
Development of Polymorphic Genic SSR Markers by Transcriptome Sequencing in the Welsh Onion (Allium fistulosum L.)

Abstract
:1. Introduction
2. Results and Discussion
2.1. Frequency and Distribution of Genic SSRs in the Welsh Onion Transcriptome

{kind=link}
{kind=link}
| SSR Motif | Repeat Number | Total | Percentage (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13–24 | |||
| Mononucleotide | 0 | 0 | 0 | 0 | 0 | 2060 | 548 | 297 | 489 | 3394 | 52.33 |
| Dinucleotide | 0 | 478 | 212 | 119 | 57 | 32 | 37 | 10 | 0 | 945 | 14.57 |
| Trinucleotide | 1328 | 510 | 188 | 47 | 0 | 0 | 0 | 0 | 0 | 2073 | 31.96 |
| Tetranucleotide | 52 | 14 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 66 | 1.02 |
| Pentanucleotide | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0.12 |
| Total | 1388 | 1002 | 400 | 166 | 57 | 2092 | 585 | 307 | 489 | 6486 | 100 |
| Percentage (%) | 21.40 | 15.45 | 6.17 | 2.56 | 0.88 | 32.25 | 9.02 | 4.73 | 7.54 | 100 | |
| Main Repeat Motif | Repeat Number | The Percentage of Each Type of SSR Repeat Motif (%) | The Percentage of Total SSR Repeat Motifs (%) |
|---|---|---|---|
| A/T | 3336 | 98.29 | 51.43 |
| C/G | 58 | 1.71 | 0.89 |
| AC/GT | 294 | 31.11 | 4.53 |
| AG/CT | 324 | 34.29 | 5.00 |
| AT/AT | 318 | 33.65 | 4.90 |
| CG/CG | 9 | 0.95 | 0.14 |
| AAC/GTT | 118 | 5.69 | 1.82 |
| AAG/CTT | 548 | 26.44 | 8.45 |
| AAT/ATT | 146 | 7.04 | 2.25 |
| ACC/GGT | 145 | 6.99 | 2.24 |
| ACG/CGT | 86 | 4.15 | 1.33 |
| ACT/AGT | 43 | 2.07 | 0.66 |
| AGC/GCT | 297 | 14.33 | 4.58 |
| AGG/CCT | 344 | 16.59 | 5.30 |
| ATC/GAT | 314 | 15.15 | 4.84 |
| CCG/CGG | 32 | 1.54 | 0.49 |
| AAAC/GTTT | 7 | 10.61 | 0.11 |
| AAAG/CTTT | 7 | 10.61 | 0.11 |
| AAAT/ATTT | 22 | 33.33 | 0.34 |
| ACAT/ATGT | 19 | 28.79 | 0.29 |
| No. | Materials | Origin |
|---|---|---|
| 1 | “Yuanzang” | Japan |
| 2 | “HanLvDaZhu” | Korea |
| 3 | “WuYeQi”1 | Hebei, China |
| 4 | “WuYeQi”2 | Tianjin, China |
| 5 | “GaoBaiQingCaiCong” | Hebei, China |
| 6 | “ZhongYingTeDa” | Henan, China |
| 7 | “SanYu” | Shandong, China |
| 8 | “YeFuYiHao” | Shandong, China |
| 9 | “ZhangQiuDaCong” | Shandong, China |
| 10 | “ErShengZi” | Shandong, China |
| 11 | “Zao134” | Shandong, China |
