Response of Differentiated Human Airway Epithelia to Alcohol Exposure and Klebsiella pneumoniae Challenge

Abstract
:1. Introduction
2. Experimental
2.1. Cell Culture
2.2. Ethanol Exposure and Bacterial Challenge
2.3. Confocal Microscopy
2.4. Transmission Electron Microscopy
2.5. Transepithelial Permeability Assay
2.6. Bacterial Viability Assay
2.7. Bacterial Binding Assay
2.8. Cytokine Measurements
2.9. Statistical Analysis
3. Results
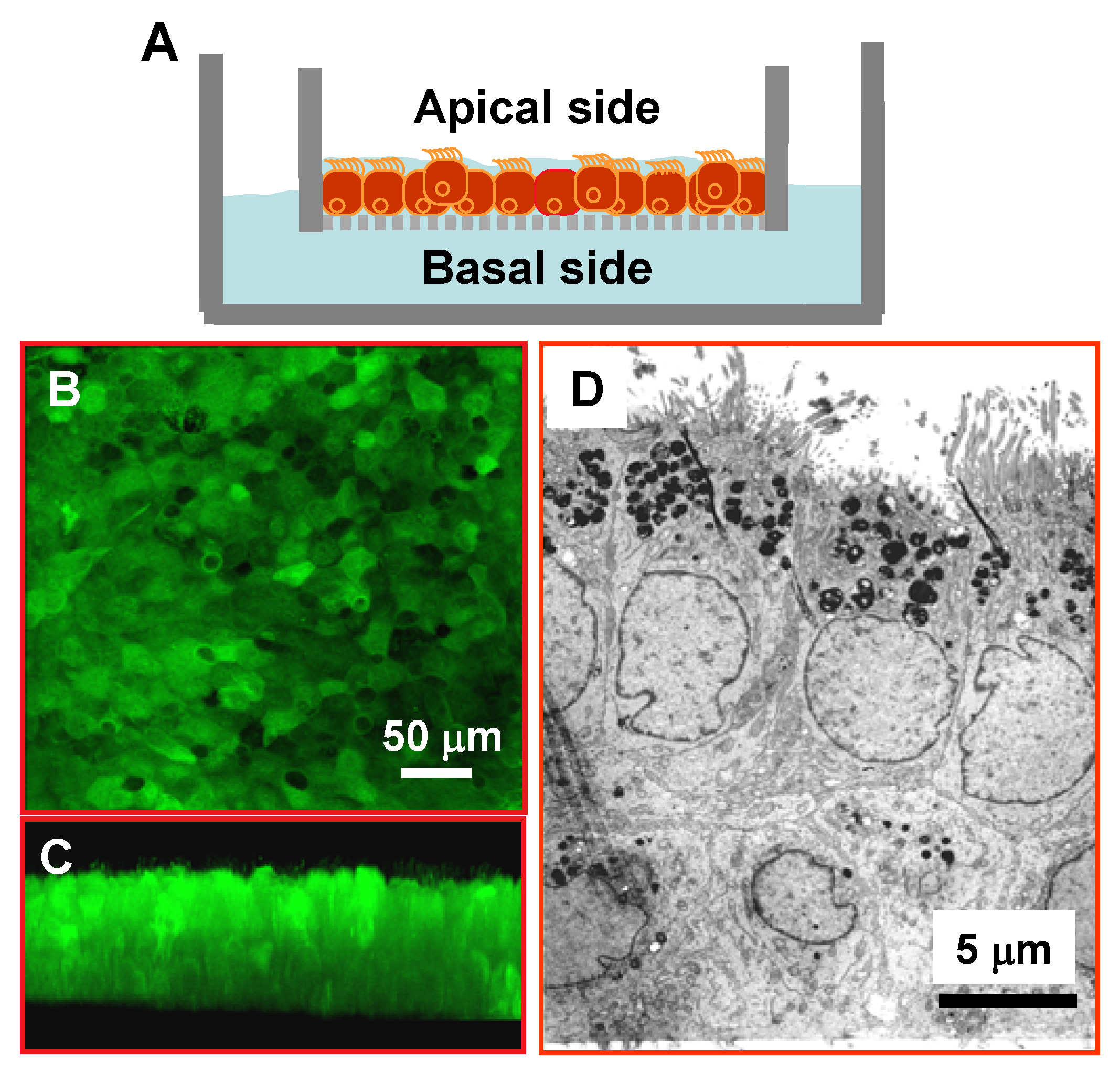
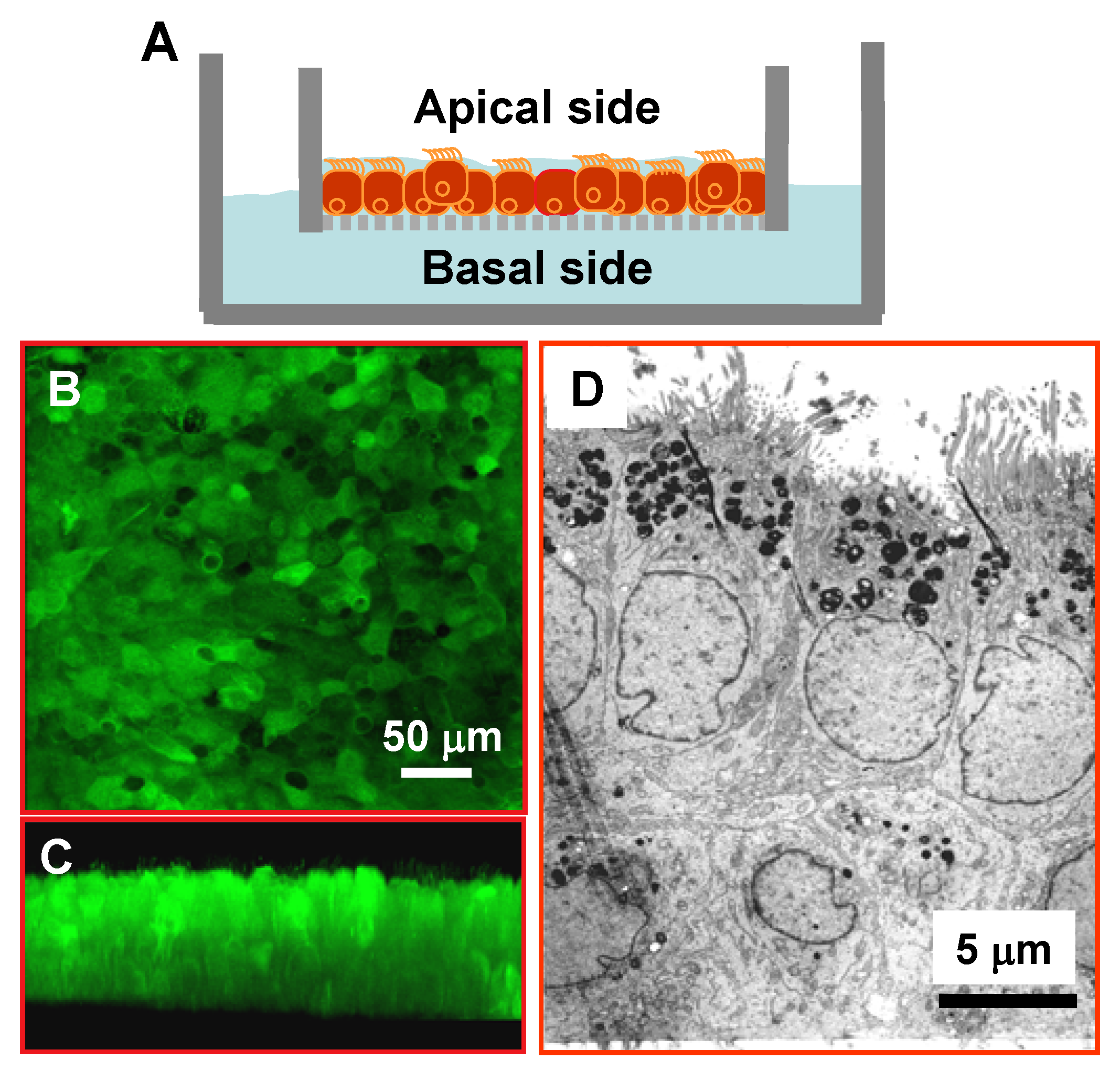
3.1. Differentiated and Polarized Human Airway Epithelia for Alcohol Studies
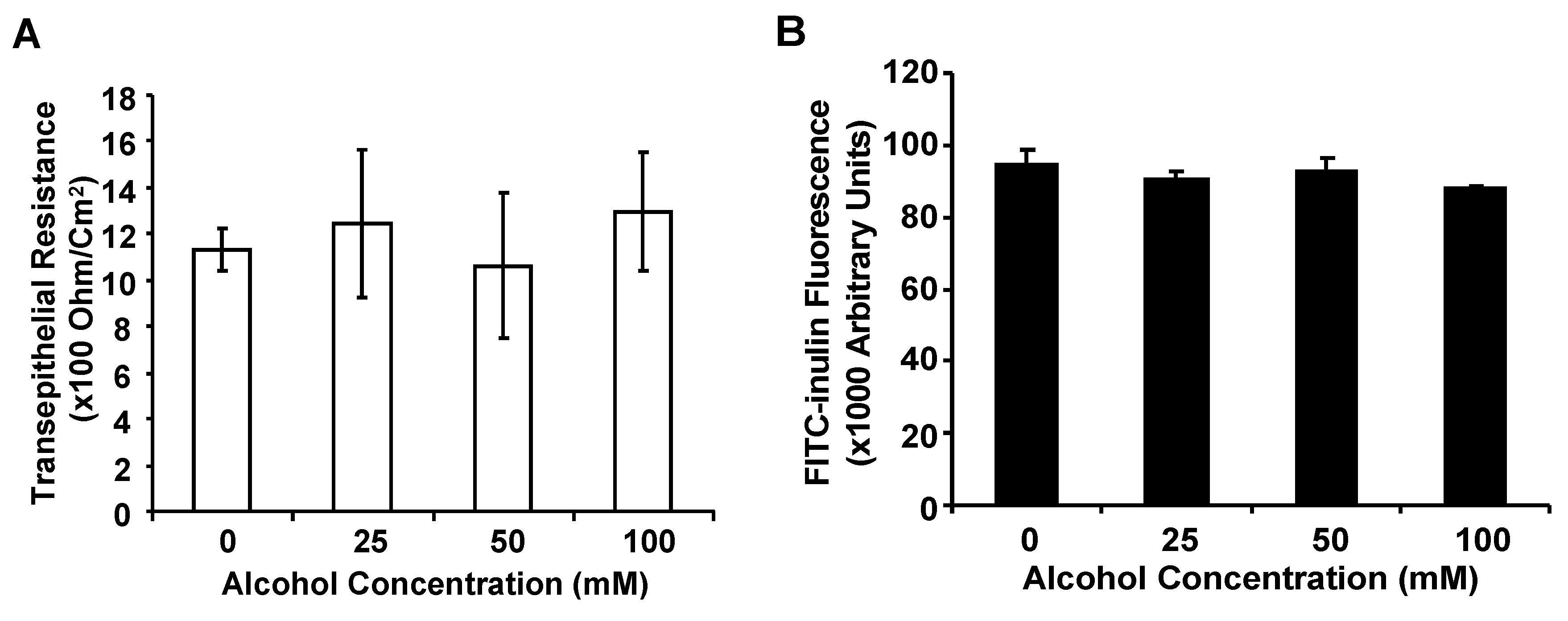
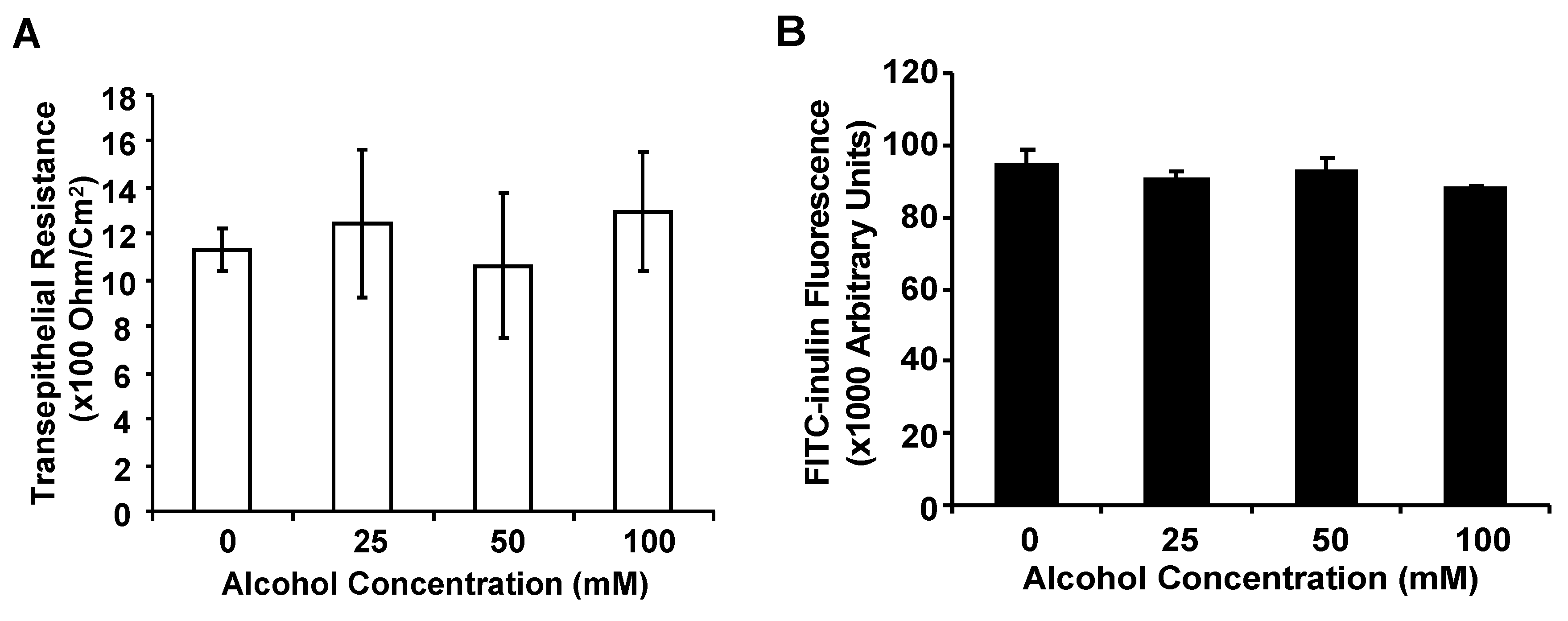
3.2. Alcohol at Clinically Relevant Concentrations Does Not Significantly Alter Airway Epithelial Barrier Function
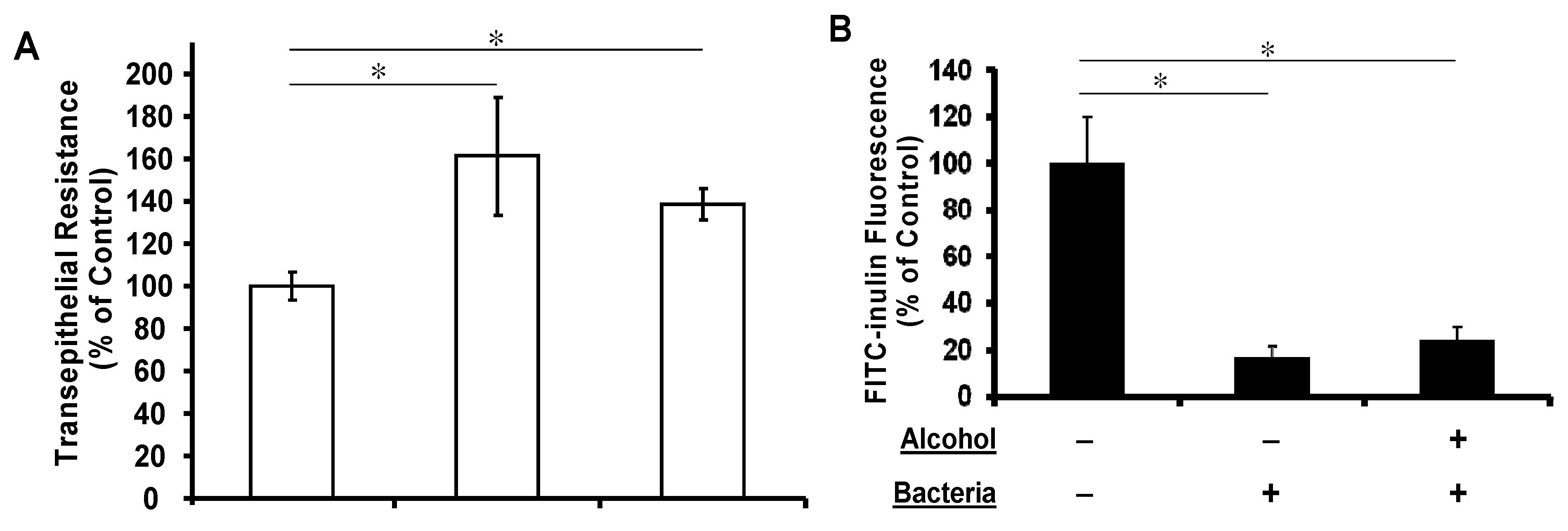
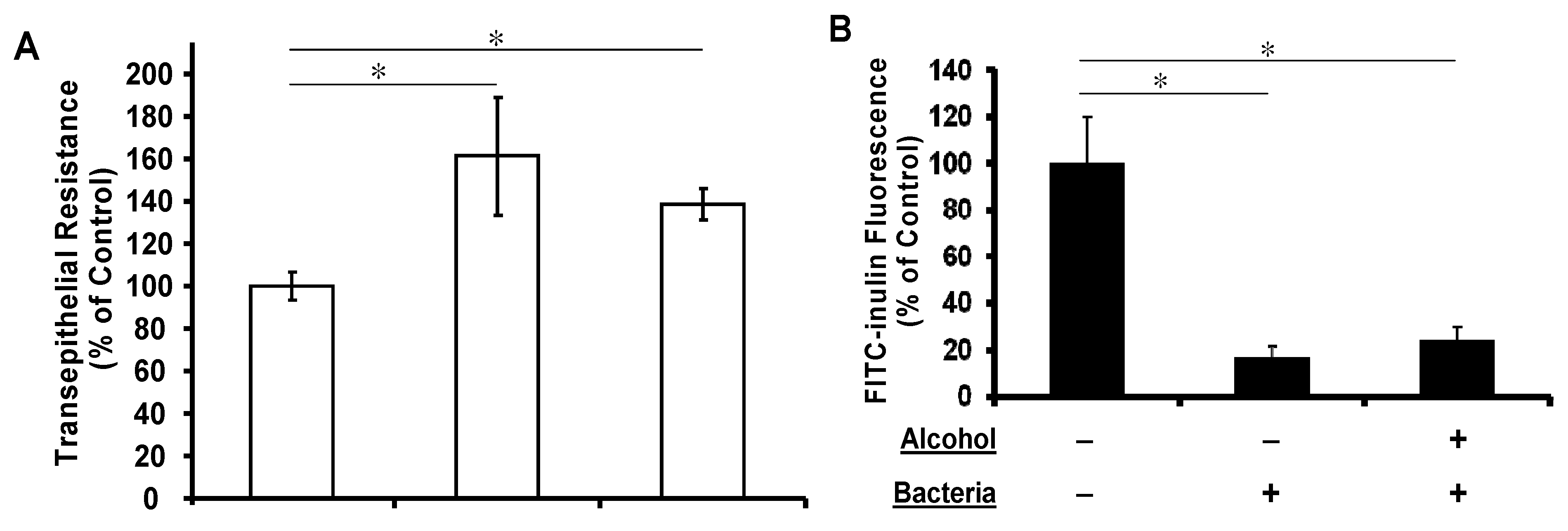
3.3. Apical Challenge with Live K. pneumoniae Enhances the Airway Epithelial Tightness Regardless of Basolateral Alcohol Presence
