Monomeric, Oligomeric and Polymeric Proteins in Huntington Disease and Other Diseases of Polyglutamine Expansion

Abstract
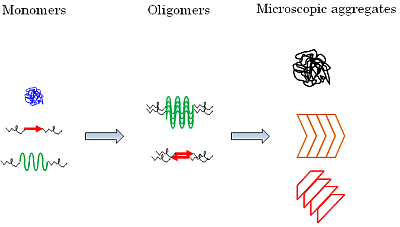
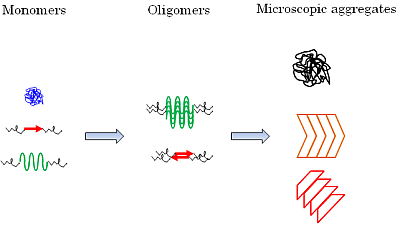
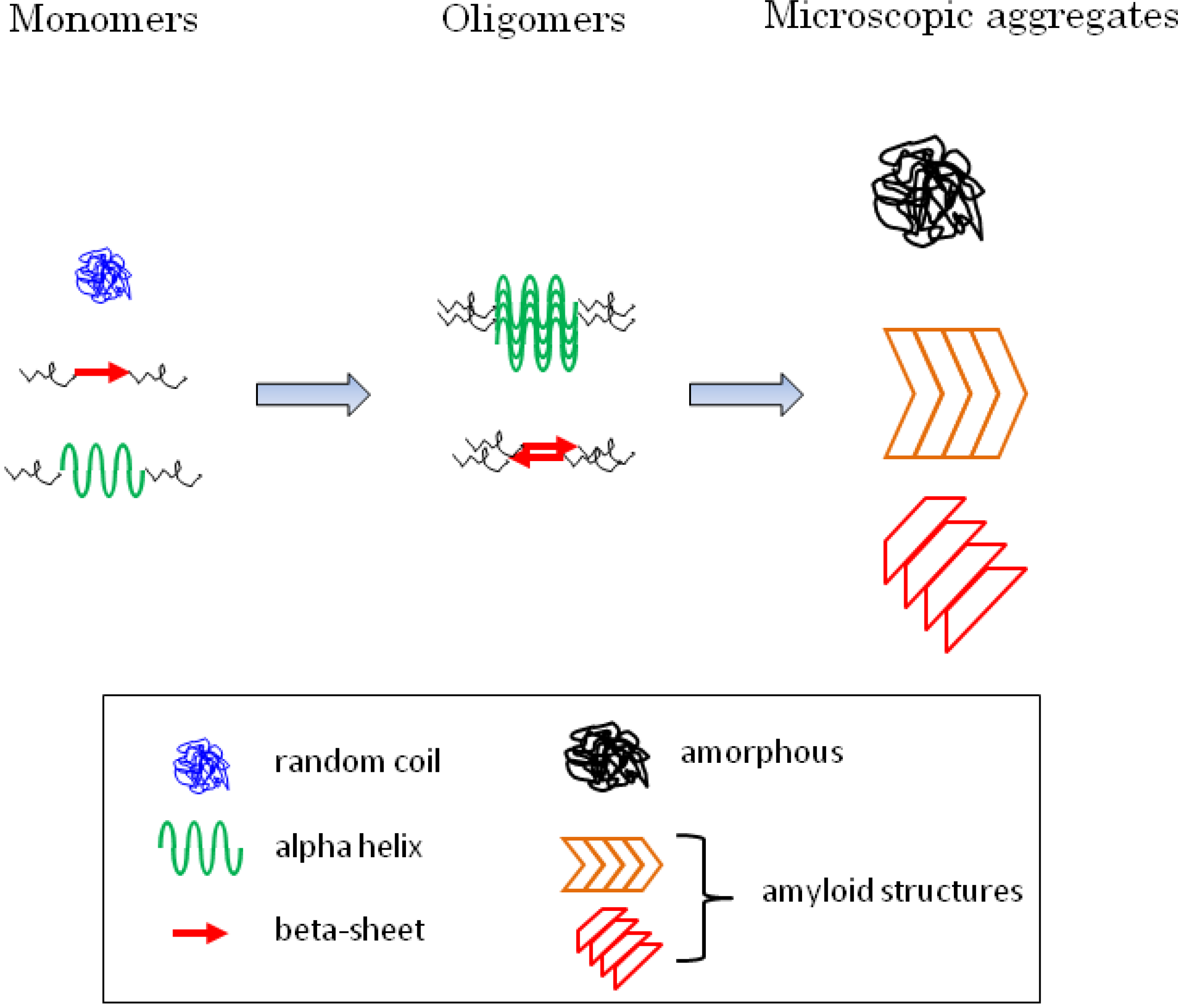
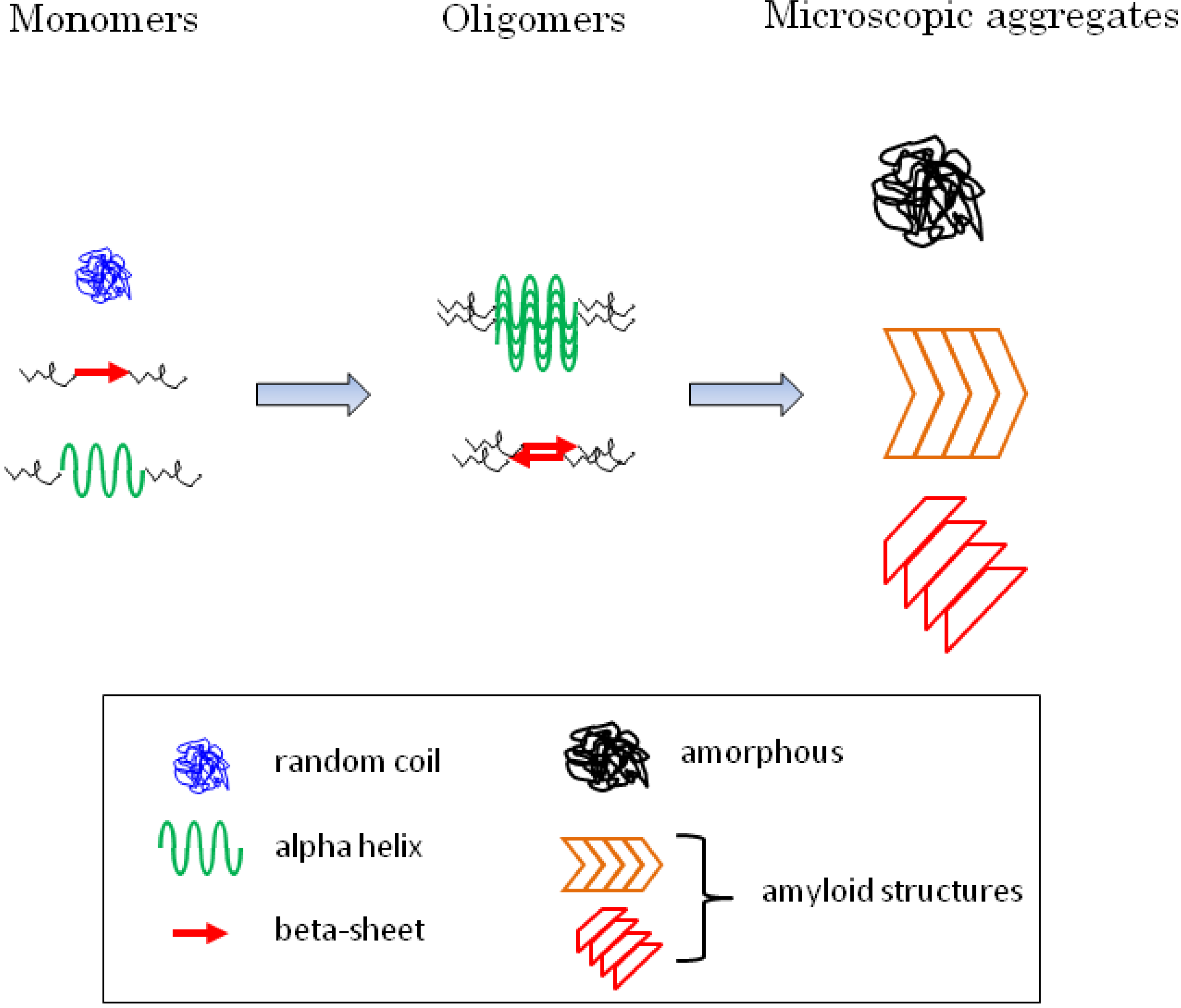
:
