1. Introduction
The rapid development of DNA sequencing techniques has resulted in explosive growth in the number of DNA primary sequences, and the analysis and comparison of biological sequences has become a topic of considerable interest in Computational Biology and Bioinformatics. The traditional measure for similarity analysis of DNA sequences is based on multiple sequence alignment, which uses dynamic programming techniques to identify the globally optimal alignment solution. However, the sequence alignment problem is NP-hard (non-deterministic polynomial-time hard), making it infeasible for dealing with large datasets [
1]. To overcome the limitation, a lot of alignment-free approaches for sequence comparison have been proposed.
The basic idea behind most alignment-free methods is to characterize DNA by certain mathematical models derived for DNA sequence, rather than by a direct comparison of DNA sequences themselves. Graphical representation is deemed to be a simple and powerful tool for the visualization and analysis of bio-sequences. The earliest attempts at the graphical representation of DNA sequences were made by Hamori and Ruskin in 1983 [
2]. Afterwards, a number of graphical representations were well developed by researchers. For instance, by assigning four directions defined by the positive/negative
x and
y coordinate axes to the four nucleic acid bases, Gates [
3], Nandy [
4,
5], and Leong and Morgenthaler [
6] introduced three different 2-D graphical representations, respectively. While Jeffrey [
7] proposed a chaos game representation (CGR) of DNA sequences, in which the four corners of a selected square are associated with the four bases respectively. In 2000, Randic
et al. [
8] generalized these 2-D graphical representations to a 3-D graphical representation, in which the center of a cube is chosen as the origin of the Cartesian (
x,y,z) coordinate system, and the four corners with coordinates (+1,−1,−1), (−1,+1,−1), (−1,−1,+1), and (+1,+1,+1) are assigned to the four bases. Some other graphical representations of bio-sequences and their applications in the field of biological science and technology can be found in [
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24].
Numerical characterization is another useful tool for sequence comparison. One way to arrive at the numerical characterization of a DNA sequence is to associate the sequence with a vector whose components are related to
k-words, including the single nucleotide, dinucleotide, trinucleotide, and so on [
25,
26,
27,
28,
29,
30]. In addition, the numerical characterization can be accomplished by associating with a graphical representation given by a curve in the space (or a plane) structural matrices, such as the Euclidean-distance matrix (ED), the graph theoretical distance matrix (GD), the quotient matrix (D/D, M/M, L/L), and their “higher order” matrices [
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
31,
32,
