subbanna, S.; Nagre, N.N.; Shivakumar, M.; Umapathy, N.S.; Psychoyos, D.; Basavarajappa, B.S. Ethanol induced acetylation of histone at G9a Exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience 2014, 258, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Shivakumar, M.; Umapathy, N.S.; Saito, M.; Mohan, P.S.; Kumar, A.; Nixonc, R.A.; Verin, A.D.; Psychoyos, D.; Basavarajappa, B.S. G9a-mediated histone methylation regulates ethanol-induced neurodegeneration in the neonatal mouse brain. Neurobiol. Dis. 2013, 54, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Suderman, M.; McGowan, P.O.; Sasaki, A.; Huang, T.C.; Hallett, M.T.; Meaney, M.J.; Turecki, G.; Szyf, M. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc. Natl. Acad. Sci. USA 2012, 109, 17266–17272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.Y.; Hellstrom, I.C.; Bagot, R.C.; Wen, X.; Diorio, J.; Meaney, M.J. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J. Neurosci. 2010, 30, 13130–13137. [Google Scholar] [CrossRef] [PubMed]
- Avery, O.T.; Macleod, C.M.; McCarty, M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: Induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J. Exp. Med. 1944, 79, 137–158. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.; Avery, O.T. Studies on the chemical nature of the substance inducing transformation on pneumococcal types; an improved method for the isolation of the transforming substance and its application to Pneumococcus Types II, III, and VI. J. Exp. Med. 1946, 83, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.D. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J. Biol. Chem. 1948, 175, 315–332. [Google Scholar] [PubMed]
- Compere, S.J.; Palmiter, R.D. DNA methylation controls the inducibility of the mouse metallothionein-I gene lymphoid cells. Cell 1981, 25, 233–240. [Google Scholar] [CrossRef]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.L.; Burdon, R.H.; Gibb, S.; McKay, E.L. DNA methylase during Xenopus laevis development. Biochim. Biophys. Acta 1981, 655, 329–334. [Google Scholar] [CrossRef]
- Adams, R.L.; McKay, E.L.; Craig, L.M.; Burdon, R.H. Mouse DNA methylase: Methylation of native DNA. Biochim. Biophys. Acta 1979, 561, 345–357. [Google Scholar] [CrossRef]
- Gruenbaum, Y.; Cedar, H.; Razin, A. Substrate and sequence specificity of a eukaryotic DNA methylase. Nature 1982, 295, 620–622. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Riggs, A.D. DNA methylation and gene function. Science 1980, 210, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Szyf, M. DNA methylation patterns. Formation and function. Biochim. Biophys. Acta 1984, 782, 331–342. [Google Scholar] [CrossRef]
- Brown, S.E.; Weaver, I.C.; Meaney, M.J.; Szyf, M. Regional-specific global cytosine methylation and DNA methyltransferase expression in the adult rat hippocampus. Neurosci. Lett. 2008, 440, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Chang, H.; Li, E.; Fan, G. Dynamic expression of de novo DNA methyltransferases DNMT3a and DNMT3b in the central nervous system. J. Neurosci. Res. 2005, 79, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Nagre, N.N.; Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Basavarajappa, B.S. CB1-receptor knockout neonatal mice are protected against ethanol-induced impairments of DNMT1, DNMT3a and DNA methylation. J. Neurochem. 2015, 132, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D.; Uzvolgyi, E.; Liang, G.; Talmadge, C.; Sumegi, J.; Gonzales, F.A.; Jones, P.A. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: Coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 1999, 27, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.K.; Stringfellow, S.A.; Glover, M.E.; Wagle, A.A.; Clinton, S.M. DNA methylation markers in the postnatal developing rat brain. Brain Res. 2013, 1533, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. DNMT1 and DNMT3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Numata, M.; Komura, J.I.; Ono, T.; Bestor, T.H.; Kondo, H. Expression of DNA methyltransferase gene in mature and immature neurons as well as proliferating cells in mice. Differentiation 1994, 56, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Nabel, C.S.; Kohli, R.M. Molecular biology. Demystifying DNA demethylation. Science 2011, 333, 1229–1230. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Kubiura, M.; Okano, M.; Kimura, H.; Kawamura, F.; Tada, M. Chromosome-wide regulation of euchromatin-specific 5mC to 5hmC conversion in mouse ES cells and female human somatic cells. Chromosome Res. 2012, 20, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. The early-life social environment and DNA methylation. Clin. Genet. 2012, 81, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. How do environments talk to genes? Nat. Neurosci. 2013, 16, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. DNA methylation, behavior and early life adversity. J. Genet. Genom. 2013, 40, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Daxinger, L.; Whitelaw, E. Transgenerational epigenetic inheritance: More questions than answers. Genome Res. 2010, 20, 1623–1628. [Google Scholar] [CrossRef] [PubMed]
- Kaminen-Ahola, N.; Ahola, A.; Maga, M.; Mallitt, K.A.; Fahey, P.; Cox, T.C.; Whitelaw, E.; Chong, S. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet 2010, 6, e1000811. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Monteggia, L.M. Role of DNA methylation and the DNA methyltransferases in learning and memory. Dialogues Clin. Neurosci. 2014, 16, 359–371. [Google Scholar] [PubMed]
- Rudenko, A.; Dawlaty, M.M.; Seo, J.; Cheng, A.W.; Meng, J.; Le, T.; Faull, K.F.; Jaenisch, R.; Tsai, L.H. TET1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 2013, 79, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Bergman, Y.; Cedar, H. DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 2013, 20, 274–281. [Google Scholar] [CrossRef] [PubMed]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. TET-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Gavin, C.F.; White, J.A.; Parrish, R.R.; Honasoge, A.; Yancey, C.R.; Rivera, I.M.; Rubio, M.D.; Rumbaugh, G.; Sweatt, J.D. Cortical DNA methylation maintains remote memory. Nat. Neurosci. 2010, 13, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases DNMT3a and DNMT3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Okano, M.; Xie, S.; Li, E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998, 19, 219–220. [Google Scholar] [PubMed]
- Hermann, A.; Goyal, R.; Jeltsch, A. The DNMT1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004, 279, 48350–48359. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by mll partner tet1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [PubMed]