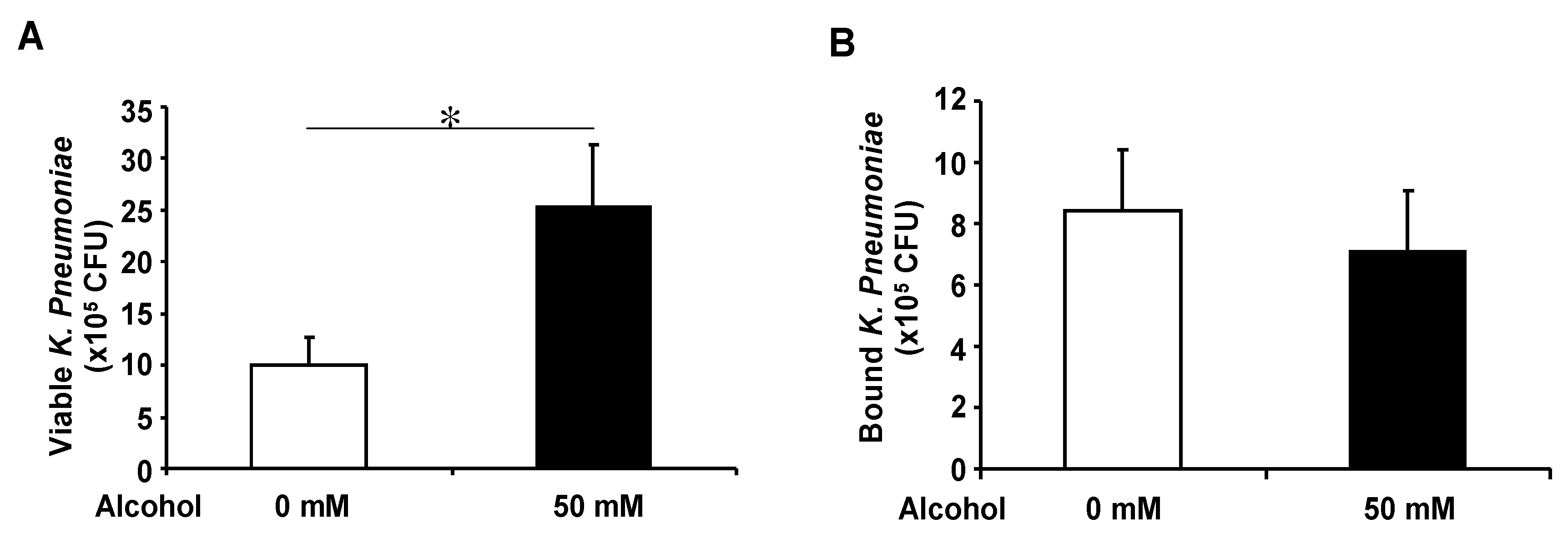
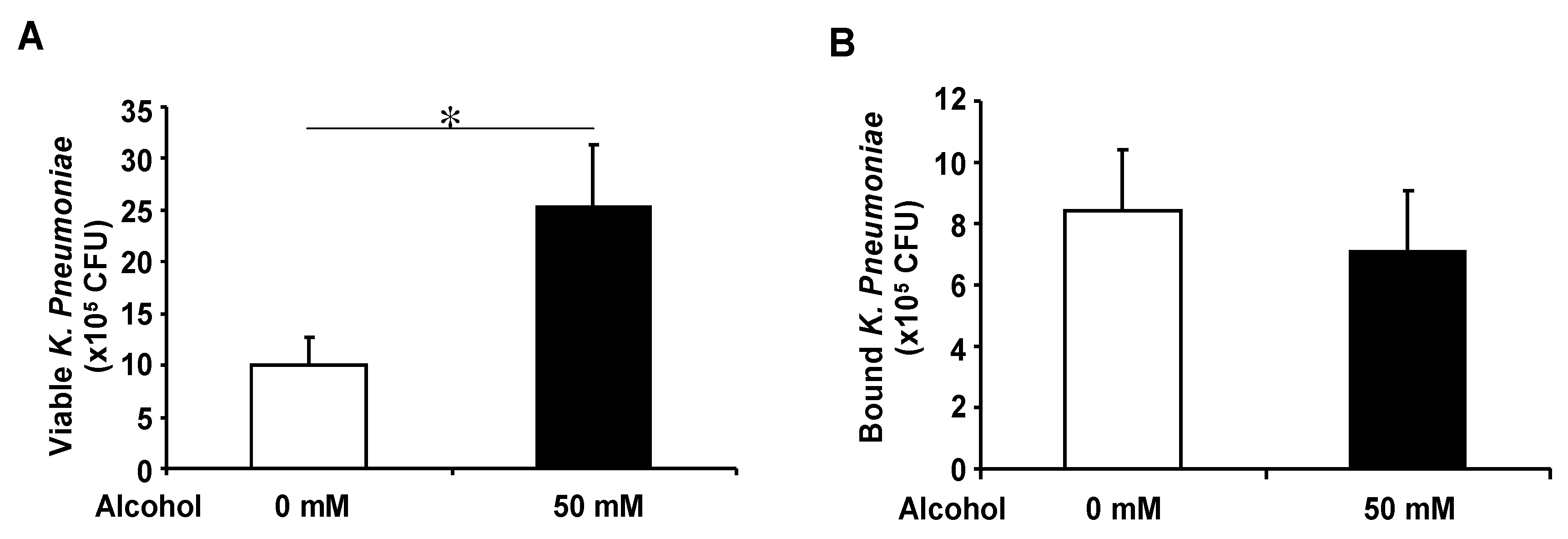
3.4. Basolateral Alcohol Exposure Facilitates Apical Bacterial Growth but Does Not Affect Apical Bacterial Binding
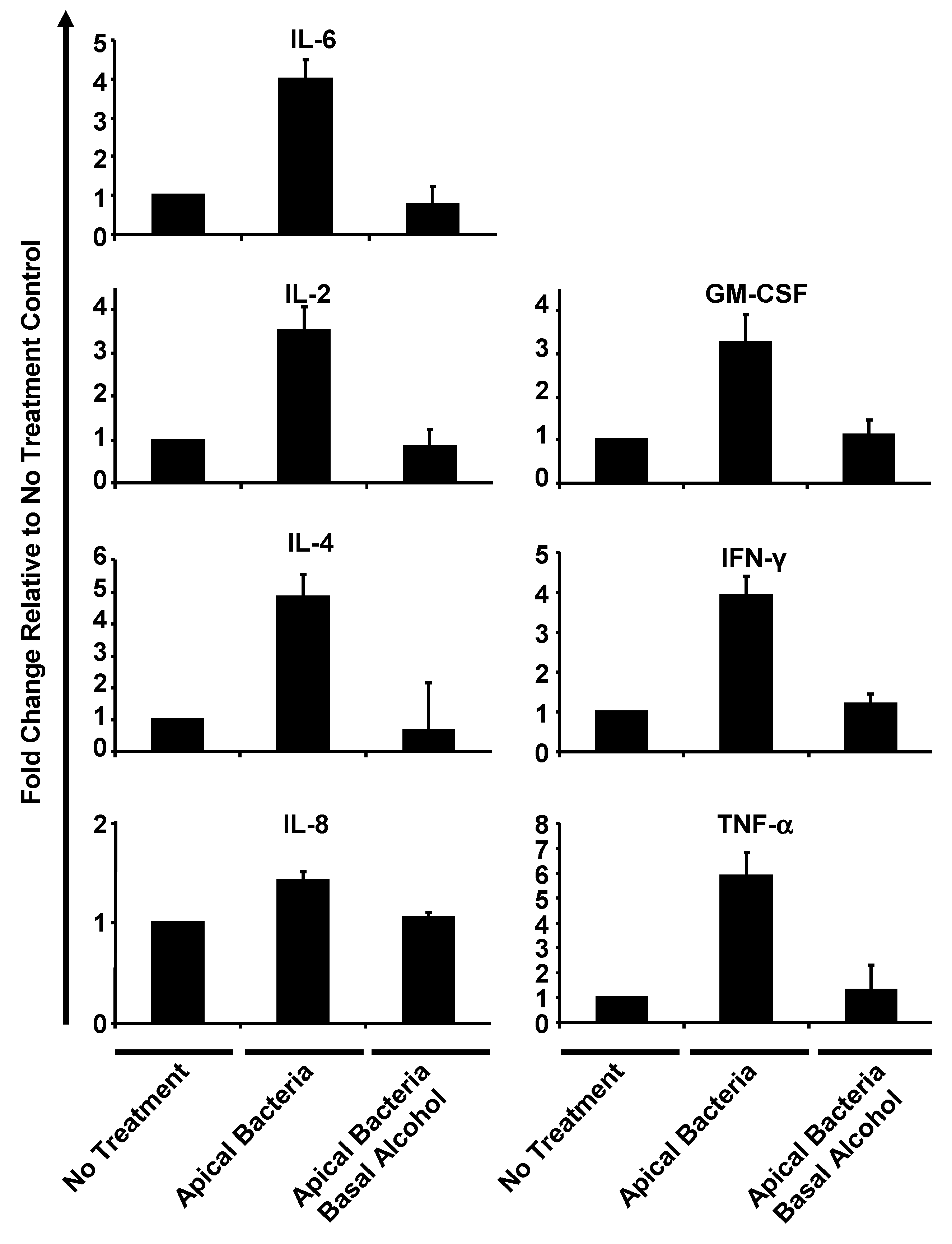
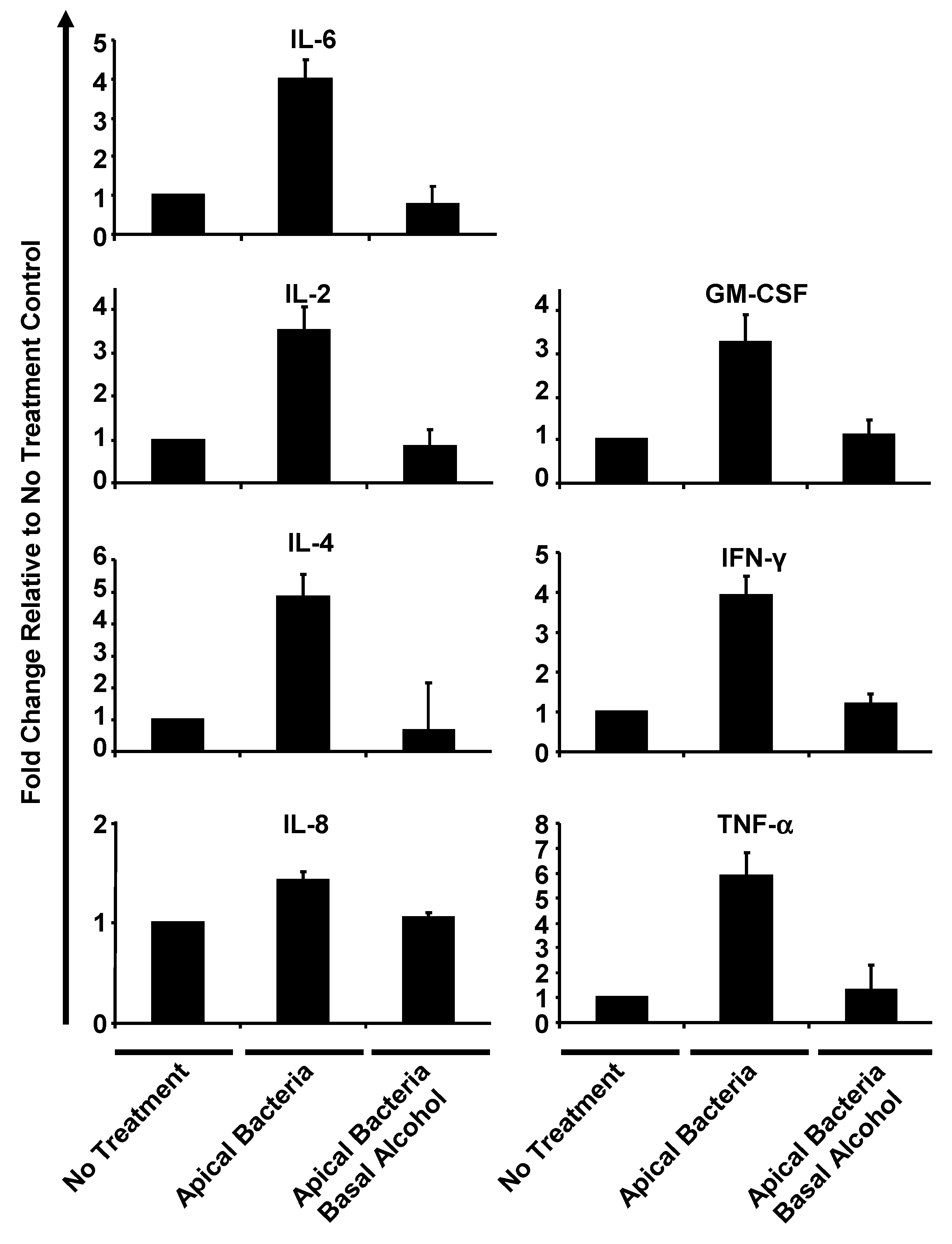
3.5. Alcohol Suppresses Human Airway Epithelial Cytokine Release under Apical Bacterial Challenge

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | Concentration in Basal Media (pg/mL) |
|---|---|
| IL-2 | 7.66 ± 1.52 |
| TNF-α | 9.33 ± 2.51 |
| IL-4 | 5.66 ± 1.51 |
| IL-6 | 767.66 ± 294.89 |
| IL-8 | 12,905.17 ± 1,915.45 |
| GM-CSF | 14.33 ± 2.30 |
| IFN-γ | 11.00 ± 2.64 |
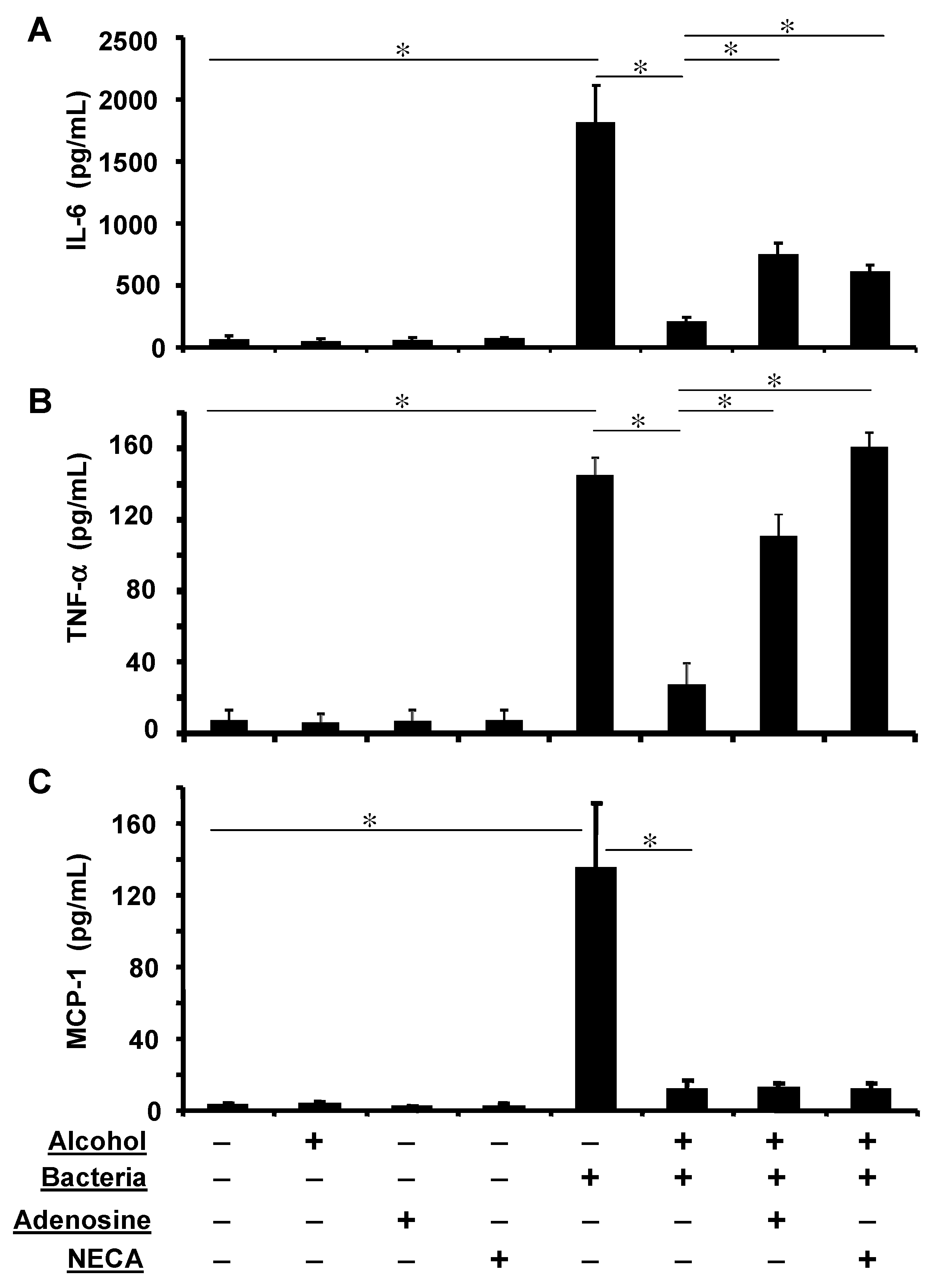
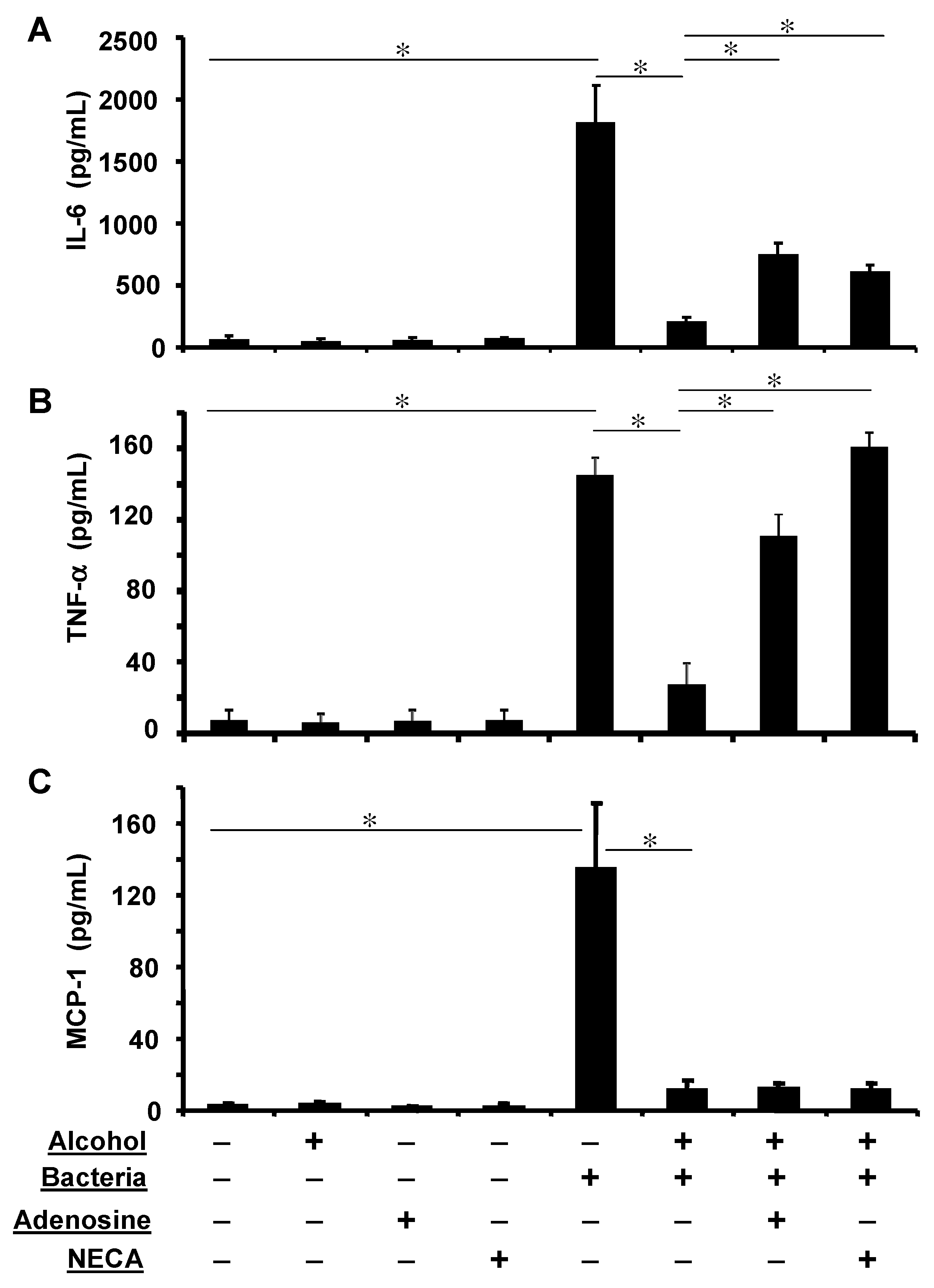
3.6. Adenosine Receptor Activation Partially Reverses Alcohol Suppressed Inflammatory Cytokine Release
4. Discussion
Acknowledgements
Conflict of Interest
References