{kind=link}
{kind=link}
1. Introduction
2. General Mechanism of PolyQ Aggregation: Formation of β-Sheet Rich Aggregates
2.1. Discovery of PolyQ Aggregation
2.2. Do Aggregates of PolyQ Possess An Amyloid Structure?
2.3. Are PolyQ β-Sheets Parallel, Antiparallel, or Both?
2.4. Nucleated Growth Polymerization
2.5. Seeded Polymerization
3. Influence of the Sequences Flanking the PolyQ on Aggregation?
3.1. PolyQ Can Cause Neurological Disease Independently of Its Protein Context
3.2. Residues Flanking the PolyQ Affect Its Solubility and Largely Condition Interactions with Heterologous Proteins
3.3. Residues N-Terminal to the PolyQ of Huntingtin Promote Aggregation
3.4. Ubiquitination and SUMOylation of the N-Terminal Sequence of Huntingtin and Their Influence on PolyQ Aggregation
3.5. The N-Terminal Sequence of Huntingtin Targets Protein Aggregation to Membranes and Affects Their Integrity
3.6. The Polyproline C-Terminal to the PolyQ of Huntingtin Inhibits Aggregation
4. Aggregates of Expanded Huntingtin Consist of N-Terminal Fragments of the Protein
4.1. Evidence of the Presence of Fragmented Huntingtin in Inclusions
4.2. Aggregating Properties and Toxicity of Fragments
4.3. Proteolytic Enzymes that Generate the Fragments
5. Microscopic Aggregates: Inclusions and Fibrils
5.1. Diversity in Morphology, Subcellular Distribution and Number of Inclusions
5.2. Diversity of Protein Secondary Structure in Microscopic Aggregates
5.3. Toxicity of Microscopic Aggregates
5.4. Influence of Protein Structure on Toxicity of Microscopic Aggregates
6. Oligomers
6.1. Discovery
6.2. Recently Revisited Tenets on the Formation of Oligomers
6.3. Oligomers Precede Microscopic Aggregates
6.4. Heterogeneity of Oligomers
6.5. Secondary Structure of the Oligomerized Proteins
6.6. Toxicity of Oligomers
7. Monomers
7.1. Water-Soluble Fragments of Monomeric Huntingtin in Affected Brain Regions
7.2. Secondary Structure of Monomeric Polyglutamine
7.3. Toxicity of Monomeric Polyglutamine
8. Conclusions
Abbreviations
| DRPLA | dentatorubral-pallidoluysian atrophy |
| FTIR spectroscopy | Fourier-transform infrared spectroscopy |
| FRET | fluorescence resonance energy transfer |
| GST | glutathione S-transferase |
| PolyQ | polyglutamine |
| SBMA | spinobulbar muscular atrophy |
| SCA | spinocerebellar ataxia |
| TIA-1 | T-cell intracellular antigen-1 |
Conflicts of Interest
References
- La Spada, A.; Wilson, E.; Lubahn, D.; Harding, A.; Fischbeck, K. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Koide, R.; Ikeuchi, T.; Onodera, O.; Tanaka, H.; Igarashi, S.; Endo, K.; Takahashi, H.; Kondo, R.; Ishikawa, A.; Hayashi, T. Unstable expansion of cag repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat. Genet. 1994, 6, 9–13. [Google Scholar] [CrossRef]
- Orr, H.; Chung, M.; Banfi, S.; Kwiatkowski, T.J.; Servadio, A.; Beaudet, A.; McCall, A.; Duvick, L.; Ranum, L.; Zoghbi, H. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef]
- Sanpei, K.; Takano, H.; Igarashi, S.; Sato, T.; Oyake, M.; Sasaki, H.; Wakisaka, A.; Tashiro, K.; Ishida, Y.; Ikeuchi, T.; et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, direct. Nat. Genet. 1996, 14, 277–284. [Google Scholar] [CrossRef]
- Pulst, S.; Nechiporuk, A.; Nechiporuk, T.; Gispert, S.; Chen, X.; Lopes-Cendes, I.; Pearlman, S.; Starkman, S.; Orozco-Diaz, G.; Lunkes, A.; et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 1996, 14, 269–276. [Google Scholar] [CrossRef]
- Imbert, G.; Saudou, F.; Yvert, G.; Devys, D.; Trottier, Y.; Garnier, J.; Weber, C.; Mandel, J.; Cancel, G.; Abbas, N.; et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 1996, 14, 285–291. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.; Amos, C.; Dobyns, W.; Subramony, S.; Zoghbi, H.; Lee, C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef]
- David, G.; Abbas, N.; Stevanin, G.; Dürr, A.; Yvert, G.; Cancel, G.; Weber, C.; Imbert, G.; Saudou, F.; Antoniou, E.; et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 1997, 17, 65–70. [Google Scholar] [CrossRef]
- Zuhlke, C.; Hellenbroich, Y.; Dalski, A.; Kononowa, N.; Hagenah, J.; Vieregge, P.; Riess, O.; Klein, C.; Schwinger, E. Different types of repeat expansion in the TATA-binding protein gene are associated with a new form of inherited ataxia. Eur J. Hum. Genet. 2001, 9, 160–164. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Leggo, J.; Coles, R.; Almqvist, E.; Biancalana, V.; Cassiman, J.J.; Chotai, K.; Connarty, M.; Crauford, D.; Curtis, A.; et al. Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am. J. Hum. Genet. 1996, 59, 16–22. [Google Scholar]
- Roze, E.; Bonnet, C.; Betuing, S.; Caboche, J. Huntington’s disease. Adv. Exp. Med. Biol. 2010, 685, 45–63. [Google Scholar] [CrossRef]
- Vonsattel, J.; Myers, R.; Stevens, T.; Ferrante, R.; Bird, E.; Richardson, E.J. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Rosas, H.D.; Salat, D.H.; Lee, S.Y.; Zaleta, A.K.; Pappu, V.; Fischl, B.; Greve, D.; Hevelone, N.; Hersch, S.M. Cerebral cortex and the clinical expression of Huntington’s disease: Complexity and heterogeneity. Brain 2008, 131, 1057–1068. [Google Scholar] [CrossRef]
- Quarrell, O.; Brewer, H.; Squitieri, F.; Barker, R.; Nance, M.; Landwehrmeyer, B. Juvenile Huntington’s Disease; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Green, H.; Djian, P. Amino acid repeats in proteins and the neurological diseases produced by polyglutamine. In Genetic Instabilities and Hereditary Neurological Diseases; Wells, R., Warren, S., Eds.; Academic Press: New York, NY, USA, 1998; pp. 739–759. [Google Scholar]