- Sarraf, S.A.; Stancheva, I. Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell 2004, 15, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Chen, R.Z.; Gribnau, J.; Rasmussen, T.P.; Fong, H.; Jaenisch, R.; Jones, E.G. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol. Dis. 2001, 8, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Hu, K.; Chang, Q.; Wu, H.; Sherman, N.E.; Martinowich, K.; Klose, R.J.; Schanen, C.; Jaenisch, R.; Wang, W.; et al. Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc. Natl. Acad. Sci. USA 2009, 106, 4882–4887. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hong, E.J.; Cohen, S.; Zhao, W.N.; Ho, H.Y.; Schmidt, L.; Chen, W.G.; Lin, Y.; Savner, E.; Griffith, E.C.; et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 2006, 52, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Gabel, H.W.; Hemberg, M.; Hutchinson, A.N.; Sadacca, L.A.; Ebert, D.H.; Harmin, D.A.; Greenberg, R.S.; Verdine, V.K.; Zhou, Z.; et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 2011, 72, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhong, X.; Chau, K.F.; Williams, E.C.; Chang, Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat. Neurosci. 2011, 14, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MeCP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [PubMed]
- Na, E.S.; Nelson, E.D.; Kavalali, E.T.; Monteggia, L.M. The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology 2013, 38, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Na, E.S.; Nelson, E.D.; Adachi, M.; Autry, A.E.; Mahgoub, M.A.; Kavalali, E.T.; Monteggia, L.M. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J. Neurosci. 2012, 32, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nature Rev. Mol. Cell Biol. 2012, 13, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef]
- Lorch, Y.; Zhang, M.; Kornberg, R.D. Histone octamer transfer by a chromatin-remodeling complex. Cell 1999, 96, 389–392. [Google Scholar] [CrossRef]
- Goll, M.G.; Bestor, T.H. Histone modification and replacement in chromatin activation. Genes Dev. 2002, 16, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Grant, P.A. A tale of histone modifications. Genome Biol. 2001, 2. [Google Scholar] [CrossRef]
- Smolle, M.; Workman, J.L. Transcription-associated histone modifications and cryptic transcription. Biochim. Biophys. Acta 2013, 1829, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Petruk, S.; Sedkov, Y.; Johnston, D.M.; Hodgson, J.W.; Black, K.L.; Kovermann, S.K.; Beck, S.; Canaani, E.; Brock, H.W.; Mazo, A. TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell 2012, 150, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Roth, S.Y. Histone acetyltransferases: Function, structure, and catalysis. Curr. Opin. Genet. Dev. 2001, 11, 155–161. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef]
- Kalkhoven, E.; Roelfsema, J.H.; Teunissen, H.; den Boer, A.; Ariyurek, Y.; Zantema, A.; Breuning, M.H.; Hennekam, R.C.; Peters, D.J. Loss of CBP acetyltransferase activity by PHD finger mutations in Rubinstein-Taybi syndrome. Hum. Mol. Genet. 2003, 12, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Naruse, I.; Hongo, T.; Xu, M.; Nakahata, T.; Maekawa, T.; Ishii, S. Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech. Dev. 2000, 95, 133–145. [Google Scholar] [CrossRef]
- Yao, T.P.; Oh, S.P.; Fuchs, M.; Zhou, N.D.; Ch’ng, L.E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef]
- Martinez-Cerdeno, V.; Lemen, J.M.; Chan, V.; Wey, A.; Lin, W.; Dent, S.R.; Knoepfler, P.S. N-Myc and GCN5 regulate significantly overlapping transcriptional programs in neural stem cells. PLoS ONE 2012, 7, e39456. [Google Scholar] [CrossRef] [PubMed]
- Bousiges, O.; Vasconcelos, A.P.; Neidl, R.; Cosquer, B.; Herbeaux, K.; Panteleeva, I.; Loeffler, J.P.; Cassel, J.C.; Boutillier, A.L. Spatial memory consolidation is associated with induction of several lysine-acetyltransferase (histone acetyltransferase) expression levels and H2B/H4 acetylation-dependent transcriptional events in the rat hippocampus. Neuropsychopharmacology 2010, 35, 2521–2537. [Google Scholar] [CrossRef] [PubMed]
- Gehrking, K.M.; Andresen, J.M.; Duvick, L.; Lough, J.; Zoghbi, H.Y.; Orr, H.T. Partial loss of Tip60 slows mid-stage neurodegeneration in a spinocerebellar ataxia type 1 (SCA1) mouse model. Hum. Mol. Genet. 2011, 20, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Levenson, J.M.; O’Riordan, K.J.; Brown, K.D.; Trinh, M.A.; Molfese, D.L.; Sweatt, J.D. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004, 279, 40545–40559. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Mizar, P.; Cassel, R.; Neidl, R.; Selvi, B.R.; Mohankrishna, D.V.; Vedamurthy, B.M.; Schneider, A.; Bousiges, O.; Mathis, C.; et al. A novel activator of CBP/p300 acetyltransferases promotes neurogenesis and extends memory duration in adult mice. J. Neurosci. 2013, 33, 10698–10712. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Coelho, C.M.; Li, X.; Marek, R.; Yan, S.; Anderson, S.; Meyers, D.; Mukherjee, C.; Sbardella, G.; Castellano, S.; et al. P300/CBP-associated factor selectively regulates the extinction of conditioned fear. J. Neurosci. 2012, 32, 11930–11941. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Sarthi, J.; Pirooznia, S.K.; Reube, W.; Elefant, F. Increasing Tip60 HAT levels rescues axonal transport defects and associated behavioral phenotypes in a Drosophila Alzheimer’s disease model. J. Neurosci. 2013, 33, 7535–7547. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, S.K.; Elefant, F. Targeting specific HATs for neurodegenerative disease treatment: Translating basic biology to therapeutic possibilities. Front. Cell. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Chatterjee, S.; Bousiges, O.; Selvi, B.R.; Swaminathan, A.; Cassel, R.; Blanc, F.; Kundu, T.K.; Boutillier, A.L. Acetyltransferases (HATs) as targets for neurological therapeutics. Neurotherapeutics 2013, 10, 568–588. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nature reviews. Genetics 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfi, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Monteggia, L.M. Unique functional roles for class I and class II histone deacetylases in central nervous system development and function. Int. J. Dev. Neurosci. 2013, 31, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.H.; Young, D.; Gelovani, J.G.; Robinson, A.; Davidson, Y.; Herholz, K.; Mann, D.M. Histone deacetylase class II and acetylated core histone immunohistochemistry in human brains with Huntington’s disease. Brain Res. 2013, 1504, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Larouche, M.; Goldowitz, D. The expression of HDAC1 and HDAC2 during cerebellar cortical development. Cerebellum 2013, 12, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Hsieh, J.; Barbosa, A.C.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc. Natl. Acad. Sci. USA 2009, 106, 7876–7881. [Google Scholar] [CrossRef] [PubMed]