33]. Once a matrix representation of a DNA sequence is given, some matrix invariants, e.g. the leading eigenvalues, can be used as descriptors of the sequence. This technique has been widely used in the field of biological science and medicine, and different types of matrices are defined to construct various invariants of DNA sequences. However, the order of these matrices is equal to
n, the length of the DNA sequence considered. A problem we must face is that the calculation of these matrix invariants will become more and more difficult with larger
n values [
17,
24,
32].
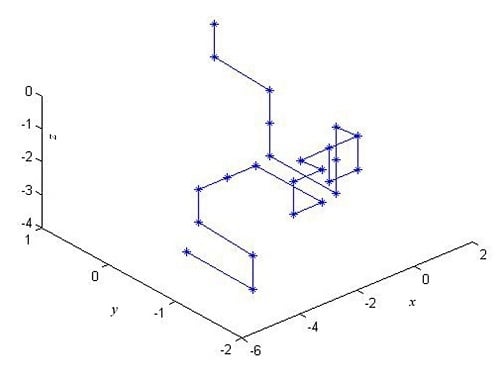
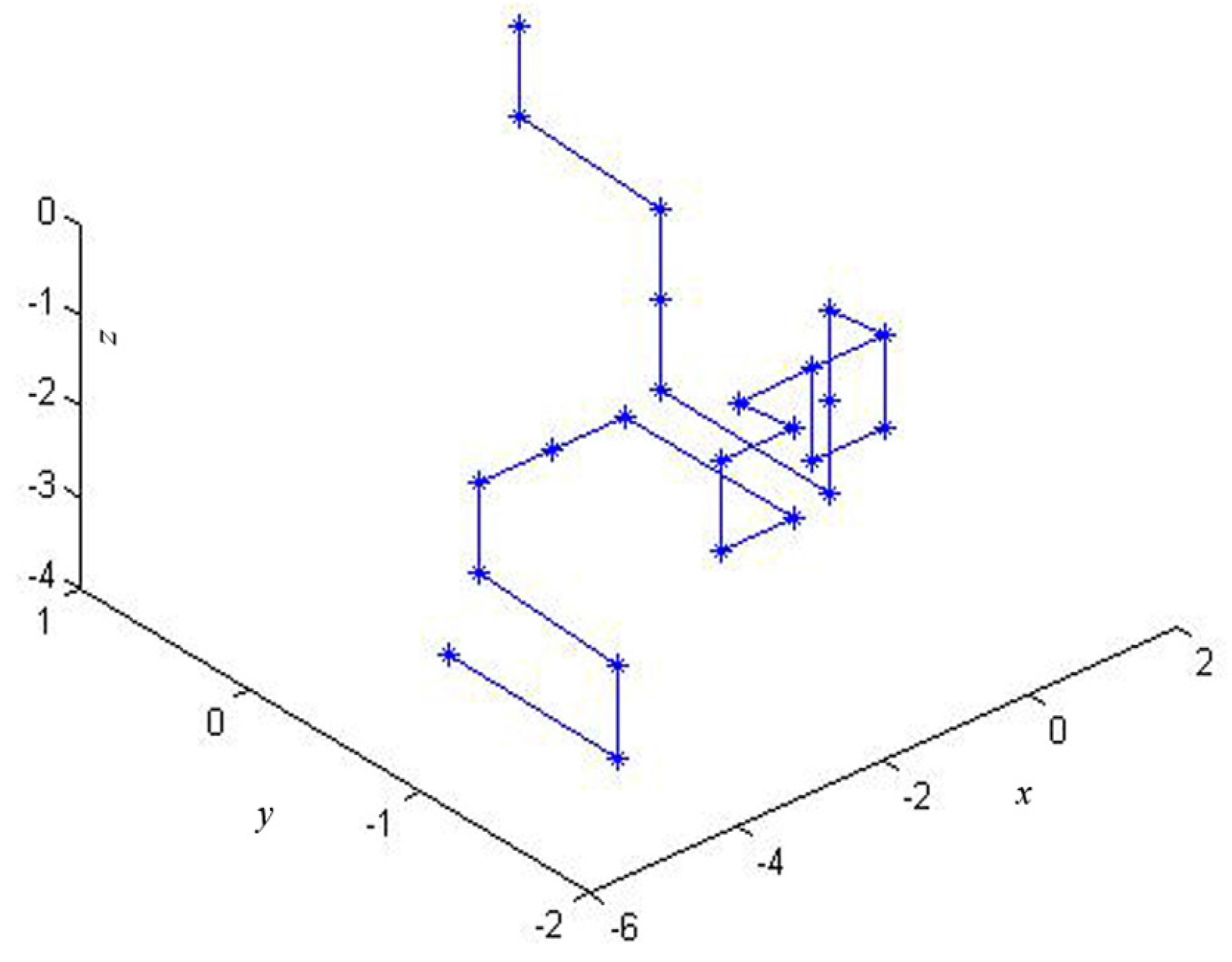
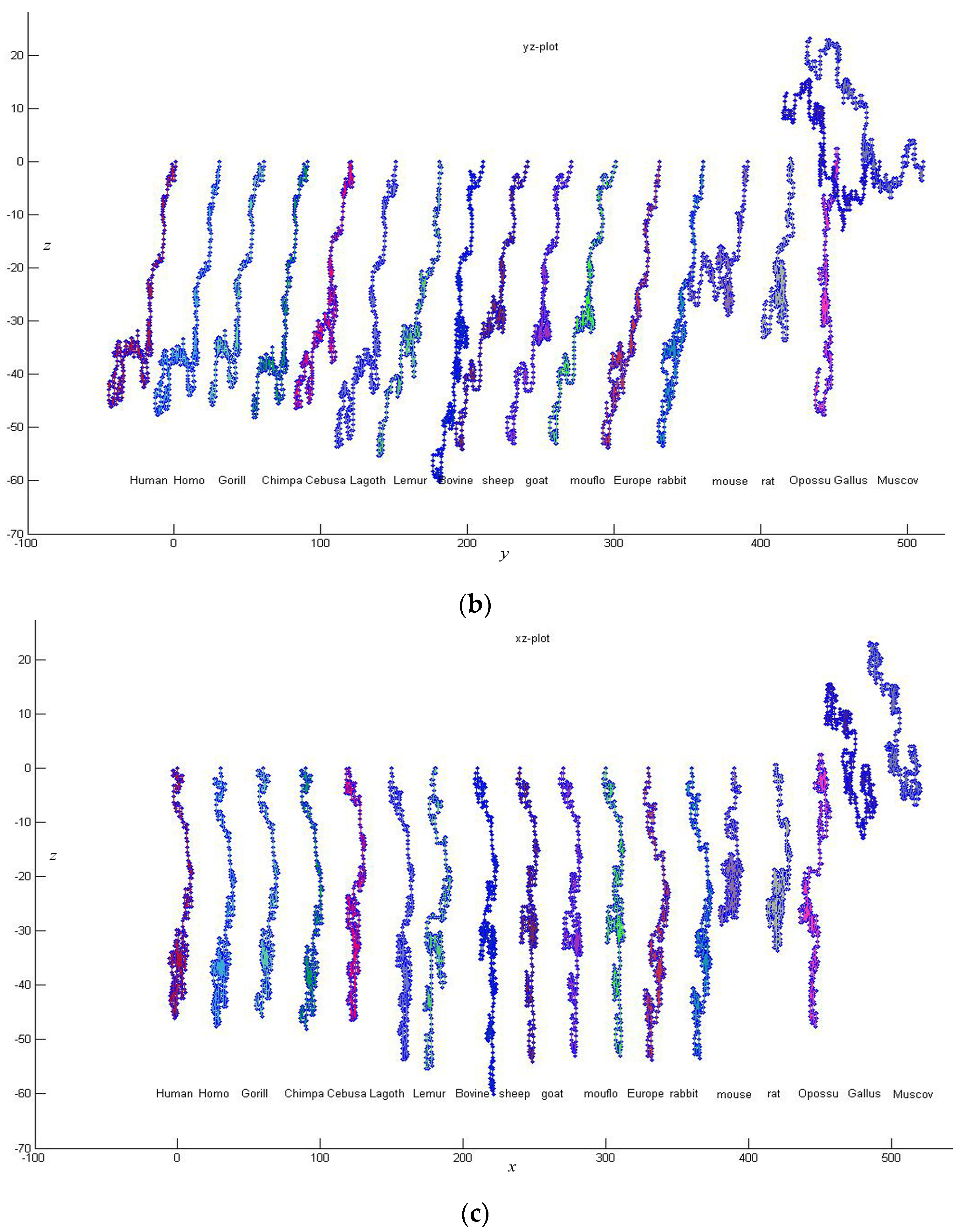
In this paper, based on all of the 2-combinations of the multiset , we propose a novel graphical representation of DNA sequences. Then, according to the idea of “piecewise function”, we describe a particular scheme that transforms the graphical representation of DNA into a cell-based descriptor vector. The introduced vector leads to more simple characterizations and comparisons of DNA sequences.
3. Results and Discussion
In this section, we will illustrate the use of the proposed cell-based descriptor
of a DNA sequence. For any two sequences
and
, suppose their descriptor vectors are
and
, respectively. Then, their similarity can be examined by the following Euclidean distance. Clearly, the smaller the Euclidean distance is, the more similar the two DNA sequences are.
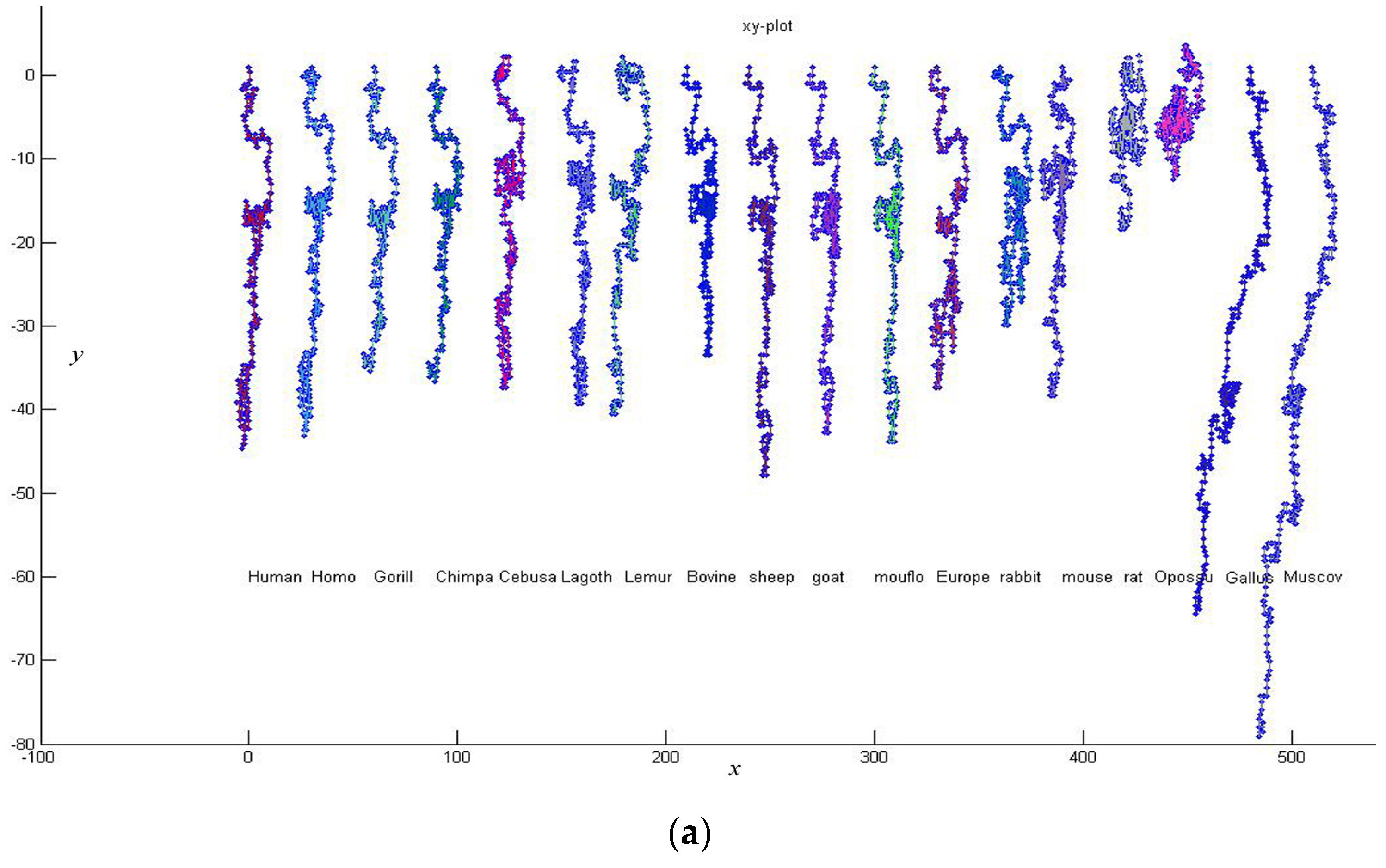