- Trottier, Y.; Devys, D.; Imbert, G.; Saudou, F.; An, I.; Lutz, Y.; Weber, C.; Agid, Y.; Hirsch, E.C.; Mandel, J.L. Cellular localization of the Huntington’s disease protein and discrimination of the normal and mutated form. Nat. Genet. 1995, 10, 104–110. [Google Scholar] [CrossRef]
- Schilling, G.; Sharp, A.H.; Loev, S.J.; Wagster, M.V.; Li, S.H.; Stine, O.C.; Ross, C.A. Expression of the Huntington’s disease (IT15) protein product in HD patients. Hum. Mol. Genet. 1995, 4, 1365–1371. [Google Scholar] [CrossRef]
- Hoffner, G.; Island, M.; Djian, P. Purification of neuronal inclusions of patients with Huntington’s disease reveals a broad range of N-terminal fragments of expanded huntingtin and insoluble polymers. J. Neurochem. 2005, 95, 125–136. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.; Davies, S.; Bates, G.; Vonsattel, J.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Perutz, M.F.; Staden, R.; Moens, L.; de Baere, I. Polar zippers. Curr. Biol. 1993, 3, 249–253. [Google Scholar] [CrossRef]
- Perutz, M.; Johnson, T.; Suzuki, M.; Finch, J. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 1994, 91, 5355–5358. [Google Scholar] [CrossRef]
- Stott, K.; Blackburn, J.; Butler, P.; Perutz, M. Incorporation of glutamine repeats makes protein oligomerize: Implications for neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 1995, 92, 6509–6513. [Google Scholar] [CrossRef]
- Perutz, M.F.; Finch, J.T.; Berriman, J.; Lesk, A. Amyloid fibers are water-filled nanotubes. Proc. Natl. Acad. Sci. USA 2002, 99, 5591–5595. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.; et al. Exon 1 of the HD gene with an expanded cag repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Davies, S.; Turmaine, M.; Cozens, B.; DiFiglia, M.; Sharp, A.; Ross, C.; Scherzinger, E.; Wanker, E.; Mangiarini, L.; Bates, G. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.; Davies, S.; Lehrach, H.; Wanker, E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef]
- Paulson, H.; Perez, M.; Trottier, Y.; Trojanowski, J.; Subramony, S.; Das, S.; Vig, P.; Mandel, J.; Fischbeck, K.; Pittman, R. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 1997, 19, 333–344. [Google Scholar] [CrossRef]
- Skinner, P.; Koshy, B.; Cummings, C.; Klement, I.; Helin, K.; Servadio, A.; Zoghbi, H.; Orr, H. Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures. Nature 1997, 389, 971–974. [Google Scholar] [CrossRef]
- Hayashi, Y.; Kakita, A.; Yamada, M.; Koide, R.; Igarashi, S.; Takano, H.; Ikeuchi, T.; Wakabayashi, K.; Egawa, S.; Tsuji, S.; et al. Hereditary dentatorubral-pallidoluysian atrophy: Detection of widespread ubiquitinated neuronal and glial intranuclear inclusions in the brain. Acta Neuropathol. 1998, 96, 547–552. [Google Scholar] [CrossRef]
- Holmberg, M.; Duyckaerts, C.; Dürr, A.; Cancel, G.; Gourfinkel-An, I.; Damier, P.; Faucheux, B.; Trottier, Y.; Hirsch, E.; Agid, Y.; et al. Spinocerebellar ataxia type 7 (sca7): A neurodegenerative disorder with neuronal intranuclear inclusions. Hum. Mol. Genet. 1998, 7, 913–918. [Google Scholar] [CrossRef]
- Ishikawa, K.; Fujigasaki, H.; Saegusa, H.; Ohwada, K.; Fujita, T.; Iwamoto, H.; Komatsuzaki, Y.; Toru, S.; Toriyama, H.; Watanabe, M.; et al. Abundant expression and cytoplasmic aggregations of α1a voltage-dependent calcium channel protein associated with neurodegeneration in spinocerebellar ataxia type 6. Hum. Mol. Genet. 1999, 8, 1185–1193. [Google Scholar] [CrossRef]
- Koyano, S.; Uchihara, T.; Fujigasaki, H.; Nakamura, A.; Yagishita, S.; Iwabuchi, K. Neuronal intranuclear inclusions in spinocerebellar ataxia type 2: Triple-labeling immunofluorescent study. Neurosci. Lett. 1999, 273, 117–120. [Google Scholar] [CrossRef]
- Nakamura, K.; Jeong, S.; Uchihara, T.; Anno, M.; Nagashima, K.; Nagashima, T.; Ikeda, S.; Tsuji, S.; Kanazawa, I. Sca17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum. Mol. Genet. 2001, 10, 1441–1448. [Google Scholar] [CrossRef]
- Fujigasaki, H.; Martin, J.; de Deyn, P.; Camuzat, A.; Deffond, D.; Stevanin, G.; Dermaut, B.; van Broeckhoven, C.; Dürr, A.; Brice, A. CAG repeat expansion in the TATA box-binding protein gene causes autosomal dominant cerebellar ataxia. Brain 2001, 124, 1939–1947. [Google Scholar] [CrossRef]
- Hazeki, N.; Tukamoto, T.; Goto, J.; Kanazawa, I. Formic acid dissolves aggregates of an N-terminal huntingtin fragment containing an expanded polyglutamine tract: Applying to quantification of protein components of the aggregates. Biochem. Biophys. Res. Commun. 2000, 277, 386–393. [Google Scholar] [CrossRef]
- Iuchi, S.; Hoffner, G.; Verbeke, P.; Djian, P.; Green, H. Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. Proc. Natl. Acad. Sci. USA 2003, 100, 2409–2414. [Google Scholar] [CrossRef]
- Zhou, H.; Cao, F.; Wang, Z.; Yu, Z.X.; Nguyen, H.P.; Evans, J.; Li, S.H.; Li, X.J. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J. Cell Biol. 2003, 163, 109–118. [Google Scholar] [CrossRef]
- Eanes, E.D.; Glenner, G.G. X-ray diffraction studies on amyloid filaments. J. Histochem. Cytochem. 1968, 16, 673–677. [Google Scholar] [CrossRef]
- Bonar, L.; Cohen, A.S.; Skinner, M.M. Characterization of the amyloid fibril as a cross-beta protein. Proc. Soc. Exp. Biol. Med. 1969, 131, 1373–1375. [Google Scholar] [CrossRef]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef]
- Fändrich, M. On the structural definition of amyloid fibrils and other polypeptide aggregates. Cell. Mol. Life Sci. 2007, 64, 2066–2078. [Google Scholar] [CrossRef]
- Greenwald, J.; Riek, R. Biology of amyloid: Structure, function, and regulation. Structure 2010, 18, 1244–1260. [Google Scholar] [CrossRef]
- Tanaka, M.; Morishima, I.; Akagi, T.; Hashikawa, T.; Nukina, N. Intra- and intermolecular β-pleated sheet formation in glutamine-repeat inserted myoglobin as a model for polyglutamine diseases. J. Biol. Chem. 2001, 276, 45470–45475. [Google Scholar] [CrossRef]