- Nelson, S.; Kolls, J.K. Alcohol, host defence and society. Nat. Rev. Immunol. 2002, 2, 205–209. [Google Scholar] [CrossRef]
- Sisson, J.H. Alcohol and airways function in health and disease. Alcohol 2007, 41, 293–307. [Google Scholar] [CrossRef]
- Zhang, P.; Bagby, G.J.; Happel, K.I.; Raasch, C.E.; Nelson, S. Alcohol abuse, immunosuppression, and pulmonary infection. Curr. Drug Abuse Rev. 2008, 1, 56–67. [Google Scholar] [CrossRef]
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef]
- Voynow, J.A.; Rubin, B.K. Mucins, mucus, and sputum. Chest 2009, 135, 505–512. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Bennett, W.D.; Zeman, K.L.; Knowles, M.R.; Tarran, R.; Boucher, R.C. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N. Engl. J. Med. 2006, 354, 241–250. [Google Scholar] [CrossRef]
- Schutte, B.C.; McCray, P.B., Jr. [beta]-Defensins in lung host defense. Annu. Rev. Physiol. 2002, 64, 709–748. [Google Scholar] [CrossRef]
- Bals, R.; Hiemstra, P.S. Innate immunity in the lung: How epithelial cells fight against respiratory pathogens. Eur. Respir. J. 2004, 23, 327–333. [Google Scholar] [CrossRef]
- Gross, C.A.; Bowler, R.P.; Green, R.M.; Weinberger, A.R.; Schnell, C.; Chu, H.W. beta2-agonists promote host defense against bacterial infection in primary human bronchial epithelial cells. BMC Pulm. Med. 2010, 10, e30. [Google Scholar] [CrossRef]
- Liu, Y.; Bartlett, J.A.; Di, M.E.; Bomberger, J.M.; Chan, Y.R.; Gakhar, L.; Mallampalli, R.K.; McCray, P.B., Jr.; Di, Y.P. SPLUNC1/BPIFA1 contributes to pulmonary host defense against Klebsiella pneumoniae respiratory infection. Am. J. Pathol. 2013, 182, 1519–1531. [Google Scholar] [CrossRef]
- Sibille, Y.; Reynolds, H.Y. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am. Rev. Respir. Dis. 1990, 141, 471–501. [Google Scholar] [CrossRef]
- Nelson, S.; Summer, W.R. Innate immunity, cytokines, and pulmonary host defense. Infect. Dis. Clin. North Am. 1998, 12, 555–567. [Google Scholar] [CrossRef]