- Jawerka, M.; Colak, D.; Dimou, L.; Spiller, C.; Lagger, S.; Montgomery, R.L.; Olson, E.N.; Wurst, W.; Gottlicher, M.; Gotz, M. The specific role of histone deacetylase 2 in adult neurogenesis. Neuron Glia Biol. 2010, 6, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Karra, A.S.; Monteggia, L.M. Histone deacetylases govern cellular mechanisms underlying behavioral and synaptic plasticity in the developing and adult brain. Behav. Pharmacol. 2010, 21, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Soriano, F.X.; Hardingham, G.E. In cortical neurons HDAC3 activity suppresses RD4-dependent SMRT export. PLoS ONE 2011, 6, e21056. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.W.; Gao, Z.; Li, S.; Farooqi, M.; Tang, T.S.; Bezprozvanny, I.; Frantz, D.E.; Hsieh, J. Small-molecule activation of neuronal cell fate. Nat Chem Biol 2008, 4, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Lai, I.L.; Lin, T.P.; Yao, Y.L.; Lin, C.Y.; Hsieh, M.J.; Yang, W.M. Histone deacetylase 10 relieves repression on the melanogenic program by maintaining the deacetylation status of repressors. J. Biol. Chem. 2010, 285, 7187–7196. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Cavalli, V. HDAC signaling in neuronal development and axon regeneration. Curr. Opin. Neurobiol. 2014, 27, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.M.; Wood, M.A. Beyond transcription factors: The role of chromatin modifying enzymes in regulating transcription required for memory. Learn. Mem. 2008, 15, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Vecsey, C.G.; Hawk, J.D.; Lattal, K.M.; Stein, J.M.; Fabian, S.A.; Attner, M.A.; Cabrera, S.M.; McDonough, C.B.; Brindle, P.K.; Abel, T.; et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J. Neurosci. 2007, 27, 6128–6140. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.H.; Lin, C.H.; Gean, P.W. Acetylation of nuclear factor-kappaB in rat amygdala improves long-term but not short-term retention of fear memory. Mol. Pharmacol. 2004, 65, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; Sweatt, J.D.; Rumbaugh, G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2010, 35, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Didonna, A.; Opal, P. The promise and perils of HDAC inhibitors in neurodegeneration. Ann. Clin. Transl. Neurol. 2015, 2, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Pena, P.; Lan, F.; Kaadige, M.R.; et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Kachirskaia, I.; Walter, K.L.; Kuo, J.H.; Lake, A.; Davrazou, F.; Chan, S.M.; Martin, D.G.; Fingerman, I.M.; Briggs, S.D.; et al. Proteome-wide analysis in Saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J. Biol. Chem. 2007, 282, 2450–2455. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, J.; Swigut, T.; Xiao, H.; Milne, T.A.; Kwon, S.Y.; Landry, J.; Kauer, M.; Tackett, A.J.; Chait, B.T.; Badenhorst, P.; et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 2006, 442, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Jing, C.; Wilson, J.R.; Walker, P.A.; Vasisht, N.; Kelly, G.; Howell, S.; Taylor, I.A.; Blackburn, G.M.; Gamblin, S.J. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature 2003, 421, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Zhou, M.M. SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cell. Mol. Life Sci. 2006, 63, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Abhiman, S.; Iyer, L.M. Natural history of the eukaryotic chromatin protein methylation system. Prog. Mol. Biol. Transl. Sci. 2011, 101, 105–176. [Google Scholar] [PubMed]
- Glaser, S.; Schaft, J.; Lubitz, S.; Vintersten, K.; van der Hoeven, F.; Tufteland, K.R.; Aasland, R.; Anastassiadis, K.; Ang, S.L.; Stewart, A.F. Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development 2006, 133, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Goo, Y.H.; Sohn, Y.C.; Kim, D.H.; Kim, S.W.; Kang, M.J.; Jung, D.J.; Kwak, E.; Barlev, N.A.; Berger, S.L.; Chow, V.T.; et al. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol. Cell. Biol. 2003, 23, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. Mll targets SET domain methyltransferase activity to hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Peters, A.H.; O’Carroll, D.; Scherthan, H.; Mechtler, K.; Sauer, S.; Schofer, C.; Weipoltshammer, K.; Pagani, M.; Lachner, M.; Kohlmaier, A.; et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 2001, 107, 323–337. [Google Scholar] [CrossRef]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Ueda, J.; Fukuda, M.; Takeda, N.; Ohta, T.; Iwanari, H.; Sakihama, T.; Kodama, T.; Hamakubo, T.; Shinkai, Y. Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., 3rd. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.H.; van Lohuizen, M. Polycomb complexes and silencing mechanisms. Curr. Opin. Cell Biol. 2004, 16, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Otte, A.P.; Kwaks, T.H. Gene repression by Polycomb group protein complexes: A distinct complex for every occasion? Curr. Opin. Genet. Dev. 2003, 13, 448–454. [Google Scholar] [CrossRef]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Jackson, M.W.; Wang, B.; Yang, M.; Chance, M.R.; Miyagi, M.; Gudkov, A.V.; Stark, G.R. Regulation of NF-kappaB by NSD1/FBXL11-dependent reversible lysine methylation of p65. Proc. Natl. Acad. Sci. USA 2010, 107, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Saddic, L.A.; West, L.E.; Aslanian, A.; Yates, J.R., 3rd; Rubin, S.M.; Gozani, O.; Sage, J. Methylation of the retinoblastoma tumor suppressor by SMYD2. J. Biol. Chem. 2010, 285, 37733–37740. [Google Scholar] [CrossRef] [PubMed]
- Kunizaki, M.; Hamamoto, R.; Silva, F.P.; Yamaguchi, K.; Nagayasu, T.; Shibuya, M.; Nakamura, Y.; Furukawa, Y. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 2007, 67, 10759–10765. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Zhang, Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011, 25, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Karachentsev, D.; Sarma, K.; Reinberg, D.; Steward, R. PR-SET7-dependent methylation of histone H4 lys 20 functions in repression of gene expression and is essential for mitosis. Genes Dev. 2005, 19, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, M.A.; Bochar, D.A.; Chenoweth, J.; Lane, W.S.; Mandel, G.; Shiekhattar, R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc. Natl. Acad. Sci. USA 2002, 99, 7420–7425. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, M.A.; Dong, Y.; Lane, W.S.; Speicher, D.W.; Shiekhattar, R. A candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexes. J. Biol. Chem. 2003, 278, 7234–7239. [Google Scholar] [CrossRef] [PubMed]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, D.N.; Su, H.; Hevi, S.; Gay, F.; Lei, H.; Bajko, J.; Xu, G.; Li, E.; Chen, T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009, 461, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Van Essen, D.; Zhu, Y.; Saccani, S. A feed-forward circuit controlling inducible NF-kappaB target gene activation by promoter histone demethylation. Mol. Cell 2010, 39, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Toumazou, C.; Tsukada, Y.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Fodor, B.D.; Kubicek, S.; Yonezawa, M.; O’Sullivan, R.J.; Sengupta, R.; Perez-Burgos, L.; Opravil, S.; Mechtler, K.; Schotta, G.; Jenuwein, T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006, 20, 1557–1562. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Yamane, K.; Bae, Y.; Zhang, D.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 2006, 442, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Adams-Cioaba, M.A.; Min, J. Structure and function of histone methylation binding proteins. Biochem. Cell Biol. 2009, 87, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Fang, J.; Bedford, M.T.; Zhang, Y.; Xu, R.M. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science 2006, 312, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Kawazu, M.; Saso, K.; Tong, K.I.; McQuire, T.; Goto, K.; Son, D.O.; Wakeham, A.; Miyagishi, M.; Mak, T.W.; Okada, H. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS ONE 2011, 6, e17830. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Sun, L.; Li, Q.; Liang, J.; Yu, W.; Yi, X.; Yang, X.; Li, Y.; Han, X.; Zhang, Y.; et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 7541–7546. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.; Agger, K.; Cloos, P.A.; Pasini, D.; Rose, S.; Sennels, L.; Rappsilber, J.; Hansen, K.H.; Salcini, A.E.; Helin, K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 2007, 128, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Iwase, S.; Lan, F.; Bayliss, P.; de la Torre-Ubieta, L.; Huarte, M.; Qi, H.H.; Whetstine, J.R.; Bonni, A.; Roberts, T.M.; Shi, Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 2007, 128, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Norman, J.; Shilatifard, A.; Shiekhattar, R. Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a polycomb-like protein. Cell 2007, 128, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Mei, P.; Fang, R.; Leonor, T.; Rutenberg, M.; Shimizu, F.; Li, J.; Rao, A.; Shi, Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 2007, 447, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Yamane, K.; Tateishi, K.; Klose, R.J.; Fang, J.; Fabrizio, L.A.; Erdjument-Bromage, H.; Taylor-Papadimitriou, J.; Tempst, P.; Zhang, Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell 2007, 25, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.G.; Song, J.; Wang, Z.; Dormann, H.L.; Casadio, F.; Li, H.; Luo, J.L.; Patel, D.J.; Allis, C.D. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature 2009, 459, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase JMJD3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Cho, Y.W.; Yu, L.R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.P.; Reinberg, D.; di Croce, L.; Shiekhattar, R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Zhu, Z.; Han, G.; Lin, H.; Xu, L.; Chen, C.D. Jmjd3 is a histone H3K27 demethylase. Cell Res. 2007, 17, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.; Ohtake, F.; Okuno, Y.; Yokota, K.; Okada, M.; Imai, Y.; Ni, M.; Meyer, C.A.; Igarashi, K.; Kanno, J.; et al. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat. Cell. Biol. 2011, 13, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Yonezawa, M.; Ye, J.; Jenuwein, T.; Grummt, I. PHF8 activates transcription of rRNA genes through H3K4me3 binding and H3K9me1/2 demethylation. Nat. Struct. Mol. Biol. 2010, 17, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Fortschegger, K.; de Graaf, P.; Outchkourov, N.S.; van Schaik, F.M.; Timmers, H.T.; Shiekhattar, R. PHF8 targets histone methylation and RNA polymerase II to activate transcription. Mol. Cell. Biol. 2010, 30, 3286–3298. [Google Scholar] [CrossRef] [PubMed]
- Kleine-Kohlbrecher, D.; Christensen, J.; Vandamme, J.; Abarrategui, I.; Bak, M.; Tommerup, N.; Shi, X.; Gozani, O.; Rappsilber, J.; Salcini, A.E.; et al. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol. Cell 2010, 38, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Shi, G.; Jia, Y.; Li, J.; Wu, M.; Dong, S.; Wong, J. The X-linked mental retardation gene PHF8 is a histone demethylase involved in neuronal differentiation. Cell Res. 2010, 20, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Li, J.; Song, T.; Lu, M.; Kan, P.Y.; Lee, M.G.; Sha, B.; Shi, X. Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J. Biol. Chem. 2010, 285, 9322–9326. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, Y.; Li, X.; Xu, L.; Wang, X.; Sun, T.; Dong, X.; Chen, L.; Mao, H.; Yu, Y.; et al. PHF8 is a histone H3K9me2 demethylase regulating rrna synthesis. Cell Res. 2010, 20, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tanasa, B.; Tyurina, O.V.; Zhou, T.Y.; Gassmann, R.; Liu, W.T.; Ohgi, K.A.; Benner, C.; Garcia-Bassets, I.; Aggarwal, A.K.; et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 2010, 466, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.R.; Upadhyay, A.K.; Qi, H.H.; Zhang, X.; Shi, Y.; Cheng, X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010, 17, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.H.; Sarkissian, M.; Hu, G.Q.; Wang, Z.; Bhattacharjee, A.; Gordon, D.B.; Gonzales, M.; Lan, F.; Ongusaha, P.P.; Huarte, M.; et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 2010, 466, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wang, Y.; Huang, S.; Wang, J.; Deng, Z.; Zhang, Q.; Wu, W.; Zhang, X.; Liu, Z.; Gong, W.; et al. Structural insights into a novel histone demethylase PHF8. Cell Res. 2010, 20, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Veazey, K.J.; Parnell, S.E.; Miranda, R.C.; Golding, M.C. Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenet. Chromatin 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Tops, B.B.; Plasterk, R.H.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Nam, J.W.; Heo, I.; Rhee, J.K.; Sohn, S.Y.; Cho, Y.; Zhang, B.T.; Kim, V.N. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 2006, 125, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Carmell, M.A.; Hannon, G.J. RNase III enzymes and the initiation of gene silencing. Nat. Struct. Mol. Biol. 2004, 11, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Impey, S.; Goodman, R.H. Role reversal: The regulation of neuronal gene expression by microRNAs. Curr. Opin. Neurobiol. 2005, 15, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S. The neuronal microRNA system. Nat. Rev. Neurosci. 2006, 7, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nature Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.J.; Gregory, R.I. MicroRNA gene regulatory pathways in the establishment and maintenance of esc identity. Cell Stem Cell 2010, 7, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Hsieh, J.; Nakashima, K.; Taira, K.; Gage, F.H. A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell 2004, 116, 779–793. [Google Scholar] [CrossRef]
- Poole, R.J.; Hobert, O. Early embryonic programming of neuronal left/right asymmetry in C. elegans. Curr. Biol. 2006, 16, 2279–2292. [Google Scholar] [CrossRef] [PubMed]
- Ronshaugen, M.; Biemar, F.; Piel, J.; Levine, M.; Lai, E.C. The Drosophila microRNA iab-4 causes a dominant homeotic transformation of halteres to wings. Genes Dev. 2005, 19, 2947–2952. [Google Scholar] [CrossRef] [PubMed]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Olde Loohuis, N.F.; Kos, A.; Martens, G.J.; van Bokhoven, H.; Nadif Kasri, N.; Aschrafi, A. MicroRNA networks direct neuronal development and plasticity. Cell. Mol. Life Sci. 2012, 69, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.H.; Cuellar, T.L.; Koch, S.M.; Barker, A.J.; Harfe, B.D.; McManus, M.T.; Ullian, E.M. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J. Neurosci. 2008, 28, 4322–4330. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, A.S.; DiRocco, D.P.; Lambert, T.J.; Garelick, M.G.; Le, J.; Nathanson, N.M.; Storm, D.R. Neuronal activity rapidly induces transcription of the CREB-regulated microRNA-132, in vivo. Hippocampus 2010, 20, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Wayman, G.A.; Davare, M.; Ando, H.; Fortin, D.; Varlamova, O.; Cheng, H.Y.; Marks, D.; Obrietan, K.; Soderling, T.R.; Goodman, R.H.; et al. An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc. Natl. Acad. Sci. USA 2008, 105, 9093–9098. [Google Scholar] [CrossRef] [PubMed]