| 12 | “ShanXiFenCong” | Shanxi, China |
| 13 | “Qi110” | Shandong, China |
| 14 | “ZhongHuaJuCong” | Shandong, China |
| 15 | “WuYeQi”3 | Shanxi, China |
| 16 | “ShanXiYunChengKeXingYiHao” | Shanxi, China |
| 17 | “HeBeiJinXianDaCong” | Hebei, China |
| 18 | “Shou1105” | Shandong, China |
| 19 | “LiaoNingChaoYangLinCong” | Liaoning, China |
| 20 | “XiYeWanShengCong” | Japan |
| 21 | “SongBenYiBenTaiCong” | Japan |
| 22 | “7722” | Guangdong, China |
2.2. Development and Detection of Genic SSR Markers
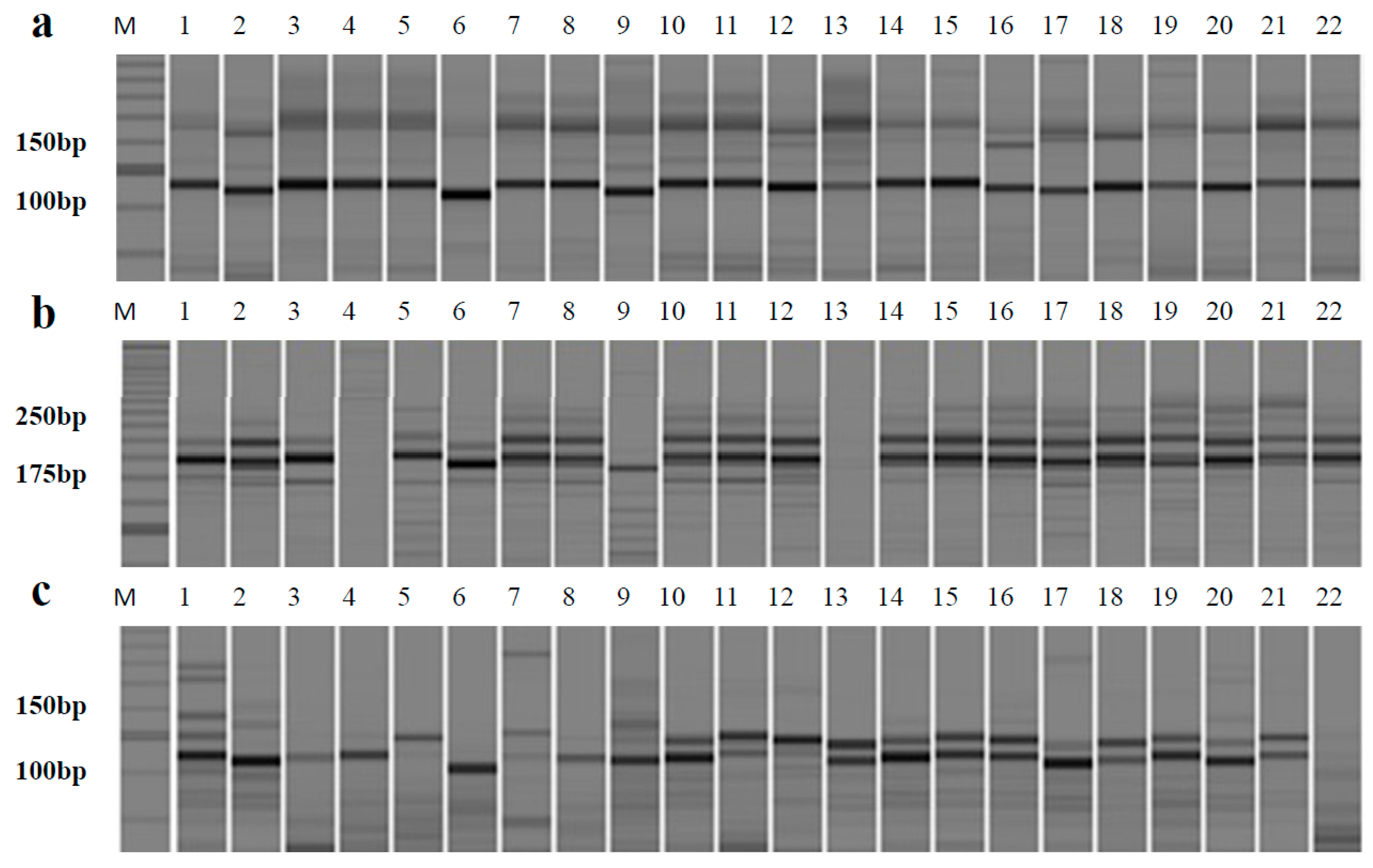
2.3. Validating the Effectiveness of Genic SSR Markers among 22 Allium Accessions
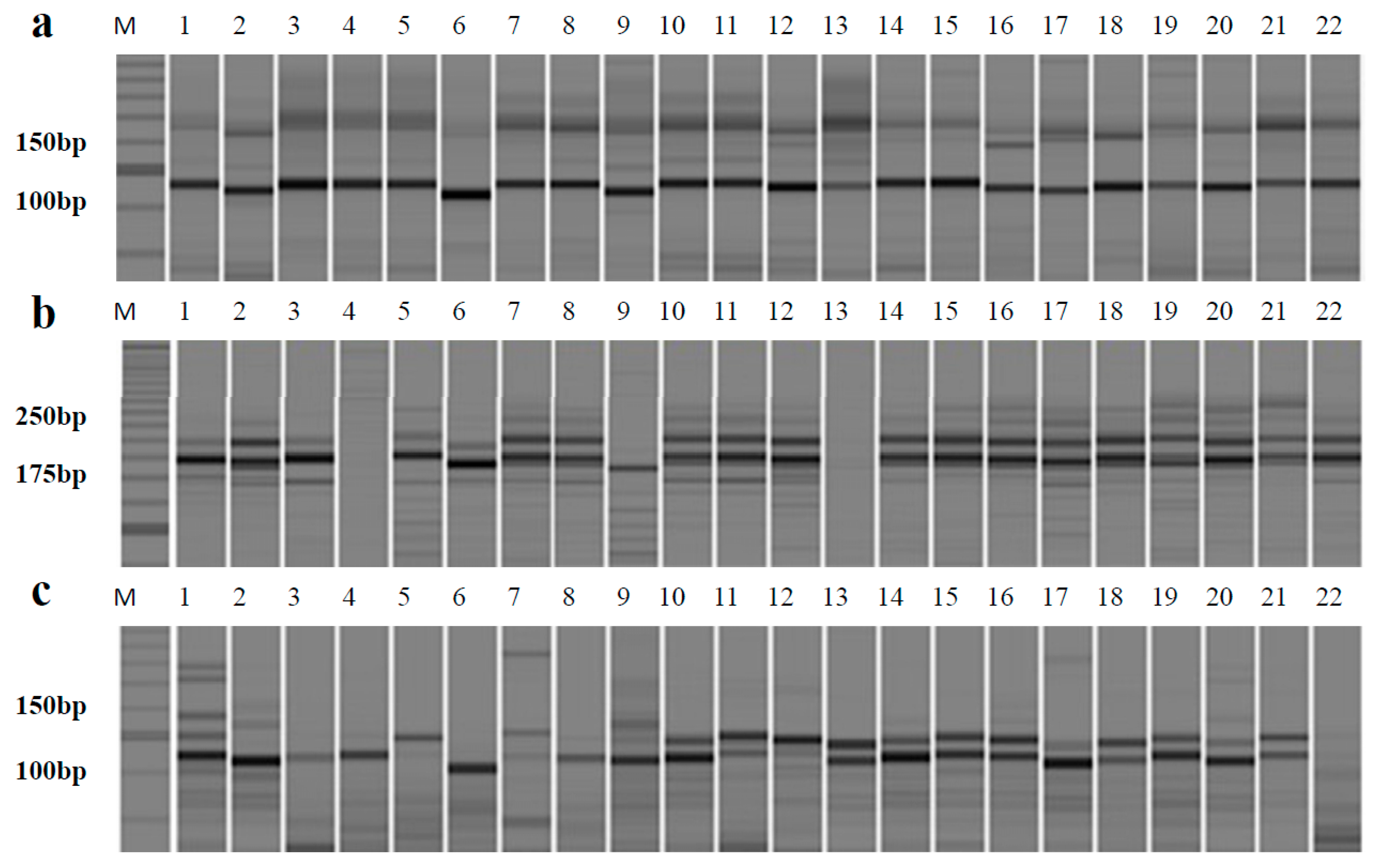
| No. | Primer | Repeat Motif | Primer Sequence (5’–3’) | Size Range (bp) | A a | Ng b | Na c | PIC d |
|---|---|---|---|---|---|---|---|---|
| 1 | MCL32 | (TG)9 | F:gatttcccatgcatcgatct | 111–117 | 0.4545 | 4 | 4 | 0.5388 |
| R:ggtcaaactgctgcatctca | ||||||||
| 2 | MCL37 | (AAG)5 | F:aaggaatgctacgccagaga | 411–461 | 0.4333 | 8 | 8 | 0.7195 |
| R:ctgaattctgctgggtctcc | ||||||||
| 3 | MCL40 | (AGA)5 | F:cggaagtagcatcggatcat | 189–226 | 0.4250 | 6 | 6 | 0.6882 |
| R:aggctcgtgctttgtgtctt | ||||||||
| 4 | MCL42 | (GCA)7 | F:cgggaacgaagagatggata | 355–497 | 0.2273 | 12 | 9 | 0.8465 |
| R:aacgaccaacaacgtccttc | ||||||||
| 5 | MCL47 | (GAT)5 | F:aggagtggaagttggtgcat | 291–298 | 0.5455 | 3 | 3 | 0.4762 |
| R:ggatcgctatccgagttcaa | ||||||||
| 6 | WC121 | (TCT)5 | F:ccgaggatctcggaactgta | 214–249 | 0.6429 | 4 | 4 | 0.4935 |
| R:tctcacagtcccatcactgc | ||||||||
| 7 | WC122 | (AGG)5 | F:attctttgaaccgcaccaac | 299–334 | 0.3750 | 7 | 8 | 0.6939 |
| R:caagacgagcaacaacctca | ||||||||
| 8 | WC126 | (TCC)6 | F:tgcctgcctcttagatgctt | 306–320 | 0.5000 | 5 | 5 | 0.6381 |
| R:caagaggggaaggtttgtca | ||||||||
| 9 | WC183 | (ATG)5 | F:aacaactgccacacaaacca | 289–304 | 0.5909 | 5 | 4 | 0.4900 |
| R:cttctggaaggcctcctctt | ||||||||
| 10 | WC193 | (CCT)5 | F:cccagtctaagcagccactc | 266–272 | 0.5882 | 3 | 3 | 0.4365 |
| R:tgttcgttcaggaggagctt | ||||||||
| 11 | WC200 | (ATC)5 | F:acagggatccatggaatgag | 158–167 | 0.4545 | 5 | 4 | 0.4938 |
| R:agtgccagactggaatggag | ||||||||
| 12 | WC208 | (CCT)5 | F:tcatctccacccttcactcc | 168–194 | 0.3409 | 7 | 6 | 0.7402 |