- Bevivino, A.E.; Loll, P.J. An expanded glutamine repeat destabilizes native ataxin-3 structure and mediates formation of parallel β-fibrils. Proc. Natl. Acad. Sci. USA 2001, 98, 11955–11960. [Google Scholar] [CrossRef]
- Chen, S.; Berthelier, V.; Hamilton, J.; O’Nuallain, B.; Wetzel, R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry 2002, 41, 7391–7399. [Google Scholar] [CrossRef]
- Huang, C.C.; Faber, P.W.; Persichetti, F.; Mittal, V.; Vonsattel, J.P.; MacDonald, M.E.; Gusella, J.F. Amyloid formation by mutant huntingtin: Threshold, progressivity and recruitment of normal polyglutamine proteins. Somat. Cell. Mol. Genet. 1998, 24, 217–233. [Google Scholar] [CrossRef]
- McGowan, D.P.; van Roon-Mom, W.; Holloway, H.; Bates, G.P.; Mangiarini, L.; Cooper, G.J.; Faull, R.L.; Snell, R.G. Amyloid-like inclusions in Huntington’s disease. Neuroscience 2000, 100, 677–680. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Dürr, A.; Cancel, G.; Brice, A. Nuclear inclusions in spinocerebellar ataxia type 1. Acta Neuropathol. 1999, 97, 201–207. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef]
- Wanderer, J.; Morton, A.J. Differential morphology and composition of inclusions in the r6/2 mouse and pc12 cell models of Huntington’s disease. Histochem. Cell Biol. 2007, 127, 473–484. [Google Scholar] [CrossRef]
- Suhr, S.T.; Senut, M.C.; Whitelegge, J.P.; Faull, K.F.; Cuizon, D.B.; Gage, F.H. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J. Cell Biol. 2001, 153, 283–294. [Google Scholar] [CrossRef]
- Hoffner, G.; Vanhoutteghem, A.; André, W.; Djian, P. Transglutaminase in epidermis and neurological disease or what makes a good cross-linking substrate. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 78, 97–160. [Google Scholar]
- Bugg, C.W.; Isas, J.M.; Fischer, T.; Patterson, P.H.; Langen, R. Structural features and domain organization of huntingtin fibrils. J. Biol. Chem. 2012, 287, 31739–31746. [Google Scholar] [CrossRef]
- Schneider, R.; Schumacher, M.C.; Mueller, H.; Nand, D.; Klaukien, V.; Heise, H.; Riedel, D.; Wolf, G.; Behrmann, E.; Raunser, S.; et al. Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy. J. Mol. Biol. 2011, 412, 121–136. [Google Scholar] [CrossRef]
- Sivanandam, V.N.; Jayaraman, M.; Hoop, C.L.; Kodali, R.; Wetzel, R.; van der Wel, P.C. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 2011, 133, 4558–4566. [Google Scholar]
- Sharma, D.; Shinchuk, L.M.; Inouye, H.; Wetzel, R.; Kirschner, D.A. Polyglutamine homopolymers having 8–45 residues form slablike β-crystallite assemblies. Proteins 2005, 61, 398–411. [Google Scholar] [CrossRef]
- Takahashi, T.; Kikuchi, S.; Katada, S.; Nagai, Y.; Nishizawa, M.; Onodera, O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 2008, 17, 345–356. [Google Scholar]
- André, W.; Sandt, C.; Dumas, P.; Djian, P.; Hoffner, G. Structure of inclusions of Huntington’s disease brain revealed by synchrotron infrared microspectroscopy: Polymorphism and relevance to cytotoxicity. Anal. Chem. 2013, 85, 3765–3773. [Google Scholar] [CrossRef]
- Feigin, A.; Leenders, K.L.; Moeller, J.R.; Missimer, J.; Kuenig, G.; Spetsieris, P.; Antonini, A.; Eidelberg, D. Metabolic network abnormalities in early Huntington’s disease: An [18F]FDG PET study. J. Nucl. Med. 2001, 42, 1591–1595. [Google Scholar]
- Clarke, G.; Collins, R.A.; Leavitt, B.R.; Andrews, D.F.; Hayden, M.R.; Lumsden, C.J.; McInnes, R.R. A one-hit model of cell death in inherited neuronal degenerations. Nature 2000, 406, 195–199. [Google Scholar] [CrossRef]
- Perutz, M.F.; Windle, A.H. Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature 2001, 412, 143–144. [Google Scholar] [CrossRef]
- Chen, S.; Ferrone, F.A.; Wetzel, R. Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. USA 2002, 99, 11884–11889. [Google Scholar] [CrossRef]
- Kar, K.; Jayaraman, M.; Sahoo, B.; Kodali, R.; Wetzel, R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat. Struct. Mol. Biol. 2011, 18, 328–336. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kaneko, K.; Matsumoto, G.; Kurosawa, M.; Nukina, N. Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J. Neurosci. 2009, 29, 5153–5162. [Google Scholar] [CrossRef]
- Gupta, S.; Jie, S.; Colby, D.W. Protein misfolding detected early in pathogenesis of transgenic mouse model of Huntington disease using amyloid seeding assay. J. Biol. Chem. 2012, 287, 9982–9989. [Google Scholar] [CrossRef]
- Tonoki, A.; Kuranaga, E.; Ito, N.; Nekooki-Machida, Y.; Tanaka, M.; Miura, M. Aging causes distinct characteristics of polyglutamine amyloids in vivo. Genes Cells 2011, 16, 557–564. [Google Scholar] [CrossRef]
- Gidalevitz, T.; Kikis, E.A.; Morimoto, R.I. A cellular perspective on conformational disease: The role of genetic background and proteostasis networks. Curr. Opin. Struct. Biol. 2010, 20, 23–32. [Google Scholar] [CrossRef]
- Gaczynska, M.; Osmulski, P.A.; Ward, W.F. Caretaker or undertaker? The role of the proteasome in aging. Mech. Ageing Dev. 2001, 122, 235–254. [Google Scholar] [CrossRef]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef]
- Scarafone, N.; Pain, C.; Fratamico, A.; Gaspard, G.; Yilmaz, N.; Filée, P.; Galleni, M.; Matagne, A.; Dumoulin, M. Amyloid-like fibril formation by polyq proteins: A critical balance between the polyQ length and the constraints imposed by the host protein. PLoS One 2012, 7, e31253. [Google Scholar] [CrossRef]
- Dumoulin, M.; Last, A.M.; Desmyter, A.; Decanniere, K.; Canet, D.; Larsson, G.; Spencer, A.; Archer, D.B.; Sasse, J.; Muyldermans, S.; et al. A camelid antibody fragment inhibits the formation of amyloid fibrils by human lysozyme. Nature 2003, 424, 783–788. [Google Scholar] [CrossRef]
