Acid Neutralization by Mining Waste Dissolution under Conditions Relevant for Agricultural Applications


1
Department of Environmental Analysis, Institute for Energy Technology, Instituttveien 18, 2007 Kjeller, Norway
2
Department of Earth Science, University of Bergen, Postboks 7803, 5020 Bergen, Norway
*
Author to whom correspondence should be addressed.
Geosciences 2018, 8(10), 380; https://doi.org/10.3390/geosciences8100380
Submission received: 20 September 2018
/
Revised: 10 October 2018
/
Accepted: 10 October 2018
/
Published: 16 October 2018
(This article belongs to the Section Geochemistry)
Abstract
:The acidification of agricultural soils in high rainfall regions is usually countered by the application of finely ground calcite or dolomite. As this carbonate dissolves, soil pH is raised, but CO2 is released. Mining activities often produce large quantities of very fine silicate rock-derived powders that are commonly deposited in stockpiles. However, the dissolution of such powders can also result in an increase in pH, without any direct release of CO2. Of particular interest are those silicate powders that have a high reactivity and higher capacity for raising pH. In this contribution, we report experimental work addressing the dissolution of various silicate rock-derived powders that were produced during mining activities in Norway under conditions that were representative of weathering in agricultural soils. Three different powders—derived from Åheim dunite, Stjernøya nepheline syenite, or Tellnes ilmenite norite—were exposed to different acids at pH 4 in unstirred flow cells, and dissolution or leaching kinetics were determined from the changes in the fluid composition. Based on these kinetics, pH neutralization rates were determined for the individual powders and compared to expected values for carbonates. Based on this comparison, it is concluded that the application of silicate rock-derived powder dissolution to replace carbonate-based liming may not be feasible due to slower reaction rates, unless larger quantities of a finer particle size than normal are used. The application of larger volumes of slower-reacting silicates may have the additional benefit of reducing the required frequency of liming.
Keywords:
mineral dissolution; weathering; liming; agriculture; soil; carbon sequestration; organic acids1. Introduction
Agricultural and natural soils, particularly in high rainfall regions, experience acidification due to cation leaching and uptake by plants, and due to the oxidation of ammonium and sulfur fertilizers. There are several agronomic reasons for mitigating acidification or increasing pH in naturally acidic soils in agriculture, including the plant availability of nutrients, variable plant tolerance to acidity, and, in some cases, reducing heavy metal availability. Another less well-known reason is the link between low soil pH and N2O emissions from soils, which is one of the main contributions of climate gas emissions from agriculture. Several studies [1,2,3,4] have shown that the N2O/N2 ratio of dentrification, which is the main N2O producing process in soils, increases substantially as pH decreases.
The most common strategy to combat soil acidification is through liming, i.e., the spreading of finely ground calcite (CaCO3), dolomite ((Ca,Mg)CO3), or burnt or slaked lime (CaO or Ca(OH)2). As the liming agent dissolves, H+ is taken up, reducing soil acidity. However, the dissolution of carbonate minerals leads to a net release of the greenhouse gas CO2 [5]. Likewise, calcining CaCO3 to create CaO or Ca(OH)2 directly releases CO2 from the calcination process, as well as from burning fuel. This release of CO2 could thus counteract the potential reduction in climate gas emissions (N2O) realized by neutralizing acidic soils.
An ubiquitous side product of mining operations is a finely ground rock-derived powder consisting mainly of those minerals that are not of commercial interest. Commonly, these powders are disposed of in stockpiles that are close to their source. However, the dissolution of the silicate minerals in these powdered rocks, under acidic conditions, will take up H+, and these powders could therefore be used as alternative liming agents for neutralizing soil pH. Taking the dissolution of olivine as an example, this pH neutralization proceeds according to the following reaction:
Furthermore, silicate dissolution does not lead to the release of CO2, and will even take up CO2 from the atmosphere [6,7,8,9], according to the following reaction (from Schuiling et al. [6]):
One additional advantage of using silicate rock powders as liming agents is that many such powders contain elements important for plant growth, such as Mg and K, which are released when these rocks dissolve. Potential problems are that silicate rocks can also contain heavy metal elements, such as Ni, Cr, Pb, U, and Th; and that there may be antagonistic effects on nutrients (e.g., lower Ca uptake in cases of high Mg-release [9]).
Whether silicate rock-derived powders produced during mining will be an effective replacement for lime in agricultural applications mainly depends on two factors: the total acid neutralization capacity of the silicate powder (how much H+ can be taken up per kg of powder), which depends on the composition of the powder, and the acid neutralization rate (how fast is H+ neutralized), which in turn depends on the dissolution or leaching rates of the minerals in the powder, the powder’s specific surface area (i.e., its fineness), and the evolution of these variables with time as a consequence of the dissolution or leaching process. The chemical dissolution rates of these rocks (or the different mineral phases within them) may also depend on the chemical composition of the liquid that surrounds them. Different acids are known to variably affect the weathering rates of some, but not necessarily all, minerals, for example due to ligand activity [8,10,11,12,13,14]. Thus, soils are a complex matrix due to the presence of organic acids (to a variable degree), variable ion concentrations, and the activities of roots, microbes, and fungi. Furthermore, microbial growth may also have a negative impact on the dissolution rate [15].