| R:tccgggagttcacctacatc | ||||||||
| 13 | WC228 | (CAC)6 | F:ccaccaccacctcaatatcc | 336–348 | 0.5455 | 5 | 5 | 0.5298 |
| R:ctagtcgaggtgcagcatca | ||||||||
| 14 | WC230 | (ATC)6 | F:ccagagcgattagatgcaca | 310–349 | 0.3000 | 10 | 7 | 0.7949 |
| R:tgaattgccaaaagggtctc | ||||||||
| 15 | TO249 | (ATTT)5 | F:acacggcttttggagacagt | 108–127 | 0.3750 | 5 | 4 | 0.6452 |
| R:gaggttccataacccgaaca | ||||||||
| 16 | TO250 | (TGTA)6 | F:tgctgctattgctgaccatc | 429–479 | 0.3684 | 6 | 6 | 0.7012 |
| R:ggaccatggaccaaagaaga | ||||||||
| 17 | TCL262 | (GA)7 | F:ccttgccgttccgtttatta | 361–374 | 0.2857 | 6 | 6 | 0.7452 |
| R:catttgccgtcaaaagaaca | ||||||||
| 18 | TCL279 | (CTG)5 | F:ttttccactcctccatcgac | 414–428 | 0.5000 | 5 | 5 | 0.6257 |
| R:actcgaaccgtggaaatgac | ||||||||
| 19 | TCL284 | (TTC)5 | F:cggtcggaaactcagctaac | 152–174 | 0.2727 | 6 | 6 | 0.7495 |
| R:cagtgggaggaattgagcat | ||||||||
| 20 | TCL285 | (GGA)6 | F:gcggctctcctatcactgac | 222–235 | 0.4000 | 5 | 5 | 0.6756 |
| R:cataacgtatccggcgatct | ||||||||
| 21 | TCL290 | (TGC)6 | F:gcctcgaccattctggtaaa | 329–340 | 0.5000 | 4 | 4 | 0.5862 |
| R:atgccccagatgaaacagag | ||||||||
| 22 | TO309 | (GGT)5 | F:ctcatgcacagaaggcacat | 186–218 | 0.6429 | 4 | 4 | 0.4503 |
| R:acgaggtgctcgatactgct | ||||||||
| Mean | 0.4440 | 5.6818 | 5.2727 | 0.6254 |
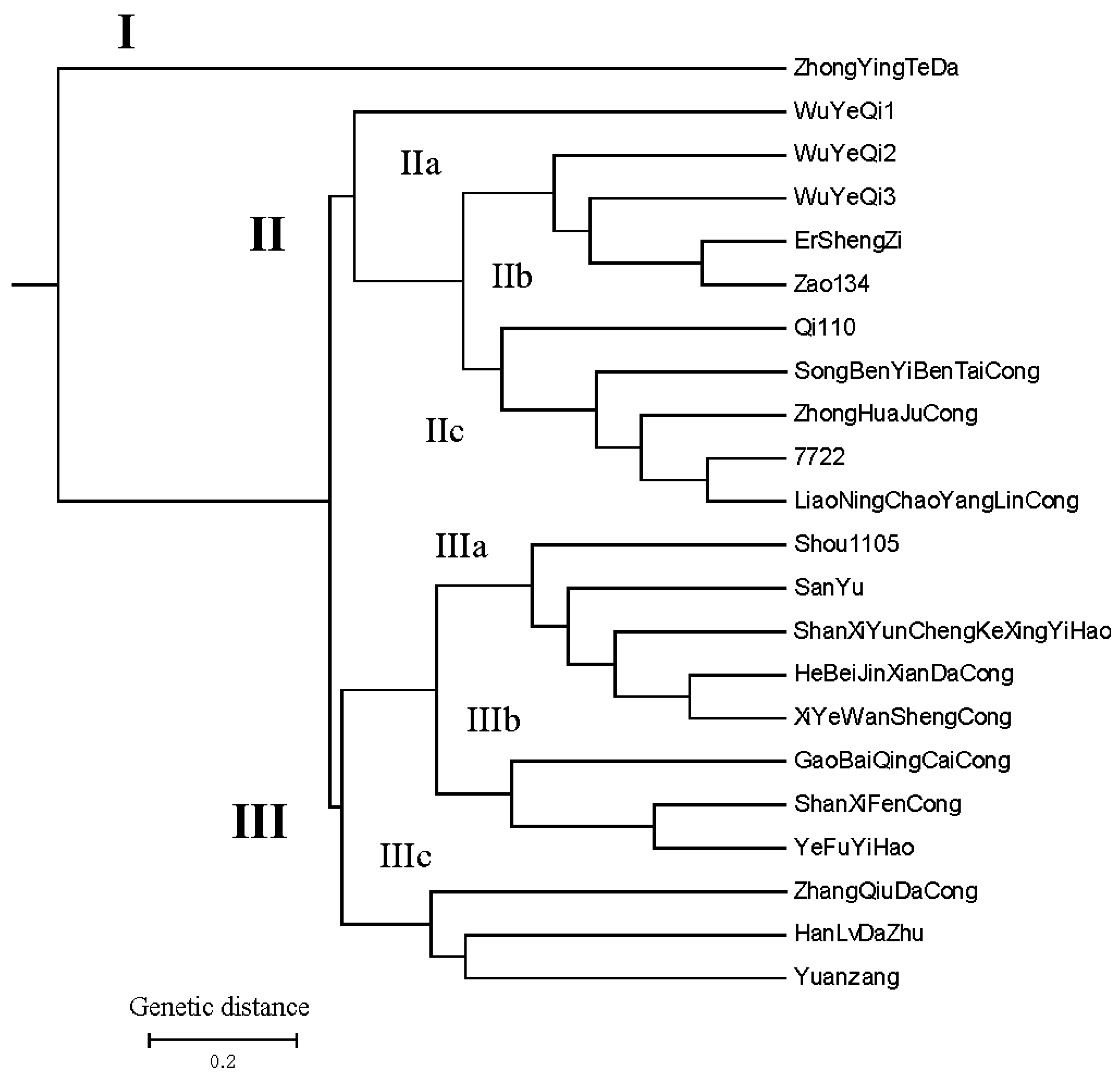
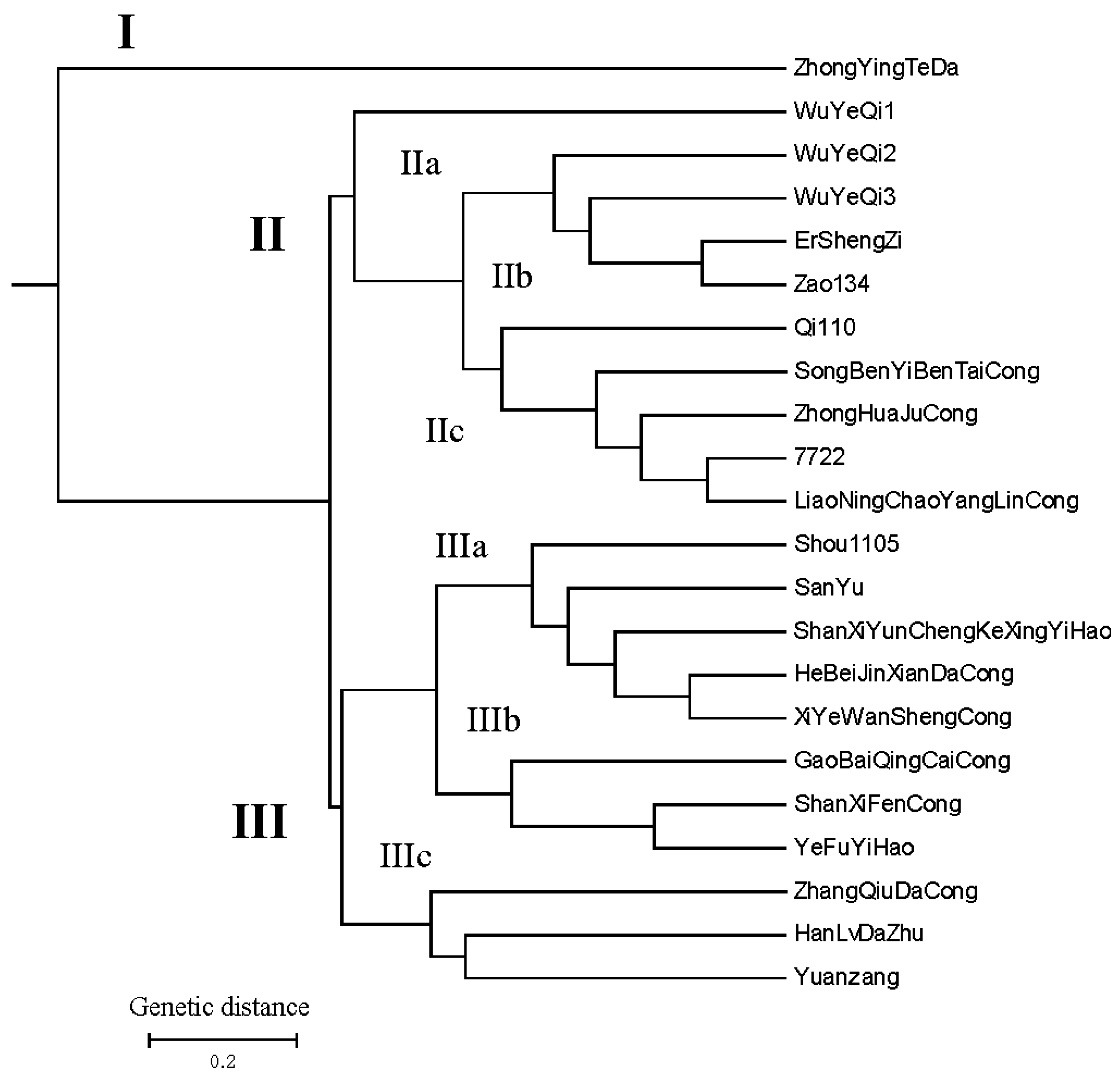
2.4. The Results of UPGMA Analysis Using 22 Highly Polymorphic Markers among 22 Allium Accessions
3. Experimental Section
3.1. Plant Materials and DNA Extraction
3.2. Identification of SSR Loci from the Welsh Onion Transcriptome and Primer Design
3.3. PCR Amplification and Screening Polymorphic Primers
3.4. Validation of Polymorphic Primers
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Takhtajan, A. Flowering Plants Origin and Dispersal; Oliver & Boyd: Edinburgh, UK, 1969; pp. 165–203. [Google Scholar]
- Ford-Lloyd, B.V.; Armstrong, S.J. Welsh onion (Allium wstulosum L.). In Genetic Improvement of Vegetable Crops; Kalloo, G., Bergh, B.O., Eds.; Pergamon Press: Oxford, UK, 1993; pp. 51–58. [Google Scholar]
- Kumazawa, S.; Katsumata, H. Negi (Japanese bunching onion). In Sosai-Engei Kakuron (Vegetable Crops); Kumazawa, S., Ed.; Yokendo Press: Tokyo, Japan, 1965; pp. 280–289. [Google Scholar]