- Chai, Y.; Wu, L.; Griffin, J.D.; Paulson, H.L. The role of protein composition in specifying nuclear inclusion formation in polyglutamine disease. J. Biol. Chem. 2001, 276, 44889–44897. [Google Scholar] [CrossRef]
- Ordway, J.; Tallaksen-Greene, S.; Gutekunst, C.; Bernstein, E.; Cearley, J.; Wiener, H.; Dure, L.T.; Lindsey, R.; Hersch, S.; Jope, R.; et al. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 1997, 91, 753–763. [Google Scholar] [CrossRef]
- Kahlem, P.; Terré, C.; Green, H.; Djian, P. Peptides containing glutamine repeats as substrates for transglutaminase-catalyzed cross-linking: Relevance to diseases of the nervous system. Proc. Natl. Acad. Sci. USA 1996, 93, 14580–14585. [Google Scholar] [CrossRef]
- Nozaki, K.; Onodera, O.; Takano, H.; Tsuji, S. Amino acid sequences flanking polyglutamine stretches influence their potential for aggregate formation. Neuroreport 2001, 12, 3357–3364. [Google Scholar] [CrossRef]
- Ignatova, Z.; Thakur, A.K.; Wetzel, R.; Gierasch, L.M. In-cell aggregation of a polyglutamine-containing chimera is a multistep process initiated by the flanking sequence. J. Biol. Chem. 2007, 282, 36736–36743. [Google Scholar] [CrossRef]
- Tam, S.; Spiess, C.; Auyeung, W.; Joachimiak, L.; Chen, B.; Poirier, M.A.; Frydman, J. The chaperonin tric blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 2009, 16, 1279–1285. [Google Scholar] [CrossRef]
- Liebman, S.W.; Meredith, S.C. Protein folding: Sticky n17 speeds huntingtin pile-up. Nat. Chem. Biol. 2010, 6, 7–8. [Google Scholar] [CrossRef]
- Thakur, A.; Jayaraman, M.; Mishra, R.; Thakur, M.; Chellgren, V.; Byeon, I.; Anjum, D.; Kodali, R.; Creamer, T.; Conway, J.; et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 2009, 16, 380–389. [Google Scholar] [CrossRef]
- Jayaraman, M.; Kodali, R.; Sahoo, B.; Thakur, A.K.; Mayasundari, A.; Mishra, R.; Peterson, C.B.; Wetzel, R. Slow amyloid nucleation via α-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J. Mol. Biol. 2012, 415, 881–899. [Google Scholar] [CrossRef]
- Mishra, R.; Jayaraman, M.; Roland, B.P.; Landrum, E.; Fullam, T.; Kodali, R.; Thakur, A.K.; Arduini, I.; Wetzel, R. Inhibiting the nucleation of amyloid structure in a huntingtin fragment by targeting α-helix-rich oligomeric intermediates. J. Mol. Biol. 2012, 415, 900–917. [Google Scholar] [CrossRef]
- Fiumara, F.; Fioriti, L.; Kandel, E.R.; Hendrickson, W.A. Essential role of coiled coils for aggregation and activity of Q/N-rich prions and polyq proteins. Cell 2010, 143, 1121–1135. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Fishman, P.S.; Thakor, N.V.; Oyler, G.A. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J. Biol. Chem. 2003, 278, 22044–22055. [Google Scholar] [CrossRef]
- Steffan, J.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.; Cattaneo, E.; et al. Sumo modification of huntingtin and Huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef]
- Atwal, R.S.; Xia, J.; Pinchev, D.; Taylor, J.; Epand, R.M.; Truant, R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 2007, 16, 2600–2615. [Google Scholar] [CrossRef]
- Rockabrand, E.; Slepko, N.; Pantalone, A.; Nukala, V.N.; Kazantsev, A.; Marsh, J.L.; Sullivan, P.G.; Steffan, J.S.; Sensi, S.L.; Thompson, L.M. The first 17 amino acids of huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum. Mol. Genet. 2007, 16, 61–77. [Google Scholar]
- Burke, K.A.; Kauffman, K.J.; Umbaugh, C.S.; Frey, S.L.; Legleiter, J. The interaction of polyglutamine peptides with lipid membranes is regulated by flanking sequences associated with huntingtin. J. Biol. Chem. 2013, 288, 14993–15005. [Google Scholar] [CrossRef]
- Duennwald, M.L.; Jagadish, S.; Muchowski, P.J.; Lindquist, S. Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. USA 2006, 103, 11045–11050. [Google Scholar] [CrossRef]
- Dehay, B.; Bertolotti, A. Critical role of the proline-rich region in huntingtin for aggregation and cytotoxicity in yeast. J. Biol. Chem. 2006, 281, 35608–35615. [Google Scholar] [CrossRef]
- Darnell, G.; Orgel, J.P.; Pahl, R.; Meredith, S.C. Flanking polyproline sequences inhibit β-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J. Mol. Biol. 2007, 374, 688–704. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Amos, W.; Leggo, J.; Goodburn, S.; Ramesar, R.S.; Old, J.; Bontrop, R.; McMahon, R.; Barton, D.E.; Ferguson-Smith, M.A. Mutational bias provides a model for the evolution of Huntington’s disease and predicts a general increase in disease prevalence. Nat. Genet. 1994, 7, 525–530. [Google Scholar] [CrossRef]
- Djian, P.; Hancock, J.M.; Chana, H.S. Codon repeats in genes associated with human diseases: Fewer repeats in the genes of nonhuman primates and nucleotide substitutions concentrated at the sites of reiteration. Proc. Natl. Acad. Sci. USA 1996, 93, 417–421. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Thakur, A.K.; Chellgren, V.M.; Thiagarajan, G.; Williams, A.D.; Chellgren, B.W.; Creamer, T.P.; Wetzel, R. Oligoproline effects on polyglutamine conformation and aggregation. J. Mol. Biol. 2006, 355, 524–535. [Google Scholar] [CrossRef]
- Darnell, G.D.; Derryberry, J.; Kurutz, J.W.; Meredith, S.C. Mechanism of cis-inhibition of polyQ fibrillation by polyP: PPII oligomers and the hydrophobic effect. Biophys. J. 2009, 97, 2295–2305. [Google Scholar] [CrossRef]
- Lakhani, V.V.; Ding, F.; Dokholyan, N.V. Polyglutamine induced misfolding of huntingtin exon1 is modulated by the flanking sequences. PLoS Comput. Biol. 2010, 6, e1000772. [Google Scholar] [CrossRef]