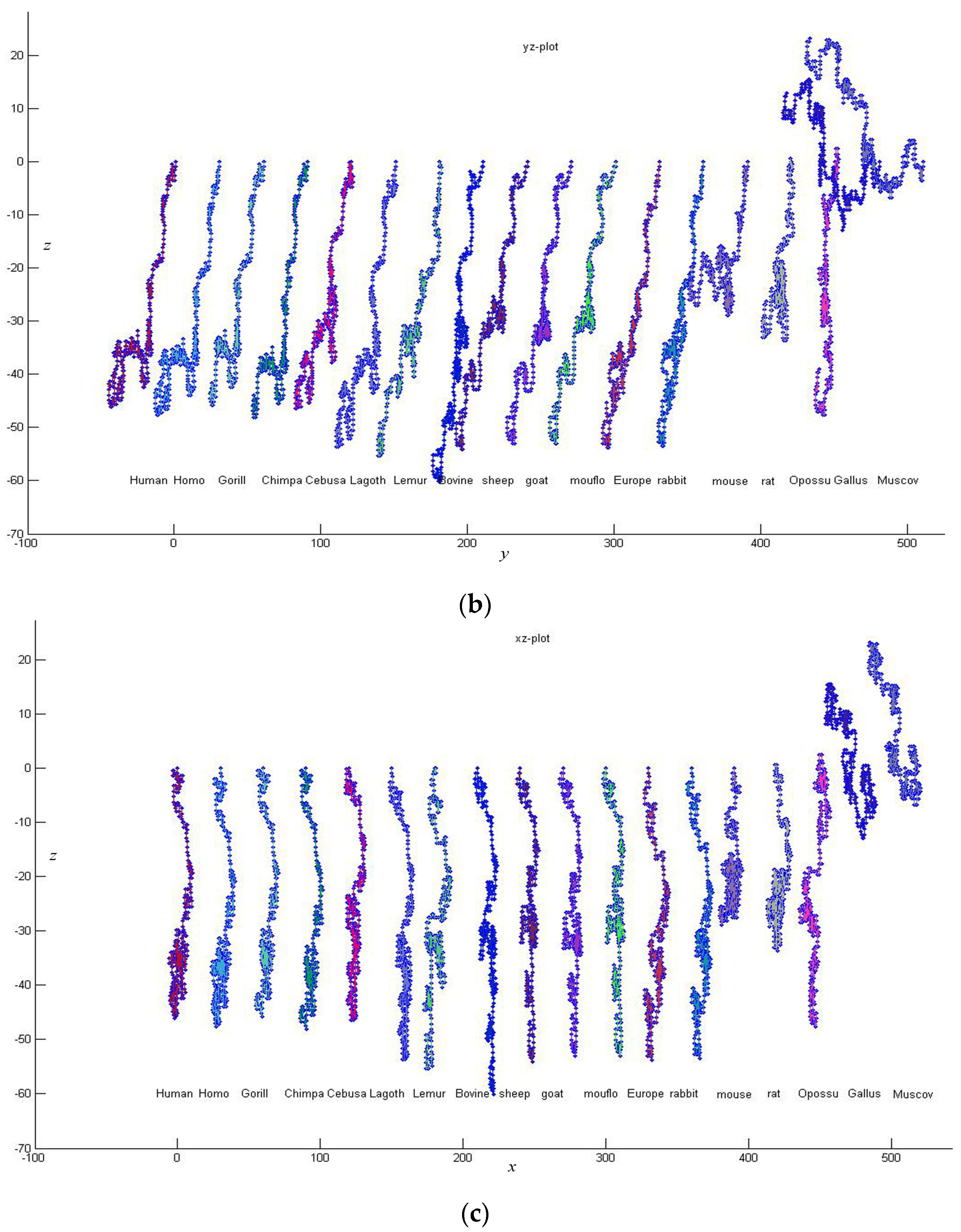
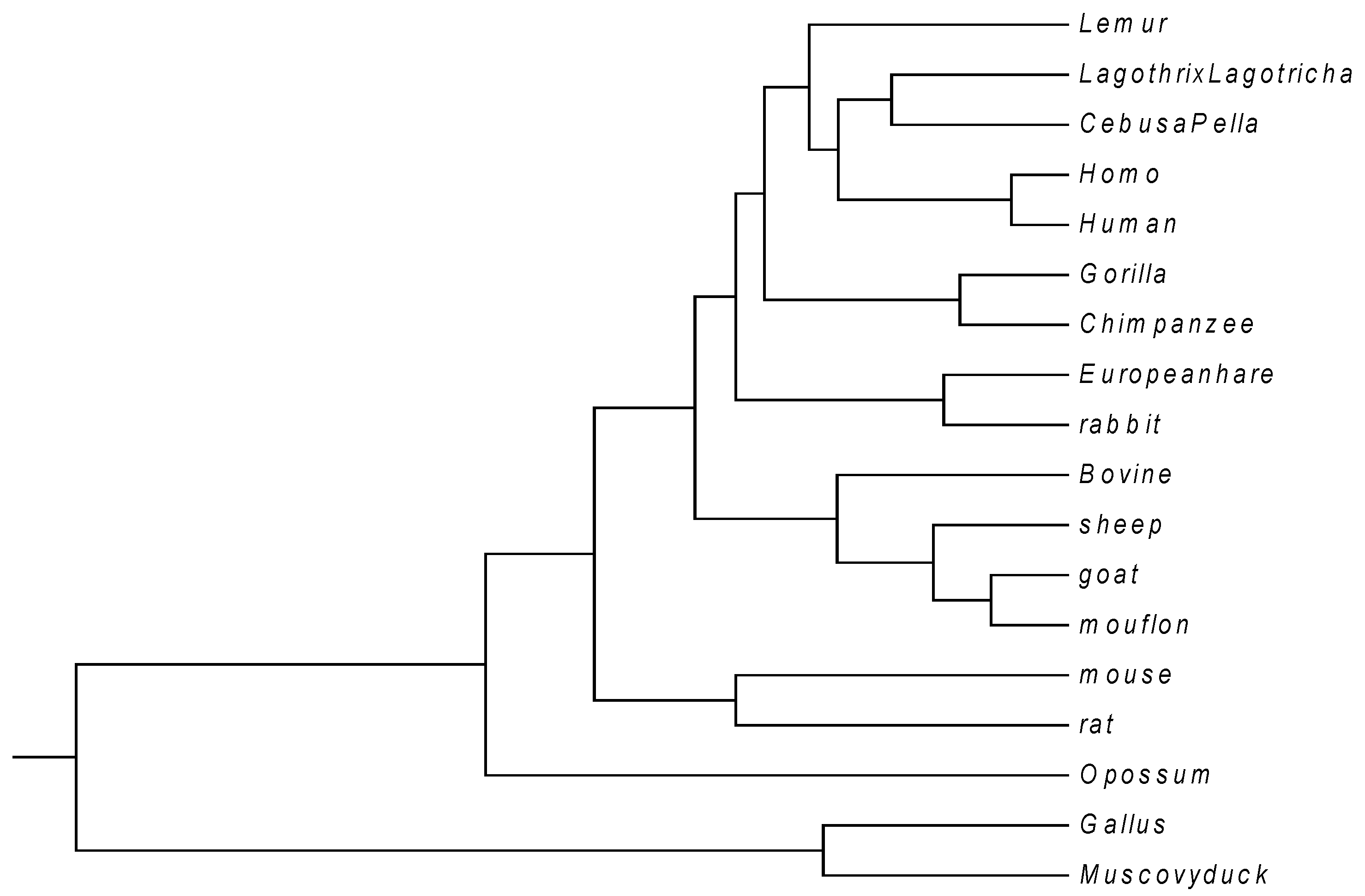
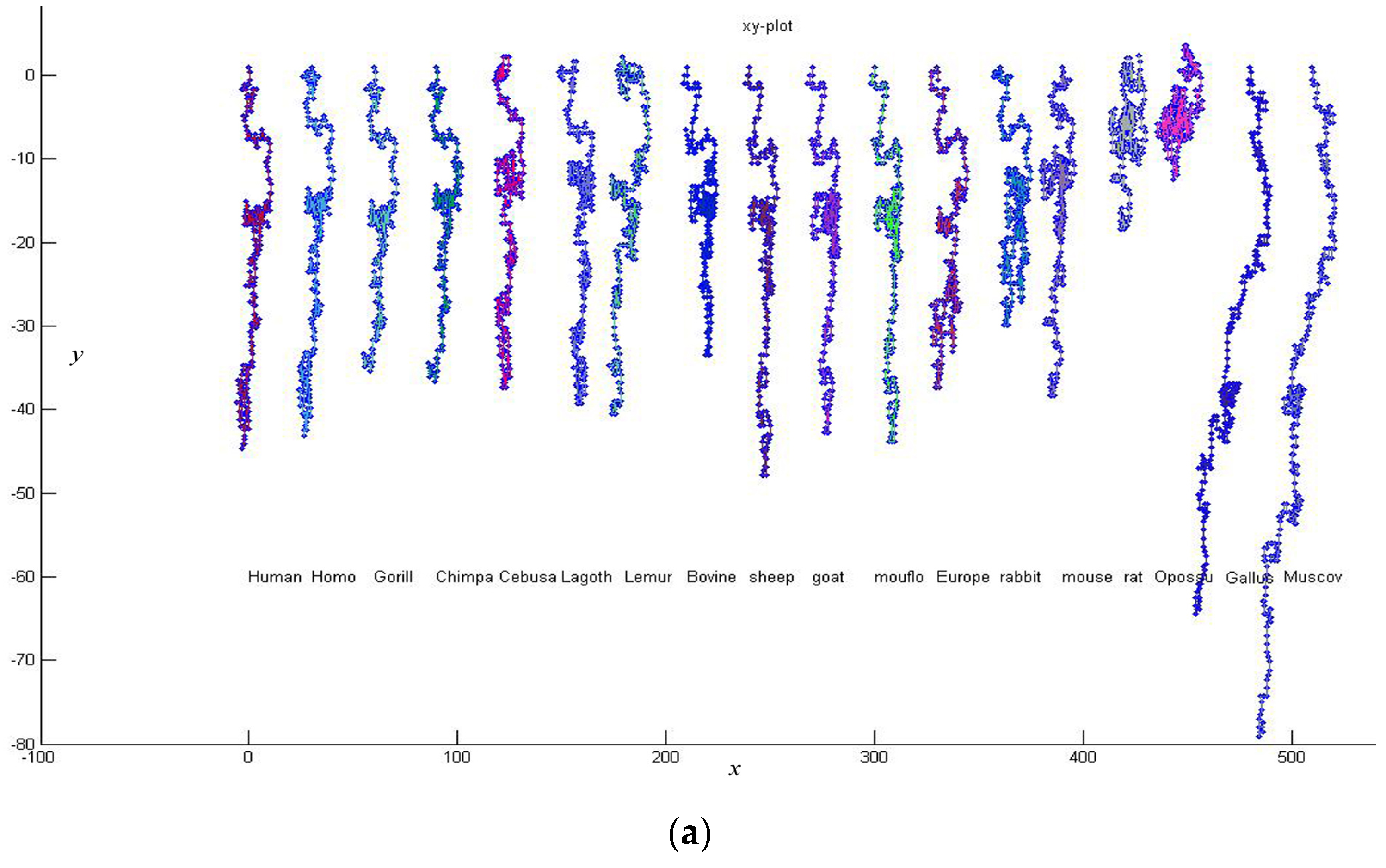
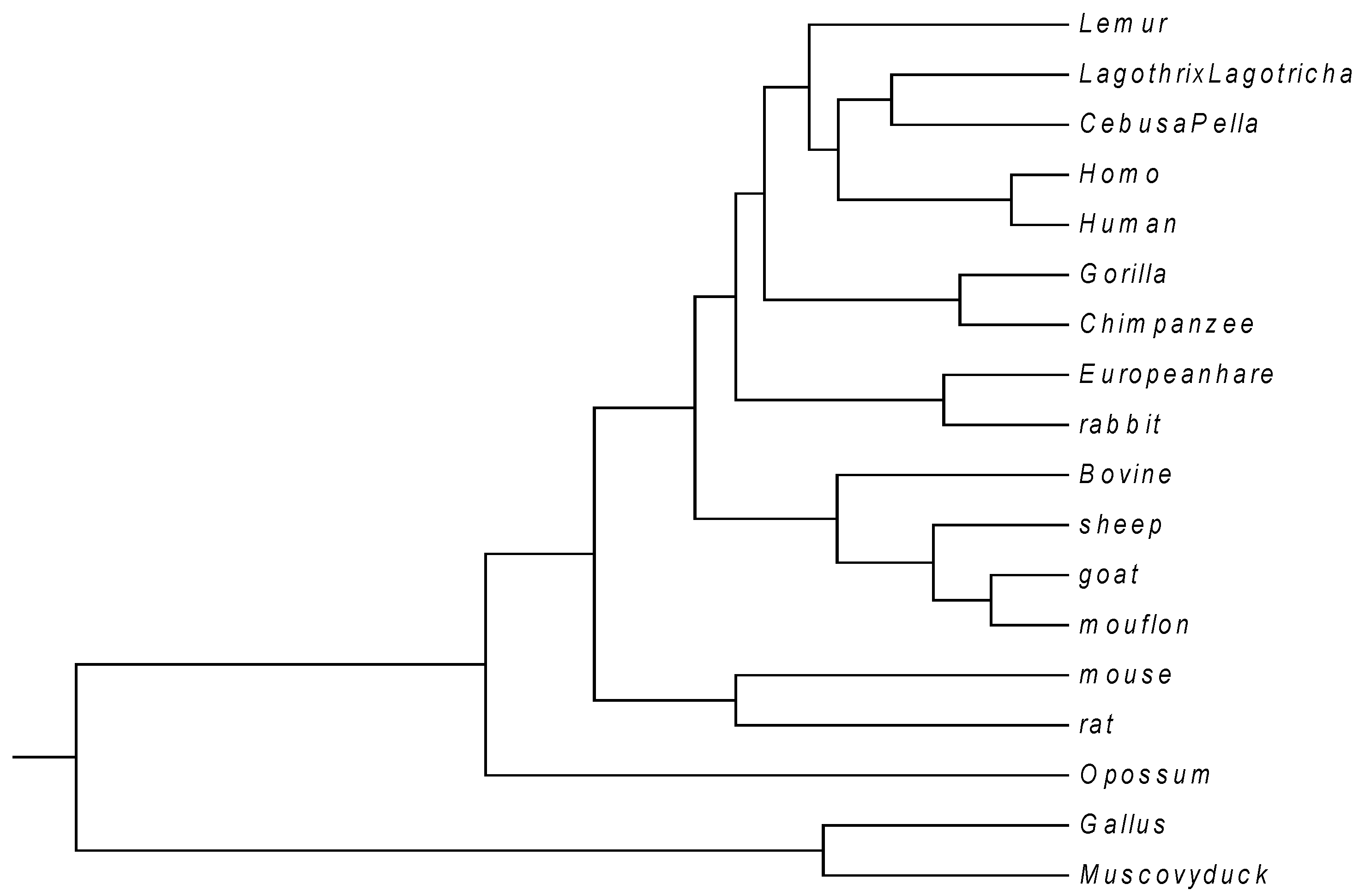
Firstly, we give a comparison for CDS (Coding DNA Sequence) of
β-globin gene of 18 species listed in
Table 3. The lengths of the 18 sequences are about 434 bp. Thus
K is taken to be 4, and each of these sequences is converted into a 12-D vector. According to Equation (7), we calculate the distance between any two of the 18 DNA sequences. Then an
real symmetric matrix
is obtained. On the basis of
, a phylogenetic tree (see
Figure 3) is constructed using UPGMA (Unweighted Pair Group Method with Arithmetic Mean) program included in MEGA4 [
34]. Observing
Figure 3, we find that the CDS are more similar for Primate group {
Gorilla, Chimpanzee, Human, Homo, CebusaPella, LagothrixLagotricha, Lemur}, Cetartiodactyla group {
bovine, sheep, goat, mouflon}, Lagomorpha group {
Rabbit, European hare}, and Rodentia group {
mouse, rat}, respectively. On the other hand, CDS of the two kinds of non-mammals {
Gallus, Muscovy duck} are very dissimilar to the mammals because they are grouped into an independent branch. This is analogous to that reported in the literature [
8,
12,
14,
31], and the relationship of these species detected by their graphical representations as well. From this result, a conclusion one can draw is that the cell-based descriptors of the new graphical representation may suffice to characterize DNA sequences.