- Muddashetty, R.S.; Nalavadi, V.C.; Gross, C.; Yao, X.; Xing, L.; Laur, O.; Warren, S.T.; Bassell, G.J. Reversible inhibition of PSD-95 mRNA translation by miR-125a, FMRP phosphorylation, and mGluR signaling. Mol. Cell 2011, 42, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Busskamp, V.; Markiewicz, I.; Stadler, M.B.; Ribi, S.; Richter, J.; Duebel, J.; Bicker, S.; Fehling, H.J.; Schubeler, D.; et al. Characterizing light-regulated retinal microRNAs reveals rapid turnover as a common property of neuronal microRNAs. Cell 2010, 141, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kwon, E.J.; Tsai, L.H. MicroRNAs in learning, memory, and neurological diseases. Learn. Mem. 2012, 19, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Shafi, G.; Aliya, N.; Munshi, A. MicroRNA signatures in neurological disorders. Can. J. Neurol. Sci. 2010, 37, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Karayiorgou, M.; Gogos, J.A. MicroRNAs in psychiatric and neurodevelopmental disorders. Brain Res. 2010, 1338, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Smith, D.W. Recognition of the fetal alcohol syndrome in early infancy. Lancet 1973, 2, 999–1001. [Google Scholar] [CrossRef]
- Alati, R.; Al Mamun, A.; Williams, G.M.; O’Callaghan, M.; Najman, J.M.; Bor, W. In utero alcohol exposure and prediction of alcohol disorders in early adulthood: A birth cohort study. Arch. Gen. Psychiatry 2006, 63, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Autti-Ramo, I.; Fagerlund, A.; Ervalahti, N.; Loimu, L.; Korkman, M.; Hoyme, H.E. Fetal alcohol spectrum disorders in Finland: Clinical delineation of 77 older children and adolescents. Am. J. med. Genet. A 2006, 140, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Ceccanti, M.; Alessandra Spagnolo, P.; Tarani, L.; Luisa Attilia, M.; Chessa, L.; Mancinelli, R.; Stegagno, M.; Francesco Sasso, G.; Romeo, M.; Jones, K.L.; et al. Clinical delineation of fetal alcohol spectrum disorders (FASD) in Italian children: Comparison and contrast with other racial/ethnic groups and implications for diagnosis and prevention. Neurosci. Biobehav. Rev. 2007, 31, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Spohr, H.L.; Willms, J.; Steinhausen, H.C. Fetal alcohol spectrum disorders in young adulthood. J. Pediatr. 2007, 150, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Streissguth, A.P.; Aase, J.M.; Clarren, S.K.; Randels, S.P.; LaDue, R.A.; Smith, D.F. Fetal alcohol syndrome in adolescents and adults. JAMA 1991, 265, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Sokol, R.J.; Delaney-Black, V.; Nordstrom, B. Fetal alcohol spectrum disorder. JAMA 2003, 290, 2996–2999. [Google Scholar] [CrossRef] [PubMed]
- Burd, L.; Klug, M.G.; Martsolf, J.T.; Kerbeshian, J. Fetal alcohol syndrome: Neuropsychiatric phenomics. Neurotoxicol. Teratol. 2003, 25, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Aragon, A.S.; Kalberg, W.O.; Buckley, D.; Barela-Scott, L.M.; Tabachnick, B.G.; May, P.A. Neuropsychological study of FASD in a sample of American Indian children: Processing simple versus complex information. Alcohol. Clin. Exp. Res. 2008, 32, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Green, C.R.; Mihic, A.M.; Nikkel, S.M.; Stade, B.C.; Rasmussen, C.; Munoz, D.P.; Reynolds, J.N. Executive function deficits in children with fetal alcohol spectrum disorders (FASD) measured using the Cambridge Neuropsychological Tests Automated Battery (CANTAB). J. Child Psychol. Psychiatry 2009, 50, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Crocker, N.; Nguyen, T.T. Fetal alcohol spectrum disorders: Neuropsychological and behavioral features. Neuropsychol. Rev. 2011, 21, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Mattson, S.N.; Roesch, S.C.; Fagerlund, A.; Autti-Ramo, I.; Jones, K.L.; May, P.A.; Adnams, C.M.; Konovalova, V.; Riley, E.P. Toward a neurobehavioral profile of fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2010, 34, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- May, P.A.; Blankenship, J.; Marais, A.S.; Gossage, J.P.; Kalberg, W.O.; Joubert, B.; Cloete, M.; Barnard, R.; de Vries, M.; Hasken, J.; et al. Maternal alcohol consumption producing fetal alcohol spectrum disorders (FASD): Quantity, frequency, and timing of drinking. Drug Alcohol Depend. 2013, 133, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Lebel, C.; Mattson, S.N.; Riley, E.P.; Jones, K.L.; Adnams, C.M.; May, P.A.; Bookheimer, S.Y.; O’Connor, M.J.; Narr, K.L.; Kan, E.; et al. A longitudinal study of the long-term consequences of drinking during pregnancy: Heavy in utero alcohol exposure disrupts the normal processes of brain development. J. Neurosci. 2012, 32, 15243–15251. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.L.; O’Brien, J.W.; Spadoni, A.D.; Tapert, S.F.; Jones, K.L.; Riley, E.P.; Mattson, S.N. A functional magnetic resonance imaging study of spatial working memory in children with prenatal alcohol exposure: Contribution of familial history of alcohol use disorders. Alcohol. Clin. Exp. Res. 2013, 37, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Sood, B.; Delaney-Black, V.; Covington, C.; Nordstrom-Klee, B.; Ager, J.; Templin, T.; Janisse, J.; Martier, S.; Sokol, R.J. Prenatal alcohol exposure and childhood behavior at age 6 to 7 years: I. dose-response effect. Pediatrics 2001, 108, E34. [Google Scholar] [CrossRef] [PubMed]
- Streissguth, A.P.; Bookstein, F.L.; Sampson, P.D.; Barr, H.M. Neurobehavioral effects of prenatal alcohol: Part III. PLS analyses of neuropsychologic tests. Neurotoxicol. Teratol. 1989, 11, 493–507. [Google Scholar] [CrossRef]
- Guerri, C.; Bazinet, A.; Riley, E.P. Foetal alcohol spectrum disorders and alterations in brain and behaviour. Alcohol Alcohol. 2009, 44, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Bailey, B.N.; Delaney-Black, V.; Covington, C.Y.; Ager, J.; Janisse, J.; Hannigan, J.H.; Sokol, R.J. Prenatal exposure to binge drinking and cognitive and behavioral outcomes at age 7 years. Am. J. Obstet. Gynecol. 2004, 191, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J.; Goodlett, C.R.; West, J.R. Blood alcohol concentration and severity of microencephaly in neonatal rats depend on the pattern of alcohol administration. Alcohol 1988, 5, 209–214. [Google Scholar] [CrossRef]
- Levitt, P.; Veenstra-VanderWeele, J. Neurodevelopment and the origins of brain disorders. Neuropsychopharmacology 2015, 40, 1–3. [Google Scholar] [PubMed]
- Giedd, J.N.; Raznahan, A.; Alexander-Bloch, A.; Schmitt, E.; Gogtay, N.; Rapoport, J.L. Child psychiatry branch of the National Institute of Mental Health longitudinal structural magnetic resonance imaging study of human brain development. Neuropsychopharmacology 2015, 40, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Hammock, E.A. Developmental perspectives on oxytocin and vasopressin. Neuropsychopharmacology 2015, 40, 24–42. [Google Scholar] [CrossRef] [PubMed]