2. Silicate Weathering
Whereas accurate laboratory measurements of dissolution rates of most rock-forming minerals are available in the literature (e.g., [16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32]), these rates are commonly measured under ideal circumstances. Flow-through cells are used to limit the changes to the overall fluid composition that occur as a result of dissolution, and stirring ensures full exposure of the powdered pure minerals to the solvent, preventing the local build-up of solutes at internal surfaces. This is done to remove any delaying artefacts from the data, in order to expose the true dissolution rate at the mineral surface. However, when these laboratory dissolution rates are directly compared to the dissolution rates determined from models for natural weathering, the laboratory dissolution rates appear to overestimate natural dissolution rates by up to several orders of magnitude [33,34]. These differences are typically ascribed to the minerals not being fully exposed to reacting fluids during natural weathering. Both in time and space, mineral grains, or parts of grains, will not be exposed to the solvent so that no dissolution can occur. Furthermore, high concentrations of solutes may build up, limiting dissolution at close to saturation conditions [34], while the dissolution of high-energy sites and the leaching of elements from dissolving mineral surfaces, along with the precipitation of new phases on dissolving surfaces can significantly diminish surface reactivity [34,35,36,37].
Hochella and Banfield ([38]—referring to [39]) and White [40] discuss microscopic observations on naturally weathered mineral surfaces. Natural weathering typically results in the formation of microporosity due to preferred dissolution at high-energy sites, such as defects and dislocations, and at defects associated with exsolution lamellae. In the case of perthite (an intergrowth of albite and K-feldspar, which is formed through the exsolution of the former from the latter), this leads to the formation of crystallographically controlled etch pits that are up to 20-µm deep, and are initiated at defects along the contacts of these lamellae. Furthermore, albite lamellae dissolve faster than the host K-feldspar, leading to additional lamella-like surface roughness development. Similar observations have been made on plagioclase feldspar, where anorthite dissolves faster than albite. On olivine, weathering has been shown to lead to the formation of channels (a few to a few tens of nm wide) that are often lined with smectite clay. Similar, often epitaxial overgrowths have been observed for various other minerals. These overgrowths commonly limit fluid access to the grain surfaces to a single water layer, thus locally inhibiting transport and dissolution rates [38]. Similar passivating layers may also form on mineral surfaces due to the leaching of cations from silicates, and the precipitation of layers of other secondary minerals, such as silica or iron(hydr)oxides.
Surface roughness studies (cf. [40]) indeed show that silicate weathering leads to a time-dependent increase in surface roughness, as grain surface area and etch pit density increase with soil age. However, this etching is not homogeneous, as some surfaces are etched while others remain pristine. In part, this is the result of crystallographic effects, but additional effects occur due to the uneven exposition of the surfaces in the soil. Furthermore, an initially rapid dissolution at high-energy sites is only transient, so once these sites have been removed, mineral dissolution rates may slow down.
Addressing the differences between the weathering rates measured in the laboratory and observed in the field, White and Brantley [34] compared laboratory plagioclase dissolution rates from freshly crushed and pre-weathered (natural) Panola granite measured over a duration of six years, and further compared these laboratory rates to field weathering rates for the same granite. When they extrapolated their slowly-declining fresh granite dissolution rates to match their weathered granite dissolution rates, this suggested that thousands of years of weathering would be required to come to the same rates, while the field rates were another order of magnitude slower. Interestingly, they show that a power function can be derived correlating weathering rates from both laboratory and field for various minerals and rocks, showing the importance of intrinsic effects (i.e., effects leading to a decrease in the accessible reactive surface area) as well as extrinsic effects on limiting field weathering rates.
Organic acids are of particular interest as agents for weathering in agricultural and other plant–soil systems. Although there are differences between species, all of the plant roots release root exudates to the rhizosphere, where small organic compounds such as organic acids are a major component [41,42,43,44,45]. Fungi have likewise been found to release organic acids, and this has also been correlated to enhanced dissolution of minerals (cf. [46]; and references therein). Root exudates and organic acids are thought to have many functions for plants [45,47,48], and it has been suggested that the mobilization of plant nutrients by organic acids is important, although this is uncertain [42,49]. The efficiency of this depends, for example, on the lifetime of the acids before they are degraded by microbes, as well as on chemical factors such as the strength of the ligand effect of the acid. Regardless of the potential benefit for the plant–microbe systems on short-term nutrient availability, the potential enhancing effect of organic acids on the weathering rates of mineral particles in the rhizosphere is interesting in the context of mineral dissolution and acid soil neutralization, as the rhizosphere is regarded to be a hotspot for microbial activity, including N and C turnover.