- Monsellier, E.; Chiti, F. Prevention of amyloid-like aggregation as a driving force of protein evolution. EMBO Rep. 2007, 8, 737–742. [Google Scholar] [CrossRef]
- Caron, N.S.; Desmond, C.R.; Xia, J.; Truant, R. Polyglutamine domain flexibility mediates the proximity between flanking sequences in huntingtin. Proc. Natl. Acad. Sci. USA 2013, 110, 14610–14615. [Google Scholar] [CrossRef]
- Sieradzan, K.A.; Mechan, A.O.; Jones, L.; Wanker, E.E.; Nukina, N.; Mann, D.M. Huntington’s disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp. Neurol. 1999, 156, 92–99. [Google Scholar] [CrossRef]
- Lunkes, A.; Lindenberg, K.S.; Ben-Haïem, L.; Weber, C.; Devys, D.; Landwehrmeyer, G.B.; Mandel, J.L.; Trottier, Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 2002, 10, 259–269. [Google Scholar] [CrossRef]
- Cooper, J.K.; Schilling, G.; Peters, M.F.; Herring, W.J.; Sharp, A.H.; Kaminsky, Z.; Masone, J.; Khan, F.A.; Delanoy, M.; Borchelt, D.R.; et al. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum. Mol. Genet. 1998, 7, 783–790. [Google Scholar] [CrossRef]
- Martindale, D.; Hackam, A.; Wieczorek, A.; Ellerby, L.; Wellington, C.; McCutcheon, K.; Singaraja, R.; Kazemi-Esfarjani, P.; Devon, R.; Kim, S.U.; et al. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 1998, 18, 150–154. [Google Scholar] [CrossRef]
- Wang, C.E.; Tydlacka, S.; Orr, A.L.; Yang, S.H.; Graham, R.K.; Hayden, M.R.; Li, S.; Chan, A.W.; Li, X.J. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington’s disease. Hum. Mol. Genet. 2008, 17, 2738–2751. [Google Scholar] [CrossRef]
- Chan, A.W.; Cheng, P.H.; Neumann, A.; Yang, J.J. Reprogramming huntington monkey skin cells into pluripotent stem cells. Cell. Reprogram. 2010, 12, 509–517. [Google Scholar] [CrossRef]
- Castiglioni, V.; Onorati, M.; Rochon, C.; Cattaneo, E. Induced pluripotent stem cell lines from Huntington’s disease mice undergo neuronal differentiation while showing alterations in the lysosomal pathway. Neurobiol. Dis. 2012, 46, 30–40. [Google Scholar] [CrossRef]
- Camnasio, S.; Delli Carri, A.; Lombardo, A.; Grad, I.; Mariotti, C.; Castucci, A.; Rozell, B.; Lo Riso, P.; Castiglioni, V.; Zuccato, C.; et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis. 2012, 46, 41–51. [Google Scholar] [CrossRef]
- Consortium, H.I. Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 2012, 11, 264–278. [Google Scholar] [CrossRef]
- Juopperi, T.A.; Kim, W.R.; Chiang, C.H.; Yu, H.; Margolis, R.L.; Ross, C.A.; Ming, G.L.; Song, H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 2012, 5. [Google Scholar] [CrossRef]
- Koch, P.; Breuer, P.; Peitz, M.; Jungverdorben, J.; Kesavan, J.; Poppe, D.; Doerr, J.; Ladewig, J.; Mertens, J.; Tüting, T.; et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature 2011, 480, 543–546. [Google Scholar]
- Jeon, I.; Lee, N.; Li, J.Y.; Park, I.H.; Park, K.S.; Moon, J.; Shim, S.H.; Choi, C.; Chang, D.J.; Kwon, J.; et al. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells 2012, 30, 2054–2062. [Google Scholar] [CrossRef]
- Goldberg, Y.P.; Nicholson, D.W.; Rasper, D.M.; Kalchman, M.A.; Koide, H.B.; Graham, R.K.; Bromm, M.; Kazemi-Esfarjani, P.; Thornberry, N.A.; Vaillancourt, J.P.; et al. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat. Genet. 1996, 13, 442–449. [Google Scholar] [CrossRef]
- Kim, Y.J.; Yi, Y.; Sapp, E.; Wang, Y.; Cuiffo, B.; Kegel, K.B.; Qin, Z.H.; Aronin, N.; DiFiglia, M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl. Acad. Sci. USA 2001, 98, 12784–12789. [Google Scholar] [CrossRef]
- Gafni, J.; Ellerby, L.M. Calpain activation in Huntington’s disease. J. Neurosci 2002, 22, 4842–4849. [Google Scholar]
- Wellington, C.L.; Ellerby, L.M.; Gutekunst, C.A.; Rogers, D.; Warby, S.; Graham, R.K.; Loubser, O.; van Raamsdonk, J.; Singaraja, R.; Yang, Y.Z.; et al. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J. Neurosci. 2002, 22, 7862–7872. [Google Scholar]
- Southwell, A.L.; Bugg, C.W.; Kaltenbach, L.S.; Dunn, D.; Butland, S.; Weiss, A.; Paganetti, P.; Lo, D.C.; Patterson, P.H. Perturbation with intrabodies reveals that calpain cleavage is required for degradation of huntingtin exon 1. PLoS One 2011, 6, e16676. [Google Scholar] [CrossRef]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef]
- Sathasivam, K.; Neueder, A.; Gipson, T.A.; Landles, C.; Benjamin, A.C.; Bondulich, M.K.; Smith, D.L.; Faull, R.L.; Roos, R.A.; Howland, D.; et al. Aberrant splicing of htt generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, 2366–2370. [Google Scholar] [CrossRef]
- Nekooki-Machida, Y.; Kurosawa, M.; Nukina, N.; Ito, K.; Oda, T.; Tanaka, M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9679–9684. [Google Scholar] [CrossRef]
- Becher, M.; Kotzuk, J.; Sharp, A.; Davies, S.; Bates, G.; Price, D.; Ross, C. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: Correlation between the density of inclusions and it15 CAG triplet repeat length. Neurobiol. Dis. 1998, 4, 387–397. [Google Scholar] [CrossRef]
- Gutekunst, C.; Li, S.; Yi, H.; Mulroy, J.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.; Hersch, S.; Li, X. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar]
- Yang, W.; Dunlap, J.; Andrews, R.; Wetzel, R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet. 2002, 11, 2905–2917. [Google Scholar] [CrossRef]
- Arrasate, M.; Mitra, S.; Schweitzer, E.; Segal, M.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef]
- Saudou, F.; Finkbeiner, S.; Devys, D.; Greenberg, M. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 1998, 95, 55–66. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Schaffar, G.; Sittler, A.; Wanker, E.E.; Hayer-Hartl, M.K.; Hartl, F.U. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 7841–7846. [Google Scholar] [CrossRef]