- Hartley, C.A.; Lee, F.S. Sensitive periods in affective development: Nonlinear maturation of fear learning. Neuropsychopharmacology 2015, 40, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.J.; Graham, D.L.; Money, K.M.; Stanwood, G.D. Developmental consequences of fetal exposure to drugs: What we know and what we still must learn. Neuropsychopharmacology 2015, 40, 61–87. [Google Scholar] [CrossRef] [PubMed]
- Washbourne, P. Synapse assembly and neurodevelopmental disorders. Neuropsychopharmacology 2015, 40, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Archer, T.; Oscar-Berman, M.; Blum, K. Epigenetics in developmental disorder: ADHD and endophenotypes. J. Genet. Syndr. Gene Ther. 2011, 2. [Google Scholar] [CrossRef]
- Sakurai, T.; Dorr, N.P.; Takahashi, N.; McInnes, L.A.; Elder, G.A.; Buxbaum, J.D. Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res. 2011, 4, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Schanen, N.C. Epigenetics of autism spectrum disorders. Hum. Mol. Genet. 2006, 15, R138–R150. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Basavarajappa, B.S. Pre-administration of G9a/GLP inhibitor during synaptogenesis prevents postnatal ethanol-induced LTP deficits and neurobehavioral abnormalities in adult mice. Exp. Neurol. 2014, 261, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Downing, C.; Johnson, T.E.; Larson, C.; Leakey, T.I.; Siegfried, R.N.; Rafferty, T.M.; Cooney, C.A. Subtle decreases in DNA methylation and gene expression at the mouse Igf2 locus following prenatal alcohol exposure: Effects of a methyl-supplemented diet. Alcohol 2011, 45, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Garro, A.J.; McBeth, D.L.; Lima, V.; Lieber, C.S. Ethanol consumption inhibits fetal DNA methylation in mice: Implications for the fetal alcohol syndrome. Alcohol Clin. Exp. Res. 1991, 15, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Haycock, P.C.; Ramsay, M. Exposure of mouse embryos to ethanol during preimplantation development: Effect on DNA methylation in the H19 imprinting control region. Biol. Reprod. 2009, 81, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Balaraman, Y.; Wang, G.; Nephew, K.P.; Zhou, F.C. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 2009, 4, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Ouko, L.A.; Shantikumar, K.; Knezovich, J.; Haycock, P.; Schnugh, D.J.; Ramsay, M. Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes: Implications for fetal alcohol spectrum disorders. Alcohol Clin. Exp. Res. 2009, 33, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.C.; Balaraman, Y.; Teng, M.; Liu, Y.; Singh, R.P.; Nephew, K.P. Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol Clin. Exp. Res. 2011, 35, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in beta-endorphin-producing pomc neurons of the hypothalamus. Alcohol. Clin. Exp. Res. 2013, 37, 1133–1142. [Google Scholar] [PubMed]
- Govorko, D.; Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol. Psychiatry 2012, 72, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Crossey, E.L.; Zhang, L.; Zucca, S.; George, O.L.; Valenzuela, C.F.; Zhao, X. Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS ONE 2011, 6, e19351. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Psychoyos, D.; Shan, X.; Basavarajappa, B.S. Postnatal ethanol exposure alters levels of 2-arachidonylglycerol-metabolizing enzymes and pharmacological inhibition of monoacylglycerol (MAGL) does not cause neurodegeneration in neonatal mice. J. Neurochem. 2015, 134, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Zhu, J.; Lv, T.; Chen, G.; Sun, H.; Yang, X.; Huang, X.; Tian, J. Ethanol and its metabolites induce histone lysine 9 acetylation and an alteration of the expression of heart development-related genes in cardiac progenitor cells. Cardiovasc. Toxicol. 2010, 10, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Balaraman, S.; Winzer-Serhan, U.H.; Miranda, R.C. Opposing actions of ethanol and nicotine on microRNAs are mediated by nicotinic acetylcholine receptors in fetal cerebral cortical-derived neural progenitor cells. Alcohol Clin. Exp. Res. 2012, 36, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Caputo, V.; Sinibaldi, L.; Fiorentino, A.; Parisi, C.; Catalanotto, C.; Pasini, A.; Cogoni, C.; Pizzuti, A. Brain derived neurotrophic factor (BDNF) expression is regulated by microRNAs miR-26a and miR-26b allele-specific binding. PLoS ONE 2011, 6, e28656. [Google Scholar] [CrossRef] [PubMed]
- Dill, H.; Linder, B.; Fehr, A.; Fischer, U. Intronic miR-26b controls neuronal differentiation by repressing its host transcript, ctdsp2. Genes Dev. 2012, 26, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Sathyan, P.; Golden, H.B.; Miranda, R.C. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: Evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J. Neurosci. 2007, 27, 8546–8557. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.R.; Pereira, P.M.; Ferreira, V.; Reverendo, M.; Simoes, J.; Bezerra, A.R.; Moura, G.R.; Santos, M.A. Ethanol exposure induces upregulation of specific microRNAs in zebrafish embryos. Toxicol. Sci. 2012, 127, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Stringer, R.L.; Laufer, B.I.; Kleiber, M.L.; Singh, S.M. Reduced expression of brain cannabinoid receptor 1 (Cnr1) is coupled with an increased complementary micro-RNA (miR-26b) in a mouse model of fetal alcohol spectrum disorders. Clin. Epigenet. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Zhang, Z.; Li, Q.; Yang, R.; Pei, X.; Xu, Y.; Wang, J.; Zhou, S.F.; Li, Y. Ethanol exposure induces differential microRNA and target gene expression and teratogenic effects which can be suppressed by folic acid supplementation. Hum. Reprod. 2009, 24, 562–579. [Google Scholar] [CrossRef] [PubMed]
- Valles, S.; Pitarch, J.; Renau-Piqueras, J.; Guerri, C. Ethanol exposure affects glial fibrillary acidic protein gene expression and transcription during rat brain development. J. Neurochem. 1997, 69, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, D.M.; Zaher, F.M.; Svinarich, D.M.; Abel, E.L. Paternal alcohol exposure affects sperm cytosine methyltransferase messenger RNA levels. Alcohol Clin. Exp. Res. 2002, 26, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Tycko, B.; Morison, I.M. Physiological functions of imprinted genes. J. Cell. Physiol. 2002, 192, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Bliek, J.; Terhal, P.; van den Bogaard, M.J.; Maas, S.; Hamel, B.; Salieb-Beugelaar, G.; Simon, M.; Letteboer, T.; van der Smagt, J.; Kroes, H.; et al. Hypomethylation of the H19 gene causes not only Silver-Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am. J. Hum. Genet. 2006, 78, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nature Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Dean, W.; Bowden, L.; Aitchison, A.; Klose, J.; Moore, T.; Meneses, J.J.; Reik, W.; Feil, R. Altered imprinted gene methylation and expression in completely ES cell-derived mouse fetuses: Association with aberrant phenotypes. Development 1998, 125, 2273–2282. [Google Scholar] [PubMed]