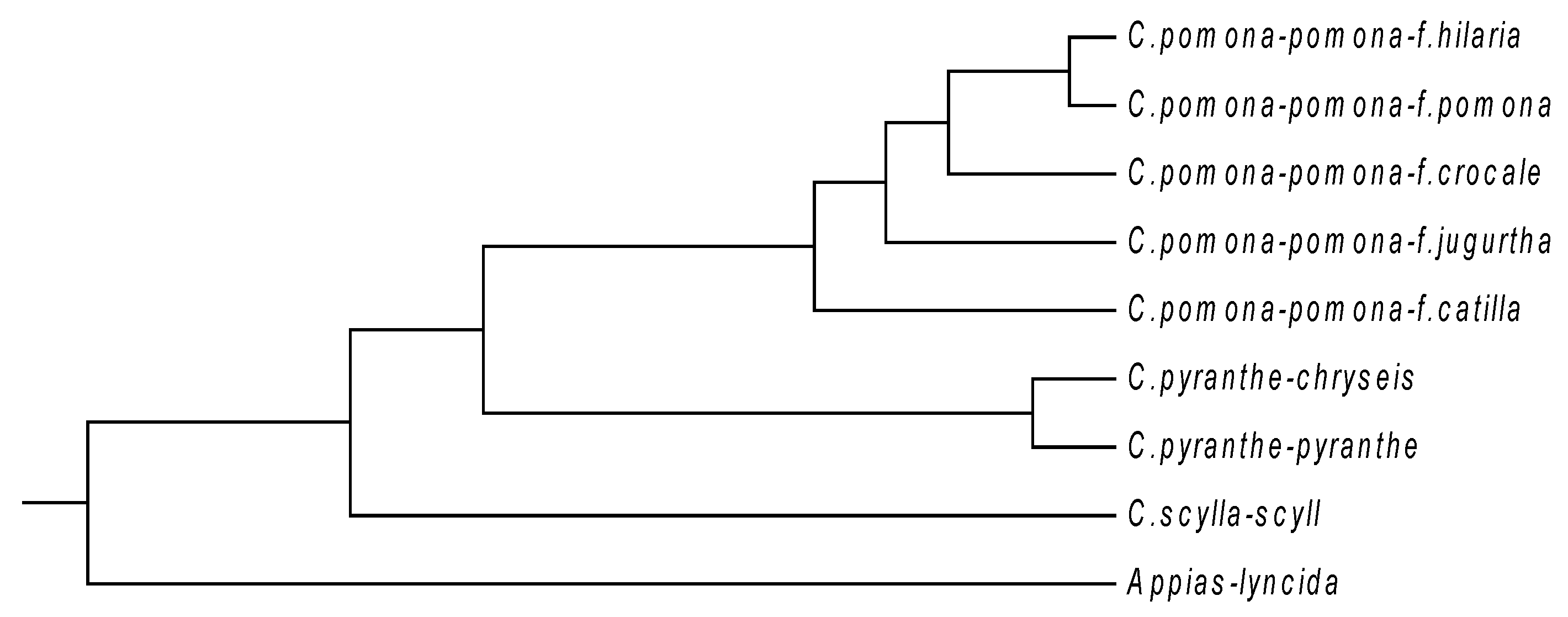
In order to further illustrate the effectiveness of our method, we test it by phylogenetic analysis on other three datasets: one consists of mitochondrial cytochrome oxidase subunit I (COI) genes of nine butterflies, another includes S segments of 32 hantaviruses (HVs), and the last is composed of 70 complete mitogenomes (mitochondrial genomes). For convenience, we denote the three datasets by COI, HV and mitogenome, respectively. In the COI dataset (see
Table 4), which is taken from Yang
et al. [
12], eight belong to the
Catopsilia genus and one belongs to
Appias genus, which is used as the out-group. The average length of these COI gene sequences is 661 bp, and thus
K, the number of cells, is calculated as 4. According to the method mentioned above, a distance matrix is constructed, and then a phylogenetic tree (see
Figure 4) is generated.
Figure 4 shows that the five
pomona sub-species have relatively high similarity with each other, while the two
pyranthe sub-species cluster together. In addition,
scylla sub-species is situated at an independent branch, whereas the
Appias lyncida stays outside of all the
Catopsilia. This result is consistent with that reported in [
12,
35].
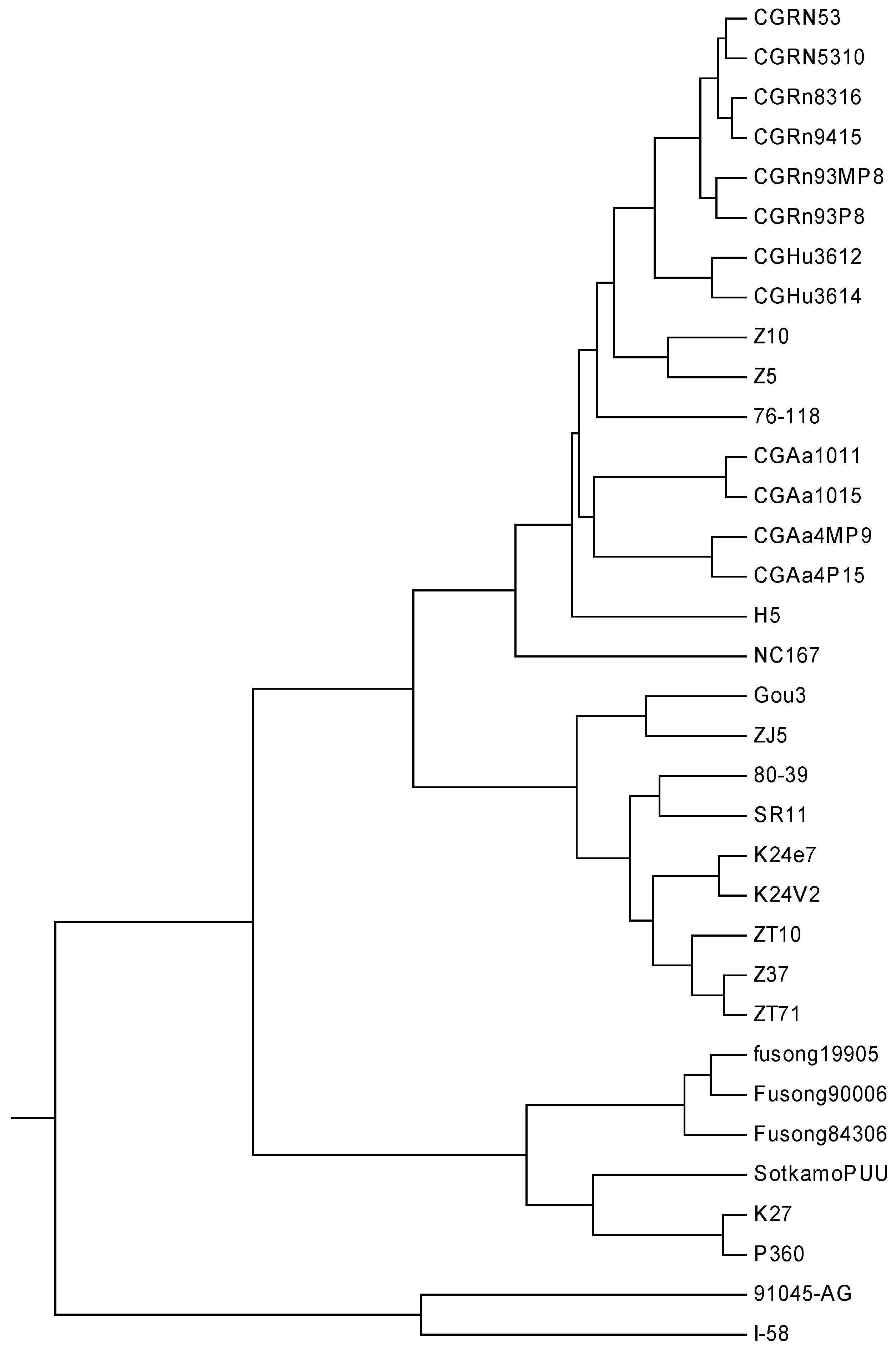
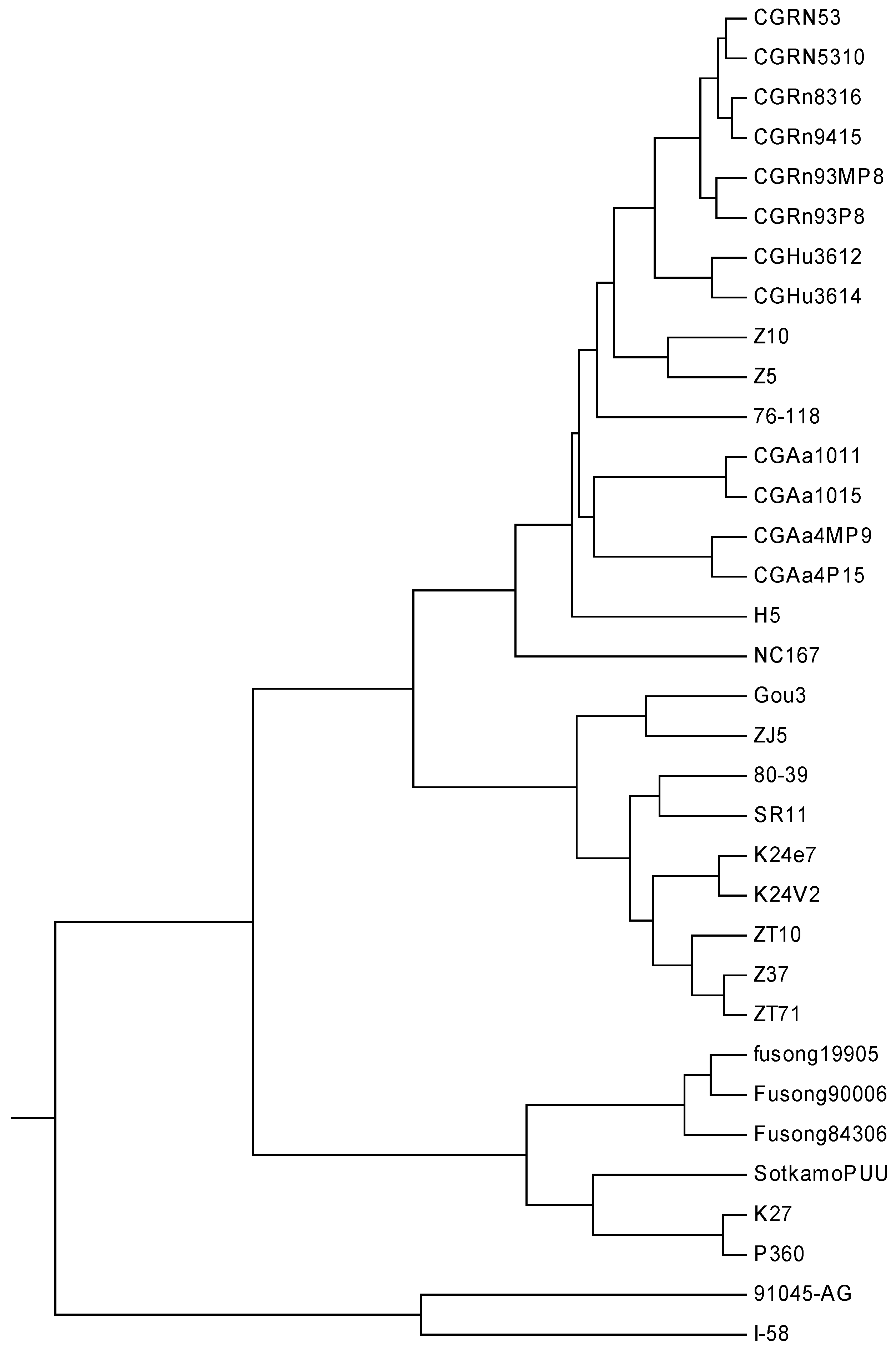
The hantavirus (HV), which is named for the Hantan River area in South Korea, is a relatively newly discovered RNA virus in the family
Bunyaviridae. This kind of virus normally infects rodents and does not cause disease in these hosts. Humans may be infected with HV, and some HV strains could cause severe, sometimes fatal, diseases in humans, such as HFRS (hantavirus hemorrhagic fever with renal syndrome) and HPS (hantavirus pulmonary syndrome). The later occurred in North and South America, while the former mainly in Eurasia [