- Morton, A.J.; Glynn, D.; Leavens, W.; Zheng, Z.; Faull, R.L.; Skepper, J.N.; Wight, J.M. Paradoxical delay in the onset of disease caused by super-long CAG repeat expansions in r6/2 mice. Neurobiol. Dis. 2009, 33, 331–341. [Google Scholar] [CrossRef]
- Thakur, A.K.; Wetzel, R. Mutational analysis of the structural organization of polyglutamine aggregates. Proc. Natl. Acad. Sci. USA 2002, 99, 17014–17019. [Google Scholar] [CrossRef]
- Zhang, Q.C.; Yeh, T.L.; Leyva, A.; Frank, L.G.; Miller, J.; Kim, Y.E.; Langen, R.; Finkbeiner, S.; Amzel, M.L.; Ross, C.A.; et al. A compact beta model of huntingtin toxicity. J. Biol. Chem. 2011, 286, 8188–8196. [Google Scholar] [CrossRef]
- Poirier, M.A.; Jiang, H.; Ross, C.A. A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: Evidence for a compact beta-sheet structure. Hum. Mol. Genet. 2005, 14, 765–774. [Google Scholar] [CrossRef]
- Poirier, M.A.; Li, H.; Macosko, J.; Cai, S.; Amzel, M.; Ross, C.A. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 2002, 277, 41032–41037. [Google Scholar]
- Mukai, H.; Isagawa, T.; Goyama, E.; Tanaka, S.; Bence, N.F.; Tamura, A.; Ono, Y.; Kopito, R.R. Formation of morphologically similar globular aggregates from diverse aggregation-prone proteins in mammalian cells. Proc. Natl. Acad. Sci. USA 2005, 102, 10887–10892. [Google Scholar] [CrossRef]
- Li, M.; Chevalier-Larsen, E.S.; Merry, D.E.; Diamond, M.I. Soluble androgen receptor oligomers underlie pathology in a mouse model of spinobulbar muscular atrophy. J. Biol. Chem. 2007, 282, 3157–3164. [Google Scholar] [CrossRef]
- Legleiter, J.; Mitchell, E.; Lotz, G.; Sapp, E.; Ng, C.; DiFiglia, M.; Thompson, L.; Muchowski, P. Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 2010, 285, 14777–14790. [Google Scholar]
- Slepko, N.; Bhattacharyya, A.M.; Jackson, G.R.; Steffan, J.S.; Marsh, J.L.; Thompson, L.M.; Wetzel, R. Normal-repeat-length polyglutamine peptides accelerate aggregation nucleation and cytotoxicity of expanded polyglutamine proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 14367–14372. [Google Scholar] [CrossRef]
- Wetzel, R. Kinetics and thermodynamics of amyloid fibril assembly. Acc. Chem. Res. 2006, 39, 671–679. [Google Scholar] [CrossRef]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu Rev. Neurosci 2003, 26, 267–298. [Google Scholar] [CrossRef]
- Natalello, A.; Frana, A.M.; Relini, A.; Apicella, A.; Invernizzi, G.; Casari, C.; Gliozzi, A.; Doglia, S.M.; Tortora, P.; Regonesi, M.E. A major role for side-chain polyglutamine hydrogen bonding in irreversible ataxin-3 aggregation. PLoS One 2011, 6, e18789. [Google Scholar] [CrossRef]
- Weiss, A.; Klein, C.; Woodman, B.; Sathasivam, K.; Bibel, M.; Régulier, E.; Bates, G.P.; Paganetti, P. Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington’s disease. J. Neurochem. 2008, 104, 846–858. [Google Scholar]
- Marcellin, D.; Abramowski, D.; Young, D.; Richter, J.; Weiss, A.; Marcel, A.; Maassen, J.; Kauffmann, M.; Bibel, M.; Shimshek, D.R.; et al. Fragments of HdhQ150 mutant huntingtin form a soluble oligomer pool that declines with aggregate deposition upon aging. PLoS One 2012, 7, e44457. [Google Scholar] [CrossRef]
- Wacker, J.L.; Zareie, M.H.; Fong, H.; Sarikaya, M.; Muchowski, P.J. Hsp70 and hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat. Struct. Mol. Biol. 2004, 11, 1215–1222. [Google Scholar] [CrossRef]
- Legleiter, J.; Lotz, G.P.; Miller, J.; Ko, J.; Ng, C.; Williams, G.L.; Finkbeiner, S.; Patterson, P.H.; Muchowski, P.J. Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J. Biol. Chem. 2009, 284, 21647–21658. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Olshina, M.A.; Angley, L.M.; Ramdzan, Y.M.; Tang, J.; Bailey, M.F.; Hill, A.F.; Hatters, D.M. Tracking mutant huntingtin aggregation kinetics in cells reveals three major populations that include an invariant oligomer pool. J. Biol. Chem. 2010, 285, 21807–21816. [Google Scholar]
- Nucifora, L.G.; Burke, K.A.; Feng, X.; Arbez, N.; Zhu, S.; Miller, J.; Yang, G.; Ratovitski, T.; Delannoy, M.; Muchowski, P.J.; et al. Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J. Biol. Chem. 2012, 287, 16017–16028. [Google Scholar] [CrossRef]
- Trottier, Y.; Lutz, Y.; Stevanin, G.; Imbert, G.; Devys, D.; Cancel, G.; Saudou, F.; Weber, C.; David, G.; Tora, L. Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature 1995, 378, 403–406. [Google Scholar] [CrossRef]
- Kahlem, P.; Green, H.; Djian, P. Transglutaminase action imitates Huntington’s disease: Selective polymerization of huntingtin containing expanded polyglutamine. Mol. Cell 1998, 1, 595–601. [Google Scholar] [CrossRef]
- Green, H. Human genetic diseases due to codon reiteration: Relationship to an evolutionary mechanism. Cell 1993, 74, 955–956. [Google Scholar] [CrossRef]
- Sathasivam, K.; Lane, A.; Legleiter, J.; Warley, A.; Woodman, B.; Finkbeiner, S.; Paganetti, P.; Muchowski, P.; Wilson, S.; Bates, G. Identical oligomeric and fibrillar structures captured from the brains of r6/2 and knock-in mouse models of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 65–78. [Google Scholar] [CrossRef]
- Slow, E.J.; Graham, R.K.; Osmand, A.P.; Devon, R.S.; Lu, G.; Deng, Y.; Pearson, J.; Vaid, K.; Bissada, N.; Wetzel, R.; et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc. Natl. Acad. Sci. USA 2005, 102, 11402–11407. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (−)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet. 2006, 15, 2743–2751. [Google Scholar] [CrossRef]
- Wong, S.L.; Chan, W.M.; Chan, H.Y. Sodium dodecyl sulfate-insoluble oligomers are involved in polyglutamine degeneration. FASEB J. 2008, 22, 3348–3357. [Google Scholar] [CrossRef]