- Reynolds, H.Y. Modulating airway defenses against microbes. Curr. Opin. Pulm. Med. 2002, 8, 154–165. [Google Scholar] [CrossRef]
- Burns, A.R.; Smith, C.W.; Walker, D.C. Unique structural features that influence neutrophil emigration into the lung. Physiol. Rev. 2003, 83, 309–336. [Google Scholar]
- Evans, S.E.; Xu, Y.; Tuvim, M.J.; Dickey, B.F. Inducible innate resistance of lung epithelium to infection. Annu. Rev. Physiol. 2010, 72, 413–435. [Google Scholar] [CrossRef]
- Polito, A.J.; Proud, D. Epithelia cells as regulators of airway inflammation. J. Allergy Clin. Immunol. 1998, 102, 714–718. [Google Scholar] [CrossRef]
- Crews, F.T.; Bechara, R.; Brown, L.A.; Guidot, D.M.; Mandrekar, P.; Oak, S.; Qin, L.; Szabo, G.; Wheeler, M.; Zou, J. Cytokines and alcohol. Alcohol. Clin. Exp. Res. 2006, 30, 720–730. [Google Scholar] [CrossRef]
- Twigg, H.L., 3rd. Pulmonary host defenses. J. Thorac. Imaging 1998, 13, 221–233. [Google Scholar] [CrossRef]