- Jiangsu New Medical College. Dictionary of traditional Chinese Medicine. In Shanghai Science and Technology; Shanghai Press: Shanghai, China, 1986; pp. 2316–2317. [Google Scholar]
- Gai, S.P.; Meng, X.D.; Xu, L.J. Studies on molecular marker-assisted selection for sterile line and maintainer line in Welsh onion (Allium fistulosum L.). Mol. Plant Breed. 2004, 2, 223–228. [Google Scholar]
- Gai, S.P. Development and Identification of DNA Molecular Markers Linked to Cytoplasmic Fertility Loci and Restore Genes and Markers Assisted Selection in Welsh Onion. Ph.D. Thesis, Shandong Agricultural University, Shandong, China, May 2002. [Google Scholar]
- Gao, L.M. The Studies of Germ Plasm among Some Allium Lines and the Analysis of Main Characters. Master’s Thesis, Shandong Agricultural University, Shandong, China, 20 May 2005. [Google Scholar]
- Tsukazaki, H.; Yamashita, K.-I.; Yaguchi, S.; Masuzaki, S.; Fukuoka, H.; Yonemaru, J.; Kanamori, H.; Kono, I.; Hang, T.T.M.; Shigyo, M.; et al. Construction of SSR-based chromosome map in bunching onion (Allium fistulosum). Theor. Appl. Genet. 2008, 117, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.-S.; Suwabe, K.; Wako, T.; Ohara, T.; Nunome, T.; Kojima, A. Development of Microsatellite Markers in Bunching Onion (Allium fistulosum L.). Breed. Sci. 2004, 54, 361–365. [Google Scholar] [CrossRef]
- Tsukazaki, H.; Yaguchi, S.; Sato, S.; Hirakawa, H.; Katayose, Y.; Kanamori, H.; Kurita, K.; Itoh, T.; Kumagai, M.; Mizuno, S.; et al. Development of transcriptome shotgun assembly-derived markers in bunching onion (Allium fistulosum). Mol. Breed. 2015, 35, 55. [Google Scholar] [CrossRef]
- Tsukazaki, H.; Yamashita, K.-I.; Kojima, A.; Wako, T. SSR-tagged breeding scheme for allogamous crops: A trial in bunching onion (Allium fistulosum). Euphytica 2009, 169, 327–334. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Tóth, G.; Gáspári, Z.; Jurka, J. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 2000, 10, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Tsukazaki, H.; Fukuoka, H.; Song, Y.-S.; Yamashita, K.; Wako, T.; Kojima, A. Considerable heterogeneity in commercial F1 varieties of bunching onion (Allium wstulosum) and the proposal of a breeding scheme for conferring genetic traceability using SSR markers. Breed. Sci. 2006, 56, 321–326. [Google Scholar] [CrossRef]