- Stefani, M. Structural features and cytotoxicity of amyloid oligomers: Implications in Alzheimer’s disease and other diseases with amyloid deposits. Prog. Neurobiol. 2012, 99, 226–245. [Google Scholar] [CrossRef]
- Pieri, L.; Madiona, K.; Bousset, L.; Melki, R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 2012, 102, 2894–2905. [Google Scholar] [CrossRef]
- Landles, C.; Sathasivam, K.; Weiss, A.; Woodman, B.; Moffitt, H.; Finkbeiner, S.; Sun, B.; Gafni, J.; Ellerby, L.M.; Trottier, Y.; et al. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J. Biol. Chem. 2010, 285, 8808–8823. [Google Scholar] [CrossRef]
- Starikov, E.B.; Lehrach, H.; Wanker, E.E. Folding of oligoglutamines: A theoretical approach based upon thermodynamics and molecular mechanics. J. Biomol. Struct. Dyn. 1999, 17, 409–427. [Google Scholar] [CrossRef]
- Altschuler, E.L.; Hud, N.V.; Mazrimas, J.A.; Rupp, B. Random coil conformation for extended polyglutamine stretches in aqueous soluble monomeric peptides. J. Pept. Res. 1997, 50, 73–75. [Google Scholar] [CrossRef]
- Masino, L.; Kelly, G.; Leonard, K.; Trottier, Y.; Pastore, A. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 2002, 513, 267–272. [Google Scholar] [CrossRef]
- Klein, F.A.; Pastore, A.; Masino, L.; Zeder-Lutz, G.; Nierengarten, H.; Oulad-Abdelghani, M.; Altschuh, D.; Mandel, J.L.; Trottier, Y. Pathogenic and non-pathogenic polyglutamine tracts have similar structural properties: Towards a length-dependent toxicity gradient. J. Mol. Biol. 2007, 371, 235–244. [Google Scholar] [CrossRef]
- Kim, M.; Chelliah, Y.; Kim, S.; Otwinowski, Z.; Bezprozvanny, I. Secondary structure of huntingtin amino-terminal region. Structure 2009, 17, 1205–1212. [Google Scholar] [CrossRef]
- Wang, X.; Vitalis, A.; Wyczalkowski, M.A.; Pappu, R.V. Characterizing the conformational ensemble of monomeric polyglutamine. Proteins 2006, 63, 297–311. [Google Scholar]
- Crick, S.L.; Jayaraman, M.; Frieden, C.; Wetzel, R.; Pappu, R.V. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. USA 2006, 103, 16764–16769. [Google Scholar] [CrossRef]
- Dougan, L.; Li, J.; Badilla, C.L.; Berne, B.J.; Fernandez, J.M. Single homopolypeptide chains collapse into mechanically rigid conformations. Proc. Natl. Acad. Sci. USA 2009, 106, 12605–12610. [Google Scholar] [CrossRef]
- Peters-Libeu, C.; Miller, J.; Rutenber, E.; Newhouse, Y.; Krishnan, P.; Cheung, K.; Hatters, D.; Brooks, E.; Widjaja, K.; Tran, T.; et al. Disease-associated polyglutamine stretches in monomeric huntingtin adopt a compact structure. J. Mol. Biol. 2012, 421, 587–600. [Google Scholar] [CrossRef]
- Bennett, M.J.; Huey-Tubman, K.E.; Herr, A.B.; West, A.P.; Ross, S.A.; Bjorkman, P.J. A linear lattice model for polyglutamine in CAG-expansion diseases. Proc. Natl. Acad. Sci. USA 2002, 99, 11634–11639. [Google Scholar] [CrossRef]
- Klein, F.A.; Zeder-Lutz, G.; Cousido-Siah, A.; Mitschler, A.; Katz, A.; Eberling, P.; Mandel, J.L.; Podjarny, A.; Trottier, Y. Linear and extended: A common polyglutamine conformation recognized by the three antibodies MW1, 1C2 and 3B5H10. Hum. Mol. Genet. 2013, 22, 4215–4223. [Google Scholar] [CrossRef]
- Nagai, Y.; Inui, T.; Popiel, H.A.; Fujikake, N.; Hasegawa, K.; Urade, Y.; Goto, Y.; Naiki, H.; Toda, T. A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol. 2007, 14, 332–340. [Google Scholar] [CrossRef]
- Miller, J.; Arrasate, M.; Brooks, E.; Libeu, C.P.; Legleiter, J.; Hatters, D.; Curtis, J.; Cheung, K.; Krishnan, P.; Mitra, S.; et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol. 2011, 7, 925–934. [Google Scholar] [CrossRef]
- Schaffar, G.; Breuer, P.; Boteva, R.; Behrends, C.; Tzvetkov, N.; Strippel, N.; Sakahira, H.; Siegers, K.; Hayer-Hartl, M.; Hartl, F.U. Cellular toxicity of polyglutamine expansion proteins: Mechanism of transcription factor deactivation. Mol. Cell 2004, 15, 95–105. [Google Scholar] [CrossRef]
- Dunah, A.W.; Jeong, H.; Griffin, A.; Kim, Y.M.; Standaert, D.G.; Hersch, S.M.; Mouradian, M.M.; Young, A.B.; Tanese, N.; Krainc, D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science 2002, 296, 2238–2243. [Google Scholar] [CrossRef]
- Busch, A.; Engemann, S.; Lurz, R.; Okazawa, H.; Lehrach, H.; Wanker, E.E. Mutant huntingtin promotes the fibrillogenesis of wild-type huntingtin: A potential mechanism for loss of huntingtin function in Huntington’s disease. J. Biol. Chem. 2003, 278, 41452–41461. [Google Scholar]
- Monoi, H.; Futaki, S.; Kugimiya, S.; Minakata, H.; Yoshihara, K. Poly-l-glutamine forms cation channels: Relevance to the pathogenesis of the polyglutamine diseases. Biophys. J. 2000, 78, 2892–2899. [Google Scholar] [CrossRef]
- Kagan, B.L.; Azimov, R.; Azimova, R. Amyloid peptide channels. J. Membr. Biol. 2004, 202, 1–10. [Google Scholar] [CrossRef]
- Singer, S.J.; Dewji, N.N. Evidence that perutz’s double-beta-stranded subunit structure for β-amyloids also applies to their channel-forming structures in membranes. Proc. Natl. Acad. Sci. USA 2006, 103, 1546–1550. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hoffner, G.; Djian, P. Monomeric, Oligomeric and Polymeric Proteins in Huntington Disease and Other Diseases of Polyglutamine Expansion. Brain Sci. 2014, 4, 91-122. https://doi.org/10.3390/brainsci4010091
Hoffner G, Djian P. Monomeric, Oligomeric and Polymeric Proteins in Huntington Disease and Other Diseases of Polyglutamine Expansion. Brain Sciences. 2014; 4(1):91-122. https://doi.org/10.3390/brainsci4010091
Chicago/Turabian StyleHoffner, Guylaine, and Philippe Djian. 2014. "Monomeric, Oligomeric and Polymeric Proteins in Huntington Disease and Other Diseases of Polyglutamine Expansion" Brain Sciences 4, no. 1: 91-122. https://doi.org/10.3390/brainsci4010091