- Wyatt, T.A.; Sisson, J.H. Chronic ethanol downregulates PKA activation and ciliary beating in bovine bronchial epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L575–L581. [Google Scholar]
- Wyatt, T.A.; Gentry-Nielsen, M.J.; Pavlik, J.A.; Sisson, J.H. Desensitization of PKA-stimulated ciliary beat frequency in an ethanol-fed rat model of cigarette smoke exposure. Alcohol. Clin. Exp. Res. 2004, 28, 998–1004. [Google Scholar] [CrossRef]
- Elliott, M.K.; Sisson, J.H.; Wyatt, T.A. Effects of cigarette smoke and alcohol on ciliated tracheal epithelium and inflammatory cell recruitment. Am. J. Respir. Cell Mol. Biol. 2007, 36, 452–459. [Google Scholar] [CrossRef]
- Boe, D.M.; Nelson, S.; Zhang, P.; Quinton, L.; Bagby, G.J. Alcohol-induced suppression of lung chemokine production and the host defense response to Streptococcus pneumoniae. Alcohol. Clin. Exp. Res. 2003, 27, 1838–1845. [Google Scholar] [CrossRef]
- NIAAA, The 10th Special Report to the US Congress on Alcohol and Health; National Institutes of Health: Bethesda, MD, USA, 2000.