12,
36]. In Eastern Asia, particularly in China and Korea, the viruses that cause HFRS mainly include Hantaan (HTN) and Seoul (SEO) viruses, while Puumala (PUU) virus is found in Western Europe, Russia and northeastern China. The HV dataset analyzed in this paper includes 32 HV sequences. Phlebovirus (PV) is another genus of the family
Bunyaviridae. Here, two PV strains KF297911 and KF297914 are used as the out-group. The name, accession number, type, and region of the 34 sequences are described in
Table 5. The lengths of these sequences are in the range of 1.30–1.88 kbp. Thus
K is calculated as 5, and each of the 34 viruses is converted into a 15-D vector. The phylogenetic tree constructed by our method is shown in
Figure 5.
From
Figure 5, we find that the two PV strains form an independent branch, which can be distinguished easily from the HV strains, while the 32 HVs are grouped into three separate branches: the strains belonging to PUUV are clearly clustered together, the strains belonging to SEOV appear to cluster together, and so do the ones belonging to HTNV. A closer look at the subtree of HTNV, all CGRn strains whose host is
Rattus norvegicus tend to cluster together, so it is with the CGHu strains whose host is Homo sapiens. In addition, all the four CGAa strains whose host is
Apodemus agrarius are grouped closely. Needless to say, the phylogeny is not only closely related to the isolated regions, but also has certain relationship with the host. This result is similar to that reported in [
12,
37].