- Tsukazaki, H.; Nunome, T.; Fukuoka, H.; Kanamori, H.; Kono, I.; Yamashita, K.; Wako, T.; Kojima, A. Isolation of 1796 SSR clones from SSR-enriched DNA libraries of bunching onion (Allium wstulosum). Euphytica 2007, 157, 83–94. [Google Scholar] [CrossRef]
- Gupta, S.K.; Bansal, R.; Gopalakrishna, T. Development and characterization of genic SSR markers for mungbean (Vigan radiate (L.) Wilczek). Euphytica 2014, 195, 245–258. [Google Scholar] [CrossRef]
- Kuhl, J.C.; Cheung, F.; Yuan, Q.; Martin, W.; Zewdie, Y.; MaCallum, J.; Catanach, A.; Rutherford, P.; Sink, K.C.; Jenderk, M.; et al. A unique set of 11,008 onion expressed sequence tags reveals expressed sequence and genomic differences between the monocot orders Asparagales and Poales. Plant Cell 2004, 16, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.J.; McCallum, J.; Shigyo, M. Genetic mapping of expressed sequences in onion and in silico comparisons with rice show scant colinearity. Mol. Genet. Genom. 2005, 274, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Duangjit, J.; Bohanec, B.; Chan, A.P.C.; Town, C.D.C.; Havey, M.J.D. Transcriptome sequencing to produce SNP-based genetic maps of onion. Theor. Appl. Genet. 2013, 127, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-J.; Wang, Y.-Y.; Xu, Y.-N.; Yan, R.-S.; Zhao, P.; Liu, W.-Z. De novo characterization of flower bud transcriptomes and the development of EST-SSR markers for the endangered tree Tapiscia sinensis. Int. J. Mol. Sci. 2015, 16, 12855–12870. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Luo, S.; Wang, R.; Zhong, Y.; Xu, X.; Lin, Y.E.; He, X.; Sun, B.; Huang, H. The first Illumina-based de novo transcriptome sequencing and analysis of pumpkin (Cucurbita moschata Duch.) and SSR marker development. Mol. Breed. 2014, 34, 1437–1447. [Google Scholar] [CrossRef]