- Schmidt, W.; de Lint, J. Causes of death of alcoholics. Q. J. Stud. Alcohol 1972, 33, 171–185. [Google Scholar]
- Martin, L.D.; Rochelle, L.G.; Fischer, B.M.; Krunkosky, T.M.; Adler, K.B. Airway epithelium as an effector of inflammation: Molecular regulation of secondary mediators. Eur. Respir. J. 1997, 10, 2139–2146. [Google Scholar] [CrossRef]
- Boe, D.M.; Nelson, S.; Zhang, P.; Bagby, G.J. Acute ethanol intoxication suppresses lung chemokine production following infection with Streptococcus pneumoniae. J. Infect. Dis. 2001, 184, 1134–1142. [Google Scholar] [CrossRef]
- Happel, K.I.; Rudner, X.; Quinton, L.J.; Movassaghi, J.L.; Clark, C.; Odden, A.R.; Zhang, P.; Bagby, G.J.; Nelson, S.; Shellito, J.E. Acute alcohol intoxication suppresses the pulmonary ELR-negative CXC chemokine response to lipopolysaccharide. Alcohol 2007, 41, 325–333. [Google Scholar] [CrossRef]
- Zhang, P.; Bagby, G.J.; Stoltz, D.A.; Summer, W.R.; Nelson, S. Granulocyte colony-stimulating factor modulates the pulmonary host response to endotoxin in the absence and presence of acute ethanol intoxication. J. Infect. Dis. 1999, 179, 1441–1448. [Google Scholar] [CrossRef]
- Jerrells, T.R.; Pavlik, J.A.; DeVasure, J.; Vidlak, D.; Costello, A.; Strachota, J.M.; Wyatt, T.A. Association of chronic alcohol consumption and increased susceptibility to and pathogenic effects of pulmonary infection with respiratory syncytial virus in mice. Alcohol 2007, 41, 357–369. [Google Scholar] [CrossRef]
- Pruett, S.B.; Schwab, C.; Zheng, Q.; Fan, R. Suppression of innate immunity by acute ethanol administration: A global perspective and a new mechanism beginning with inhibition of signaling through TLR3. J. Immunol. 2004, 173, 2715–2724. [Google Scholar]
- Pruett, S.B.; Zheng, Q.; Fan, R.; Matthews, K.; Schwab, C. Acute exposure to ethanol affects Toll-like receptor signaling and subsequent responses: An overview of recent studies. Alcohol 2004, 33, 235–239. [Google Scholar] [CrossRef]
- Pruett, S.B.; Zheng, Q.; Fan, R.; Matthews, K.; Schwab, C. Ethanol suppresses cytokine responses induced through Toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol 2004, 33, 147–155. [Google Scholar]
- Pruett, B.S.; Pruett, S.B. An explanation for the paradoxical induction and suppression of an acute phase response by ethanol. Alcohol 2006, 39, 105–110. [Google Scholar] [CrossRef]
- Bailey, K.L.; Wyatt, T.A.; Romberger, D.J.; Sisson, J.H. Alcohol functionally upregulates Toll-like receptor 2 in airway epithelial cells. Alcohol. Clin. Exp. Res. 2009, 33, 499–504. [Google Scholar] [CrossRef]
- Bhatty, M.; Jan, B.L.; Tan, W.; Pruett, S.B.; Nanduri, B. Role of acute ethanol exposure and TLR4 in early events of sepsis in a mouse model. Alcohol 2011, 45, 795–803. [Google Scholar] [CrossRef]
- Karp, P.H.; Moninger, T.O.; Weber, S.P.; Nesselhauf, T.S.; Launspach, J.L.; Zabner, J.; Welsh, M.J. An in vitro model of differentiated human airway epithelia. Methods Mol. Biol. 2002, 188, 115–137. [Google Scholar]
- Gomez, M.; Raju, S.V.; Viswanathan, A.; Painter, R.G.; Bonvillain, R.; Byrne, P.; Nguyen, D.H.; Bagby, G.J.; Kolls, J.K.; Nelson, S.; et al. Ethanol upregulates glucocorticoid-induced leucine zipper expression and modulates cellular inflammatory responses in lung epithelial cells. J. Immunol. 2010, 184, 5715–5722. [Google Scholar] [CrossRef]
- Wang, G.; Davidson, B.L.; Melchert, P.; Slepushkin, V.A.; van Es, H.H.; Bodner, M.; Jolly, D.J.; McCray, P.B., Jr. Influence of cell polarity on retrovirus-mediated gene transfer to differentiated human airway epithelia. J. Virol. 1998, 72, 9818–9826. [Google Scholar]
- Wang, G.; Zabner, J.; Deering, C.; Launspach, J.; Shao, J.; Bodner, M.; Jolly, D.J.; Davidson, B.L.; McCray, P.B., Jr. Increasing epithelial junction permeability enhances gene transfer to airway epithelia in vivo. Am. J. Respir. Cell Mol. Biol. 2000, 22, 129–138. [Google Scholar] [CrossRef]
- Simet, S.M.; Wyatt, T.A.; DeVasure, J.; Yanov, D.; Allen-Gipson, D.; Sisson, J.H. Alcohol increases the permeability of airway epithelial tight junctions in Beas-2B and NHBE cells. Alcohol. Clin. Exp. Res. 2012, 36, 432–442. [Google Scholar] [CrossRef]
- Teplin, L.A.; Abram, K.M.; Michaels, S.K. Blood alcohol level among emergency room patients: A multivariate analysis. J. Stud. Alcohol 1989, 50, 441–447. [Google Scholar]
- Spicuzza, L.; di Maria, G.; Polosa, R. Adenosine in the airways: Implications and applications. Eur. J. Pharmacol. 2006, 533, 77–88. [Google Scholar] [CrossRef]
- Button, B.; Boucher, R.C. Role of mechanical stress in regulating airway surface hydration and mucus clearance rates. Respir. Physiol. Neurobiol. 2008, 163, 189–201. [Google Scholar] [CrossRef]
- Valls, M.D.; Cronstein, B.N.; Montesinos, M.C. Adenosine receptor agonists for promotion of dermal wound healing. Biochem. Pharmacol. 2009, 77, 1117–1124. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, F.; Sun, F.; Huang, P. Adenosine promotes IL-6 release in airway epithelia. J. Immunol. 2008, 180, 4173–4181. [Google Scholar]
- Sitaraman, S.V.; Merlin, D.; Wang, L.; Wong, M.; Gewirtz, A.T.; Si-Tahar, M.; Madara, J.L. Neutrophil-epithelial crosstalk at the intestinal lumenal surface mediated by reciprocal secretion of adenosine and IL-6. J. Clin. Invest. 2001, 107, 861–869. [Google Scholar] [CrossRef]
- Sun, C.X.; Zhong, H.; Mohsenin, A.; Morschl, E.; Chunn, J.L.; Molina, J.G.; Belardinelli, L.; Zeng, D.; Blackburn, M.R. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J. Clin. Invest. 2006, 116, 2173–2182. [Google Scholar] [CrossRef]