- Khosla, S.; Dean, W.; Brown, D.; Reik, W.; Feil, R. Culture of preimplantation mouse embryos affects fetal development and the expression of imprinted genes. Biol. Reprod. 2001, 64, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, K.D.; Young, L.E.; Wilmut, I.; McEvoy, T.G. In-utero overgrowth in ruminants following embryo culture: Lessons from mice and a warning to men. Hum. Reprod. 2000, 15 (Suppl. 5), 68–86. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.D.; Middleton, F.A.; Miller, M.W. Ethanol-induced methylation of cell cycle genes in neural stem cells. J. Neurochem. 2010, 114, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.C.; Chen, Y.; Love, A. Cellular DNA methylation program during neurulation and its alteration by alcohol exposure. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ozturk, N.C.; Zhou, F.C. DNA methylation program in developing hippocampus and its alteration by alcohol. PLoS ONE 2013, 8, e60503. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.H.; Williams, J.K.; Thomas, J.D. Choline supplementation attenuates learning deficits associated with neonatal alcohol exposure in the rat: Effects of varying the timing of choline administration. Brain Res. 2008, 1237, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Biane, J.S.; O’Bryan, K.A.; O’Neill, T.M.; Dominguez, H.D. Choline supplementation following third-trimester-equivalent alcohol exposure attenuates behavioral alterations in rats. Behav. Neurosci. 2007, 121, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Otero, N.K.; Thomas, J.D.; Saski, C.A.; Xia, X.; Kelly, S.J. Choline supplementation and DNA methylation in the hippocampus and prefrontal cortex of rats exposed to alcohol during development. Alcohol. Clin. Exp. Res. 2012, 36, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rezzoug, F.; Kaikaus, J.; Greene, R.M.; Pisano, M.M. Alcohol modulates expression of DNA methyltranferases and methyl CpG-/CpG domain-binding proteins in murine embryonic fibroblasts. Reprod. Toxicol. 2013, 37, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Shivakumar, M.; Psychoyos, D.; Xie, S.; Basavarajappa, B.S. Anandamide-CB1 receptor signaling contributes to postnatal ethanol-induced neonatal neurodegeneration, adult synaptic and memory deficits. J. Neuosci. 2013, 33, 6350–6366. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Lehmann, C.; Lawrence, R.C.; Kelly, S.J. Alcohol exposure during development: Impact on the epigenome. Int. J. Dev. Neurosci. 2013, 31, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.R.; Ho, M.F.; Vega, M.C.; Burne, T.H.; Chong, S. Prenatal ethanol exposure alters adult hippocampal VGLUT2 expression with concomitant changes in promoter DNA methylation, H3K4 trimethylation and miR-467b-5p levels. Epigenet. Chromatin 2015, 8. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Blewitt, M.E.; Druker, R.; Preis, J.I.; Whitelaw, E. Metastable epialleles in mammals. Trends Genet. 2002, 18, 348–351. [Google Scholar] [CrossRef]
- Wolff, G.L. Influence of maternal phenotype on metabolic differentiation of agouti locus mutants in the mouse. Genetics 1978, 88, 529–539. [Google Scholar] [PubMed]
- Khalid, O.; Kim, J.J.; Kim, H.S.; Hoang, M.; Tu, T.G.; Elie, O.; Lee, C.; Vu, C.; Horvath, S.; Spigelman, I.; et al. Gene expression signatures affected by alcohol-induced DNA methylomic deregulation in human embryonic stem cells. Stem Cell Res. 2014, 12, 791–806. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, A.K.; Khan, I.A. Modulation of DNA methylation machineries in Japanese rice fish (Oryzias latipes) embryogenesis by ethanol and 5-azacytidine. Comp. Biochemistry Physiol. Toxicol. Pharmacol. 2016, 179, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Laufer, B.I.; Mantha, K.; Kleiber, M.L.; Diehl, E.J.; Addison, S.M.; Singh, S.M. Long-lasting alterations to DNA methylation and ncRNAs could underlie the effects of fetal alcohol exposure in mice. Dis. Models Mech. 2013, 6, 977–992. [Google Scholar] [CrossRef] [PubMed]
- Portales-Casamar, E.; Mah, S.M.; MacIsaac, J.L.; Barhdadi, A.; Provost, S.; Dube, M.P.; Reynolds, J.N.; Pavlidis, P.; Kobor, M.S. DNA methylation changes in fetal alcohol spectrum disorder. Int. J. Dev. Neurosci. 2015, 47. [Google Scholar] [CrossRef]
- Wood, M.A.; Attner, M.A.; Oliveira, A.M.; Brindle, P.K.; Abel, T. A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn. Mem. 2006, 13, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, F.; Lewin, J.; Cortese, R.; Rakyan, V.K.; Attwood, J.; Burger, M.; Burton, J.; Cox, T.V.; Davies, R.; Down, T.A.; et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006, 38, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Rishi, V.; Bhattacharya, P.; Chatterjee, R.; Rozenberg, J.; Zhao, J.; Glass, K.; Fitzgerald, P.; Vinson, C. CpG methylation of half-CRE sequences creates C/EBPalpha binding sites that activate some tissue-specific genes. Proc. Natl. Acad. Sci. USA 2010, 107, 20311–20316. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.E.; Cramer, J.A.; West, J.R.; Sohrabji, F. Alcohol exposure during the first two trimesters equivalent alters granule cell number and neurotrophin expression in the developing rat olfactory bulb. J. Neurobiol. 1999, 41, 414–423. [Google Scholar] [CrossRef]
- Hu, Y.; Willett, K.L.; Khan, I.A.; Scheffler, B.E.; Dasmahapatra, A.K. Ethanol disrupts chondrification of the neurocranial cartilages in medaka embryos without affecting aldehyde dehydrogenase 1A2 (Aldh1A2) promoter methylation. Comp. Biochem. Physiol. Toxicol. Pharmacol. 2009, 150, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, M.; Gerwe, B.A.; Scharer, C.D.; Sahasranaman, V.; Eilertson, C.D.; Nash, R.J.; Usta, S.N.; Kelly, S.; Rose, M.; Peraza, R.; et al. Ethanol alters proliferation and differentiation of normal and chromosomally abnormal human embryonic stem cell-derived neurospheres. Birth Defects Res. B Dev. Reprod. Toxicol. 2013, 98, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Tyler, C.R.; Allan, A.M. Prenatal alcohol exposure alters expression of neurogenesis-related genes in an ex vivo cell culture model. Alcohol 2014, 48, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Marjonen, H.; Sierra, A.; Nyman, A.; Rogojin, V.; Grohn, O.; Linden, A.M.; Hautaniemi, S.; Kaminen-Ahola, N. Early maternal alcohol consumption alters hippocampal DNA methylation, gene expression and volume in a mouse model. PLoS ONE 2015, 10, e0124931. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, A.K.; Khan, I.A. DNA methyltransferase expressions in Japanese rice fish (Oryzias latipes) embryogenesis is developmentally regulated and modulated by ethanol and 5-azacytidine. Comp. Biochem. Physiol. Toxicol. Pharmacol. 2015, 176–177, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ramadoss, J.; Liao, W.X.; Chen, D.B.; Magness, R.R. High-throughput caveolar proteomic signature profile for maternal binge alcohol consumption. Alcohol 2010, 44, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Peng, C.; Zheng, M.; Gao, W.; Zhu, J.; Lv, T.; Liu, L.; Liu, Z.; Li, H.; Xv, Y.; et al. Prenatal alcohol exposure causes the over-expression of dhand and ehand by increasing histone H3K14 acetylation in C57 BL/6 mice. Toxicol. Lett. 2014, 228, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zhu, J.; Sun, H.C.; Huang, X.P.; Zhao, W.A.; Zheng, M.; Liu, L.J.; Tian, J. Inhibition of histone H3K9 acetylation by anacardic acid can correct the over-expression of Gata4 in the hearts of fetal mice exposed to alcohol during pregnancy. PLoS ONE 2014, 9, e104135. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Idrus, N.M.; Monk, B.R.; Dominguez, H.D. Prenatal choline supplementation mitigates behavioral alterations associated with prenatal alcohol exposure in rats. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo-Carter, D.L.; Balaraman, S.; Sathyan, P.; Carter, E.S.; Chen, W.J.; Miranda, R.C. Suppression and epigenetic regulation of miR-9 contributes to ethanol teratology: Evidence from zebrafish and murine fetal neural stem cell models. Alcohol. Clin. Exp. Res. 2013, 37, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Mantha, K.; Laufer, B.I.; Singh, S.M. Molecular changes during neurodevelopment following second-trimester binge ethanol exposure in a mouse model of fetal alcohol spectrum disorder: From immediate effects to long-term adaptation. Dev. Neurosci. 2014, 36, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Ignacio, C.; Mooney, S.M.; Middleton, F.A. Effects of acute prenatal exposure to ethanol on microRNA expression are ameliorated by social enrichment. Front. Pediatrics 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Balaraman, S.; Lunde, E.R.; Sawant, O.; Cudd, T.A.; Washburn, S.E.; Miranda, R.C. Maternal and neonatal plasma microRNA biomarkers for fetal alcohol exposure in an ovine model. Alcohol. Clin. Exp. Res. 2014, 38, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Parchem, R.J.; Moore, N.; Fish, J.L.; Parchem, J.G.; Braga, T.T.; Shenoy, A.; Oldham, M.C.; Rubenstein, J.L.; Schneider, R.A.; Blelloch, R. MiR-302 is required for timing of neural differentiation, neural tube closure, and embryonic viability. Cell Rep. 2015, 12, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, J.; Feng, W.K.; Wu, X.; Chen, S.Y. MiR-125b protects against ethanol-induced apoptosis in neural crest cells and mouse embryos by targeting Bak 1 and PUMA. Exp. Neurol. 2015, 271, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Kleiber, M.L.; Laufer, B.I.; Stringer, R.L.; Singh, S.M. Third trimester-equivalent ethanol exposure is characterized by an acute cellular stress response and an ontogenetic disruption of genes critical for synaptic establishment and function in mice. Dev. Neurosci. 2014, 36, 499–519. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alcohol Exposure | Tissue Examined | Effects |
|---|---|---|
| 1. GD 9–11 | GD 12 fetus | Reduced DNA methylation [215] |
| 2. GD | Cultured astrocytes | BDNF gene hypermethylation [260] |
| 3. GD 1–18 | PD 10 | Hypermethylation of the GFAP gene [232] |
| 4. Oryzias latipes embryos | 2–6 dpf | No change in Aldh1A2 gene promoter methylation [261] |
| 5. Embryo (E8.25) cultures | Embryo cultures | Both hyper/hypomethylation of gene promoter [217] |
| 6. Neural stem cells in culture | NSCs culture | Hypermethylation of cell cycle genes [240] |
| 7. Neural stem cells in culture | NSCs culture | Hypomethylation of NSC genes [219] |
| 8. GD 9 | E-15 and 17 | Decreased Igf2 gene DNA methylation [214] |
| 9. PD 2–10 | PD 21 | Increased DNA methylation [245] |
| 10. GD 1–22 | PD 21 | Enhanced DNMTs activity [248] |
| 11. hESC | Cell lines | No change in gene methylation [262] |
| 12. hESC | Cell lines | Hypermethylation of many regions of chromosomes [252] |
| 13. GD 1–17 | Cultured neural progenitor cells | Disrupt DNA methylation machinery and delays the maturation of dentate gyrus [242] |
| 14. Murine embryonic fibroblasts | Fibroblasts | Impaired DNA methylation and DNMT1, DNMT3a and DNMT3b proteins [246] |
| 15. GD 7–21 | PD 60–65 | Increased DNMT1protein and Pomc gene methylation [220] |
| 16. PD 7 | PD 7 | Reduced DNA methylation, DNMT1and DNMT3a proteins [23] |
| 17. GD 0.5–8.5 | PD 28 | Decreased mRNA levels of Dnmt1 and Dnmt3a genes [263] |
| 18. GD 0.5–8.5 | PD 28 | Both up/down regulation of DNA methylation [264] |
| 19. GD 0.5–8.5 | PD 87 | Decreased Slc17a6 gene promoter [249] |
| 20. Oryzias latipes embryos | 2–6 dpf | Altered DNMT1 mRNA [265] |
| Alcohol Exposure | Tissue Examined | Effects |
|---|---|---|
| 1. Gestational Day (GD) 7 | GD 17 | Increased H3K9me2, H3K9ace and decreased H3K27me3 [162] Altered G9a, Setdb1, Kdm1a, Kdm4c, Uhrf1, Ezh2 and Dnmt1 mRNA levels [162] |
| 2. Postnatal Days (PD) 2–12 | PD 2–12 | Decreased AcH3, AcH4, H3K23ace and increased HAT (CBP) [222] |
| 3. GD 7–21 | PD 60–80 from F1–F3 generation | Decreased H3K4me2, H3K4me3, H3K9ace, pH3S10 and mRNA Set7/9 [220]; Increased mRNA G9a, setdb1 H3K9me2 [220] Decreased H3K4me2,H3K4me3, H3K9ace and Decreased mRNA levels of Set7/9; Increased G9a mRNA H3K9me2 and HDAC2 [221] |
| 4. GD 1–14.5 | Embryonic days (ED) 7.0–14.5 | Increased H3K14ace [267] |
| 5. PD 7 | PD 7 | Increased H4K8ace [4], H3K14ace [5] Decreased H3K9me2, H3K27me2 [4] Decreased H3K9me2, H3K27me2 [6], H3K9me2 [213] Increased G9a mRNA and Protein [6] |
| 6. ED 8.5–E16.5 | ED14.5–PD 7 | Increased p300 and SRC1 protein. No Change in HDAC. Increased HAT (CBP and PACF) [268] |
| 7. ED 8.5–16.5 | ED 14.5–16.5 | Increased H3K14ace [267] |
| 8. Days of post cotium (dpc) 0.5–8.5 | PD 87 | Increased H3K4me3 and Slcl7a6 gene expression [249] |
| Alcohol Exposure | Tissue Examined | Effects |
|---|---|---|
| 1. GD 12.5 | Neurosphere cultures | Reduced the expression of miR-9, 21, 335 and -135 [228] |
| 2. GD 6–15 | GD 17 embryo culture | Increased miR-10a, -9, -145, -30a-3p and -152. Also decreased miR-200a, -496, -296, -30e-5p, -362, -339, -29c and -154 [231] |
| 3. Zebrafish | Embryos (4–96 hpf) | Enhanced miR-153a, -30d, -736, -183 and reduced -23a [229] |
| 4. GD 12.5 | Neurosphere cultures | Reduced the expression of miR-140-3p [225] |
| 5. PD 7 | PD 60 | Increased expression of miR-26b [230] |
| 6. Zebrafish | Embryos (24–72 hpf) | Suppressed miR-9a and increased the accumulation of pre-miR-9-3 [270] |
| 7. PD 7 | PD 60 | Enhanced miR-302c [271] |
| 8. GD 1–22 | PD 42 | Decreased miR-874-5p, 1843a-3p, -221-5p, -29c-3p, -384-5p, -412-3p, 129-1, -138-2, -322-2, -496, -9a-2. Increased miR-155, -34c, -let-7c-1, -let-7c-2-3p, -542-1 [272] |
| 9. GD 0.5–8.5 | PD 87 | Enhanced miR-135a, -135b, -467b-5b and -487b [249] |
| 10. GD 4–132 (Ewes) | Plasma (GD 147) | Decreased miR-572, -720, -9, -15b, -17-92 and increased miR-34b [273] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basavarajappa, B.S.; Subbanna, S. Epigenetic Mechanisms in Developmental Alcohol-Induced Neurobehavioral Deficits. Brain Sci. 2016, 6, 12. https://doi.org/10.3390/brainsci6020012
Basavarajappa BS, Subbanna S. Epigenetic Mechanisms in Developmental Alcohol-Induced Neurobehavioral Deficits. Brain Sciences. 2016; 6(2):12. https://doi.org/10.3390/brainsci6020012
Chicago/Turabian StyleBasavarajappa, Balapal S., and Shivakumar Subbanna. 2016. "Epigenetic Mechanisms in Developmental Alcohol-Induced Neurobehavioral Deficits" Brain Sciences 6, no. 2: 12. https://doi.org/10.3390/brainsci6020012