- Chen, X.-B.; Xie, Y.-H.; Sun, X.-M. Development and Characterization of polymorphic Genic-SSR markers in Larix kaempferi. Molecules 2015, 20, 6060–6067. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Bachmann, K. Onion microsatellites for germplasm analysis and their use in assessing intra- and interspecific relatedness within the subgenus Rhizirideum. Theor. Appl. Genet. 2000, 101, 153–164. [Google Scholar] [CrossRef]
- Yao, L.H.; Zheng, X.Y.; Cai, D.Y.; Gao, Y.; Wang, K.; Cao, Y.F.; Teng, Y.W. Exploitation of Malus EST-SSRs and the utility in evaluation of genetic diversity in Malus and Pyrus. Genet. Resour. Crop. Evol. 2010, 57, 841–851. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Li, J.; Luo, Z.X.; Huang, L.F.; Chen, X.L.; Fang, B.P.; Li, Y.J.; Chen, J.Y.; Zhang, X.J. Characterization and development of EST-derived SSR markers in cultivated sweetpotato (Ipomoeabatatas). BMC Plant Biol. 2011, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.L.; Xu, L.; Wang, Y.; Cheng, H.; Chen, Y.L.; Gong, Y.Q.; Liu, L.W. Novel and useful genic-SSR markers from de novo transcriptome sequencing of radish (Raphanus sativus L.). Mol. Breed. 2014, 33, 611–642. [Google Scholar] [CrossRef]
- Jena, S.N.; Srivastava, A.; Rai, K.M.; Ranjan, A.; Singh, S.K.; Nisar, T.; Srivastava, M.; Bag, S.K.; Mantri, S.; Asif, M.H.; et al. Development and characterization of genomic and expressed SSRs for levant cotton (Gossypium herbaceum L.). Theor. Appl. Genet. 2012, 124, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.W.; Samuels, T.D.; Wu, Y.Q. Development of 1030 genomic SSR markers in switchgrass. Theor. Appl. Genet. 2011, 122, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Kumawat, G.; Singh, B.P.; Gupta, D.K.; Singh, S.; Dogra, V.; Gaikwad, K.; Sharma, T.R.; Raje, R.S.; Bandhopadhya, T.K.; et al. Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea (Cajanus cajan (L.) Millspaugh). BMC Plant Biol. 2011, 11, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.Y.; Wei, L.B.; Miao, H.M.; Zhang, T.; Wang, C.Y. Development and validation of genic-SSR markers in sesame by RNA-Seq. BMC Genomics 2012, 13, 316. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.I.; Martins, A.M.; Gouvea, E.G.; Pessoa-Filho, M.; Ferreira, M.E. Development and validation of microsatellite markers for Brachiaria ruziziensis obtained by partial genome assembly of Illumina single-end reads. BMC Genomics 2013, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Iorizzo, M.; Senalik, D.A.; Grzebelus, D.; Bowman, M.; Cavagnaro, P.F.; Matvienko, M.; Ashrafi, H.; Deynze, A.V.; Simon, P.W. De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genom. 2011, 12, 389. [Google Scholar] [CrossRef] [PubMed]
- Ricroch, A.; Yockteng, R.; Brown, S.C.; Nadot, S. Evolution of genome size across some cultivated Allium species. Genome 2005, 48, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Yadav, H.K.; Ranjan, A.; Asif, M.H.; Mantri, S.; Sawant, S.V.; Tuli, R. EST-derived SSR markers in Jatropha curcas L.: Development, characterization, polymorphism, and transferability across the species/genera. Tree Genet. Genomes 2011, 7, 207–219. [Google Scholar] [CrossRef]
- Yu, L.J.; Luo, Y.F.; Liao, B.; Xie, L.J.; Chen, L.; Xiao, S.; Li, J.T.; Hu, S.N.; Shu, W.S. Comparative transcriptome analysis of transporters, phytohormone and lipid metabolism pathways in response to arsenic stress in rice (Oryza sativa). New Phytol. 2012, 195, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.Y.; Liu, G.Q.; Zong, Y.; Teng, Y.W.; Cai, D.Y. Development of genic SSR markers from transcriptome sequence of pear buds. J. Zhejiang Univ. Sci. B 2014, 15, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Gopalakrishna, T. Development of unigene-derived SSR markers in cowpea (Vigna unguiculata) and their transferability to other Vigna species. Genome 2010, 53, 508–523. [Google Scholar] [PubMed]
- Barchi, L.; Lanteri, S.; Portis, E.; Acquadro, A.; Vale, G.; Toppino, L.; Rotino, G.L. Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genomics 2011, 12, 304. [Google Scholar] [CrossRef] [PubMed]
- Triwitayakorn, K.; Chatkulkawin, P.; Kanjanawattanawong, S.; Sraphet, S.; Yoocha, T.; Sangsrakru, D.; Chanprasert, J.; Ngamphiw, C.; Jomchai, N.; Therawattanasuk, K.; et al. Transcriptome sequencing of Hevea brasiliensis for development of microsatellite markers and construction of a genetic linkage map. DNA Res. 2011, 18, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Brandon, S.; Diego, F.; Tierney, B.; Eric, W.; James, P.; Nicholi, V.; Shawn, S.; Juan, Z. Development and validation of 697 novel polymorphic genomic and EST-SSR markers in the American Cranberry (Vaccinium macrocarpon Ait.). Molecules 2015, 20, 2001–2013. [Google Scholar]