- Zhong, H.; Belardinelli, L.; Maa, T.; Feoktistov, I.; Biaggioni, I.; Zeng, D. A(2B) adenosine receptors increase cytokine release by bronchial smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 118–125. [Google Scholar] [CrossRef]
- Goral, J.; Karavitis, J.; Kovacs, E.J. Exposure-dependent effects of ethanol on the innate immune system. Alcohol 2008, 42, 237–247. [Google Scholar] [CrossRef]
- Adler, K.B.; Cheng, P.W.; Kim, K.C. Characterization of guinea pig tracheal epithelial cells maintained in biphasic organotypic culture: Cellular composition and biochemical analysis of released glycoconjugates. Am. J. Respir. Cell Mol. Biol. 1990, 2, 145–154. [Google Scholar] [CrossRef]
- Adler, K.B.; Fischer, B.M.; Wright, D.T.; Cohn, L.A.; Becker, S. Interactions between respiratory epithelial cells and cytokines: Relationships to lung inflammation. Ann. NY Acad. Sci. 1994, 725, 128–145. [Google Scholar]
- Adler, K.B.; Li, Y. Airway epithelium and mucus: Intracellular signaling pathways for gene expression and secretion. Am. J. Respir. Cell Mol. Biol. 2001, 25, 397–400. [Google Scholar] [CrossRef]
- Mandrekar, P.; Bala, S.; Catalano, D.; Kodys, K.; Szabo, G. The opposite effects of acute and chronic alcohol on lipopolysaccharide-induced inflammation are linked to IRAK-M in human monocytes. J. Immunol. 2009, 183, 1320–1327. [Google Scholar] [CrossRef]
- Brown, L.A.; Cook, R.T.; Jerrells, T.R.; Kolls, J.K.; Nagy, L.E.; Szabo, G.; Wands, J.R.; Kovacs, E.J. Acute and chronic alcohol abuse modulate immunity. Alcohol. Clin. Exp. Res. 2006, 30, 1624–1631. [Google Scholar] [CrossRef]
- Happel, K.I.; Nelson, S. Alcohol, immunosuppression, and the lung. Proc. Am. Thorac. Soc. 2005, 2, 428–432. [Google Scholar] [CrossRef]
- Pruett, S.B.; Fan, R. Ethanol inhibits LPS-induced signaling and modulates cytokine production in peritoneal macrophages in vivo in a model for binge drinking. BMC Immunol. 2009, 10, e49. [Google Scholar] [CrossRef]
- Mandrekar, P.; Jeliazkova, V.; Catalano, D.; Szabo, G. Acute alcohol exposure exerts anti-inflammatory effects by inhibiting IkappaB kinase activity and p65 phosphorylation in human monocytes. J. Immunol. 2007, 178, 7686–7693. [Google Scholar]
- Burnham, E.L.; Kovacs, E.J.; Davis, C.S. Pulmonary cytokine composition differs in the setting of alcohol use disorders and cigarette smoking. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L873–L882. [Google Scholar] [CrossRef]
- Ballinger, M.N.; Paine, R., 3rd; Serezani, C.H.; Aronoff, D.M.; Choi, E.S.; Standiford, T.J.; Toews, G.B.; Moore, B.B. Role of granulocyte macrophage colony-stimulating factor during gram-negative lung infection with Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 2006, 34, 766–774. [Google Scholar]
- Quinton, L.J.; Mizgerd, J.P. NF-kappaB and STAT3 signaling hubs for lung innate immunity. Cell Tissue Res. 2011, 343, 153–165. [Google Scholar] [CrossRef]
- Fredholm, B.B.; AP, I.J.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar]
- Feoktistov, I.; Biaggioni, I. Adenosine A2b receptors evoke interleukin-8 secretion in human mast cells. J. Clin. Invest. 1995, 96, 1979–1986. [Google Scholar] [CrossRef]
- Link, A.A.; Kino, T.; Worth, J.A.; McGuire, J.L.; Crane, M.L.; Chrousos, G.P.; Wilder, R.L.; Elenkov, I.J. Ligand-activation of the adenosine A2a receptors inhibits IL-12 production by human monocytes. J. Immunol. 2000, 164, 436–442. [Google Scholar]
- Zaynagetdinov, R.; Ryzhov, S.; Goldstein, A.E.; Yin, H.; Novitskiy, S.V.; Goleniewska, K.; Polosukhin, V.V.; Newcomb, D.C.; Mitchell, D.; Morschl, E.; et al. Attenuation of chronic pulmonary inflammation in A2B adenosine receptor knockout mice. Am. J. Respir. Cell Mol. Biol. 2010, 42, 564–571. [Google Scholar] [CrossRef]
- Driver, A.G.; Kukoly, C.A.; Ali, S.; Mustafa, S.J. Adenosine in bronchoalveolar lavage fluid in asthma. Am. Rev. Respir. Dis. 1993, 148, 91–97. [Google Scholar] [CrossRef]
- Huszar, E.; Vass, G.; Vizi, E.; Csoma, Z.; Barat, E.; Molnar Vilagos, G.; Herjavecz, I.; Horvath, I. Adenosine in exhaled breath condensate in healthy volunteers and in patients with asthma. Eur. Respir. J. 2002, 20, 1393–1398. [Google Scholar] [CrossRef]
- Blackburn, M.R.; Lee, C.G.; Young, H.W.; Zhu, Z.; Chunn, J.L.; Kang, M.J.; Banerjee, S.K.; Elias, J.A. Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J. Clin. Invest. 2003, 112, 332–344. [Google Scholar]
- Ma, B.; Blackburn, M.R.; Lee, C.G.; Homer, R.J.; Liu, W.; Flavell, R.A.; Boyden, L.; Lifton, R.P.; Sun, C.X.; Young, H.W.; et al. Adenosine metabolism and murine strain-specific IL-4-induced inflammation, emphysema, and fibrosis. J. Clin. Invest. 2006, 116, 1274–1283. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Raju, S.V.; Painter, R.G.; Bagby, G.J.; Nelson, S.; Wang, G. Response of Differentiated Human Airway Epithelia to Alcohol Exposure and Klebsiella pneumoniae Challenge. Med. Sci. 2013, 1, 2-19. https://doi.org/10.3390/medsci1010002
Raju SV, Painter RG, Bagby GJ, Nelson S, Wang G. Response of Differentiated Human Airway Epithelia to Alcohol Exposure and Klebsiella pneumoniae Challenge. Medical Sciences. 2013; 1(1):2-19. https://doi.org/10.3390/medsci1010002
Chicago/Turabian StyleRaju, Sammeta V., Richard G. Painter, Gregory J. Bagby, Steve Nelson, and Guoshun Wang. 2013. "Response of Differentiated Human Airway Epithelia to Alcohol Exposure and Klebsiella pneumoniae Challenge" Medical Sciences 1, no. 1: 2-19. https://doi.org/10.3390/medsci1010002