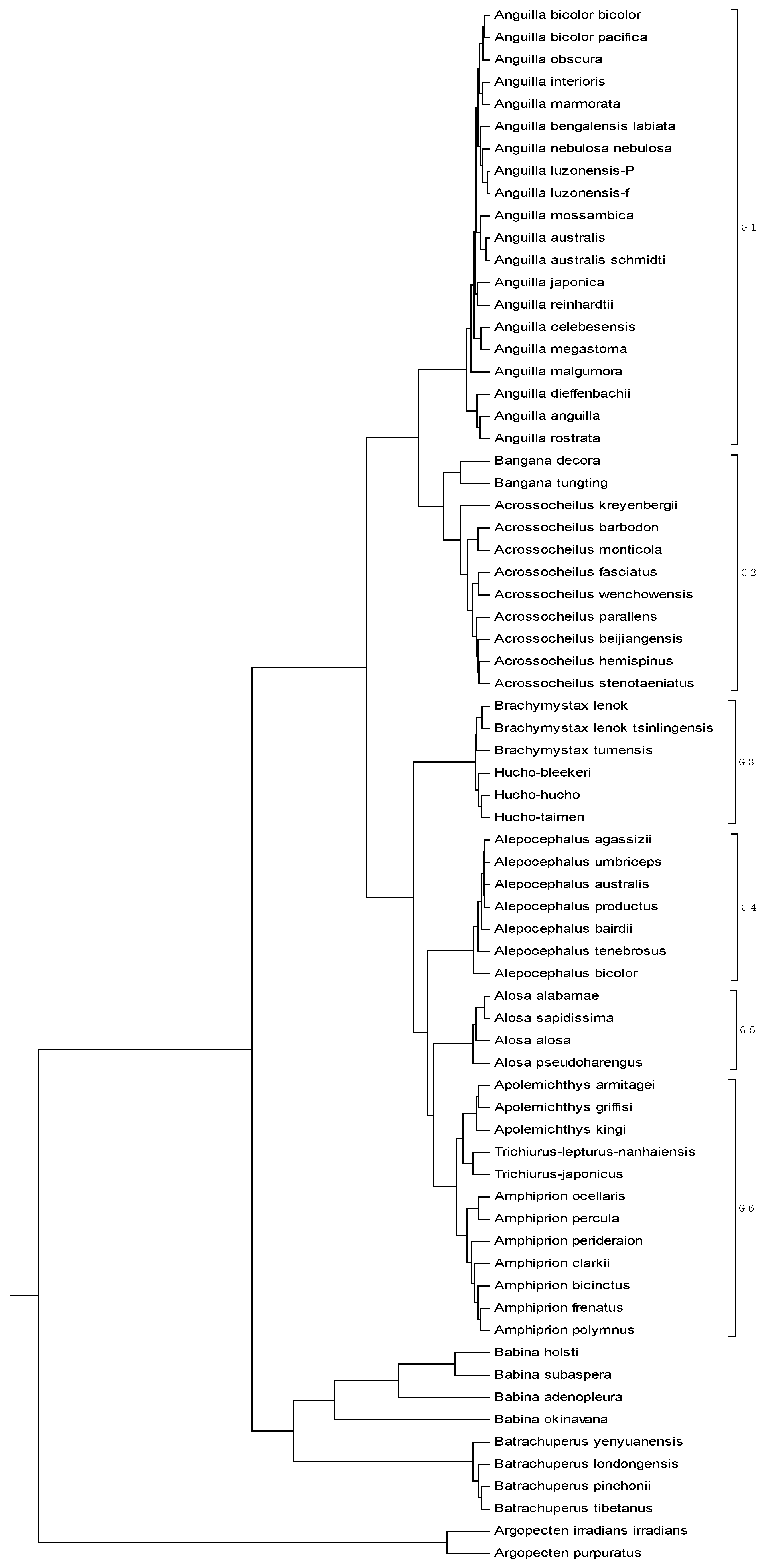
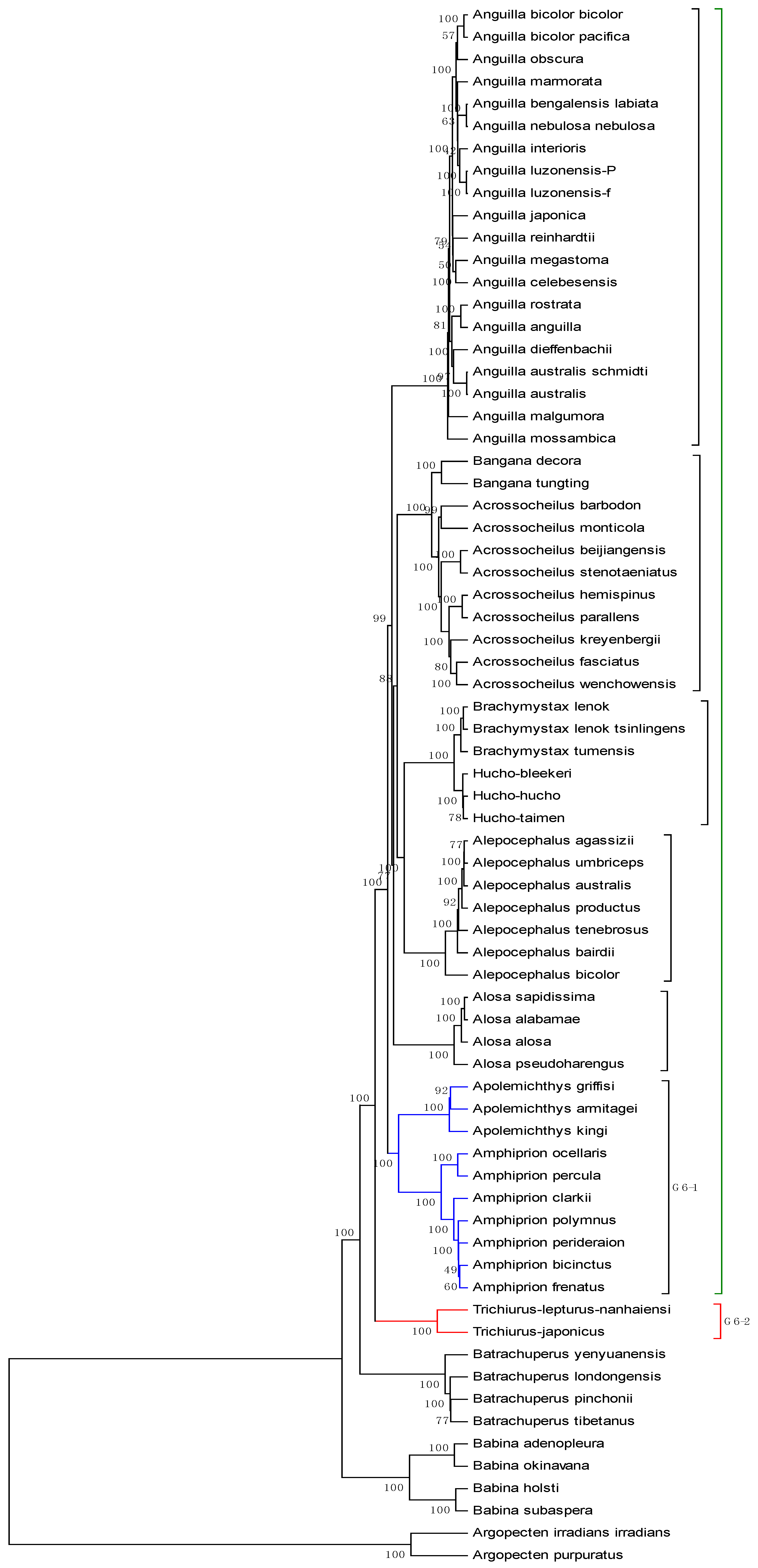
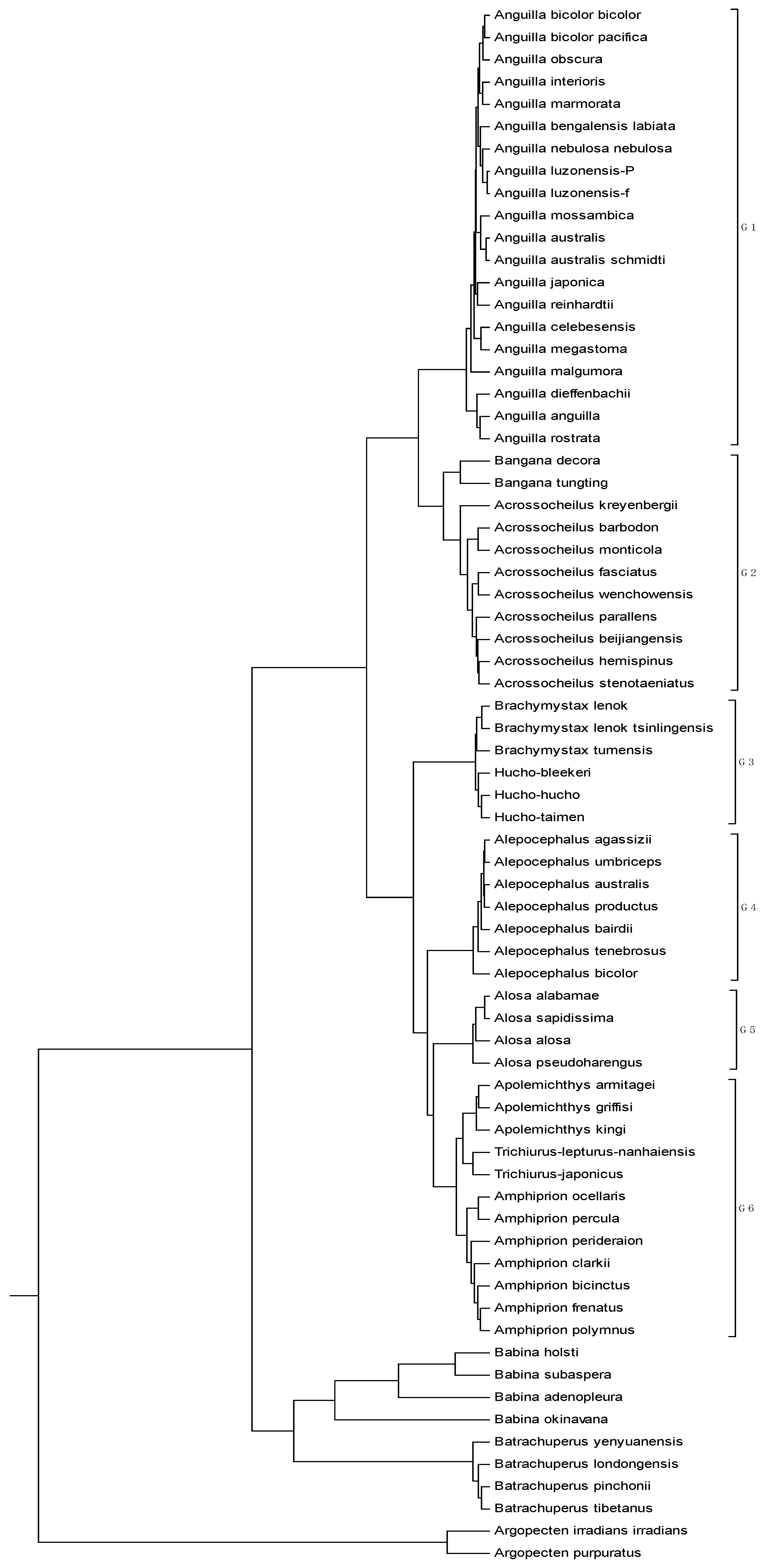
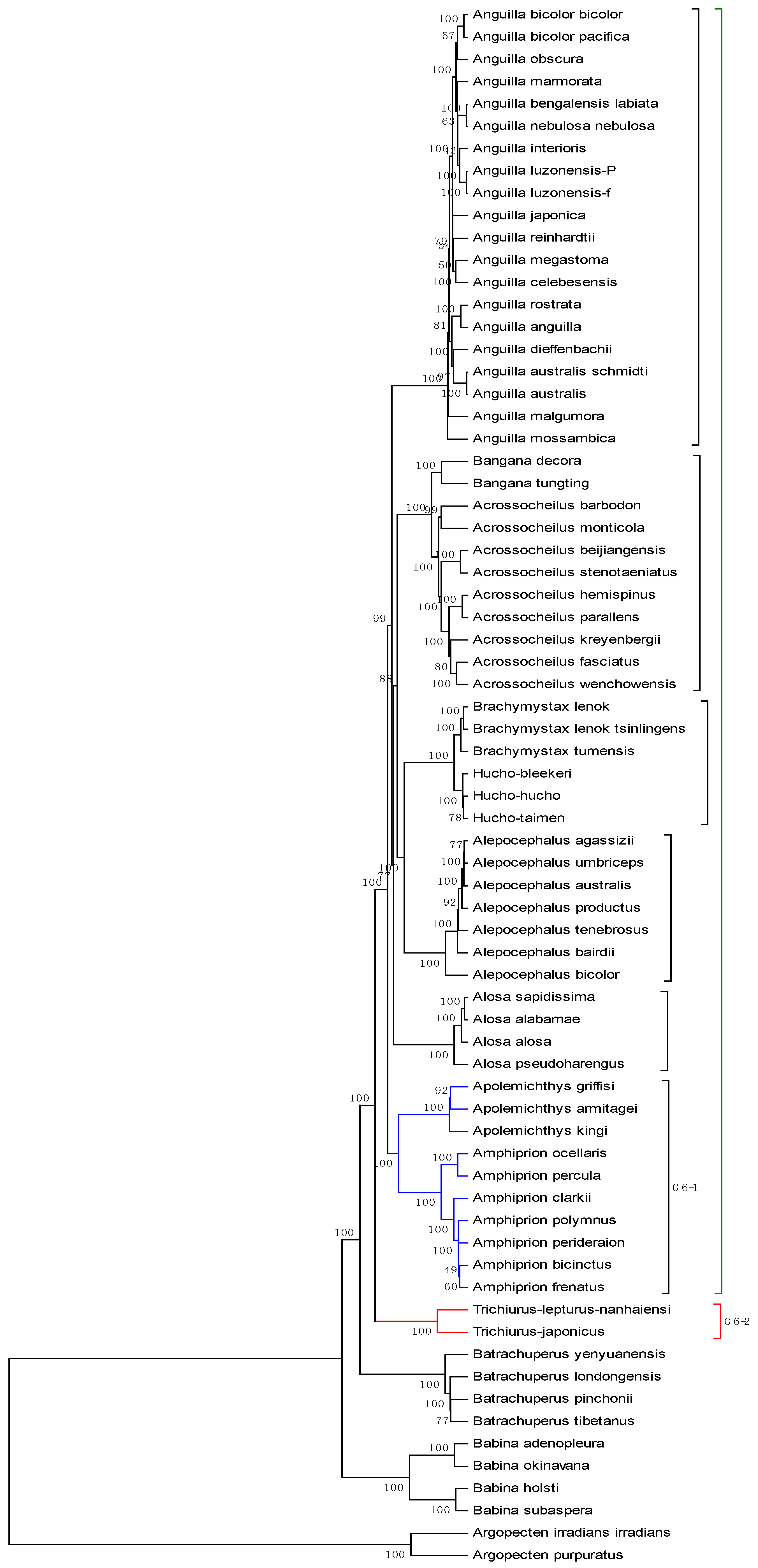
The mitogenome dataset comprises 70 complete mitochondrial genomes of Eukaryota. The name, accession number, and genome length are listed in
Table 6. Among them, two species (
Argopecten irradians irradians and
Argopecten purpuratus) belong to family Pectinidae are used as the out-group. Four species belong to the Order Caudata under the Class Amphibia, while four species belong to the Order Anura under the same Class. The remaining belongs to the Class Actinopterygii. The average length of the 70 genome sequences is about 16817 bp. Thus,
K is calculated as 6, and each of these genome sequences is converted into an 18-D vector. The phylogenetic tree constructed by our method is shown in
Figure 6. It is easy to see from
Figure 6 that the two
Pectinidae species stay outside of the others, while the four
Hynobiidae species and four
Ranidae species form an independent branch. In the subtree of the Class Actinopterygii, the 60 genomes are separated into six groups: group 1 corresponds to genus
Anguilla under family Anguillidae; group 2 includes genera
Bangana and
Acrossocheilus under family Cyprinidae; group 3 includes genera
Brachymystax and
Hucho under family Salmonidae; group 4 is genus
Alepocephalus under family Alepocephalidae; group 5 is the family of Clupeidae; group 6 includes genera
Trichiurus,
Amphiprion and
Apolemichthys under Acanthomorphata. This result agrees well with the established taxonomic groups. In addition, we make a comparison for the 70 genome sequences by using ClustalX2.1 [
38], and the corresponding tree is shown in
Figure 7. Observing
Figure 7, we find that the tree includes four branches: the outside is the
Argopecten branch, the following is
Babina, then
Batrachuperus, and the subtree consisting of the other 60 species. A closer look at the subtree shows that
Trichiurus is distinguished from the remaining, which seems to be a disappointing phenomenon in the evolutionary sense.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}