- Zeng, S.H.; Xiao, G.; Guo, J.; Fei, Z.J.; Xu, Y.Q.; Roe, B.A.; Wang, Y. Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC Genomics 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.N.; Liang, S.; Duan, J.L.; Wang, J.; Chen, S.L.; Cheng, Z.S.; Zhang, Q.; Liang, X.Q.; Li, Y.R. De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in peanut (Arachis hypogaea L.). BMC Genomics 2012, 13, 90. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.C.; Wen, C.L.; Zhao, H.; Zhang, L.Y.; Wang, J.; Wang, Y.Q. RNA-Seq Reveals Leaf Cuticular Wax-Related Genes in Welsh Onion. PLoS ONE 2014, 9, e113290. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.Y.; Han, G.F.; Yan, Y.J.; Zhang, F.; Liu, Z.C.; Li, X.M.; Li, W.R.; Ma, Y.; Li, H.; Liu, Y.X.; et al. Transcript assembly and quantification by RNA-Seq reveals differentially expressed genes between Soft-Endocarp and Hard-Endocarp Hawthorns. PLoS ONE 2013, 8, e72910. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Huo, N.; Gu, Y.Q.; Luo, M.-C.; Ma, Y.; Hane, D.; Lazo, G.R.; Dvorak, J.; Anderson, O.D. BatchPrimer3: A high throughput web application for PCR and sequencing primer design. BMC Bioinformatics 2008, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Wen, C.; Zhao, H.; Liu, Q.; Yang, J.; Liu, L.; Wang, Y. Development of Polymorphic Genic SSR Markers by Transcriptome Sequencing in the Welsh Onion (Allium fistulosum L.). Appl. Sci. 2015, 5, 1050-1063. https://doi.org/10.3390/app5041050
Yang L, Wen C, Zhao H, Liu Q, Yang J, Liu L, Wang Y. Development of Polymorphic Genic SSR Markers by Transcriptome Sequencing in the Welsh Onion (Allium fistulosum L.). Applied Sciences. 2015; 5(4):1050-1063. https://doi.org/10.3390/app5041050
Chicago/Turabian StyleYang, Liuyi, Changlong Wen, Hong Zhao, Qianchun Liu, Jingjing Yang, Lecheng Liu, and Yongqin Wang. 2015. "Development of Polymorphic Genic SSR Markers by Transcriptome Sequencing in the Welsh Onion (Allium fistulosum L.)" Applied Sciences 5, no. 4: 1050-1063. https://doi.org/10.3390/app5041050