Mineral dissolution rates can be influenced by the presence of organic ligands, as such ligands may bind metal cations in solution, or may form complexes on the mineral surface that enhance dissolution rates. However, the exact effects are strongly dependent on ligand and mineral species, and on pH. Studying forsterite dissolution, Declerq et al. [13] reported a negligible influence from 13 different ligands at 25 °C and pH 3. Performing dissolution experiments at 25 °C and pH 3.6, Wolff-Boenisch et al. ([8]) similarly reported no effect of oxalate or citrate on the rate of Si-release from peridotite, but an increased Si-release rate from basalt, which was likely at least partially due to enhanced pyroxene dissolution in the presence of these ligands [50]. In contrast, at pH > 4.2, an oxalate concentration of 0.001 m was reported to result in an increase in the forsterite dissolution rate by approximately six times [51]. This effect decreased at a lower pH, which was most likely as a result of the oxalate ion speciation. The dissolution rates of various plagioclase minerals are also enhanced by organic ligands, in particular those that preferentially dissolve Al relative to Si, by up to an order of magnitude, although this effect decreases with decreasing pH [11,52]. Finally, Bray et al. [14] reported that the presence of low concentrations of organic ligands (up to 0.0005 M) enhanced the release of certain cations (in particular Al and Fe) from biotite, but did not significantly affect the overall (Si-based) dissolution rate.
To study the applicability of various powdered rock-derived wastes, we have performed flow-through tests on three such powders obtained from mining operations in Norway. The powders studied are a powdered dunite from Åheim, a powder obtained in the mining and processing of nepheline syenite from Stjernøya, and a powder obtained in the mining and processing of ilmenite norite from Tellnes. These powders have been dissolved over a period in excess of four months, in acidic solutions with a pH of 4, under unstirred flow-through conditions, at room temperature. These conditions were chosen to approximate natural weathering conditions, including the potential to form concentration gradients in the liquid phase, and limiting particle collisions that may abrade leached or precipitated layers. Such leached or precipitated layers may form on the particles during weathering in agricultural fields (cf. [38,40]). During the experiment, the acid type that was used was changed to directly observe the effects of different acids. After the experiment, SEM was used to study the effects of dissolution on the particles. The effects of the activities of roots, microbes [15], and fungi where not addressed directly in these experiments, and should be considered in a future work.
3. Materials and Methods
In order to investigate whether the rock-derived powders that were produced during mining operations in Norway have sufficiently rapid dissolution/leaching kinetics to be used as replacements for common liming agents based on calcite or dolomite, we have performed unstirred flow-through dissolution experiments on three selected powdered, rock-derived waste products that are currently produced in significant volumes. The experiments performed here were aimed at studying dissolution and acid neutralization due to weathering effects under conditions that approached natural conditions as encountered in agricultural fields. ICP-MS analyses were performed on the solutions produced to determine the dissolution rates, and SEM was used to observe the effects of dissolution on our particles. Here, we will describe our starting materials and the methods that were used in this work.
3.1. Materials
We used three different rock powder waste products from mining operations in Norway. First, all of the powders were sieved dry to obtain the 200–300 µm fraction. Next, these fractions were washed in demineralized water, and then cleaned ultrasonically, in water and in ethanol, to remove all of the fines attached to the surfaces. The powders were then dried at 80 °C for several days. The three sieved and cleaned rock powder samples were analyzed using whole rock XRF, XRD, and BET to determine their bulk chemical compositions, mineral compositions, and specific surface areas. The results of these analyses are given in Table 1, Table 2 and Table 3.
The first sample used in this study was a powdered dunite from Åheim (Norway), supplied by Sibelco Nordic AS. XRD analysis identified the main mineral phase to be olivine, with minor chlorite and orthopyroxene. Based on the bulk chemical composition and loss on ignition (LOI), the powder sample consisted of 95.1% olivine (with composition Mg1.86Fe0.14SiO4), with 3.0% orthopyroxene and 1.9% chlorite.
The second sample used here was a powdered waste material produced during nepheline syenite mining operations on Stjernøya (Finnmark, Norway). This waste material was depleted in nepheline and feldspar compared to the natural rock, and has been marketed as an agricultural supplement under the brand name Altagro. Based on our analyses, which were supported by mineral chemical composition analyses reported by Strand [53] and Mjelde [54], we inferred the mineral composition of our sample to be: 32.2% feldspar, consisting of both Ca-bearing albite and perthite (with an overall estimated composition of Na0.52K0.29Ca0.19Al1.19Si2.81O8), 24.5% pyroxene (Mg0.24Fe0.30Ca0.46SiO3—composition based on Mjelde, [54]), 20.7% biotite, 17.5% nepheline ((Na0.78K0.22)AlSiO4), 2.9% calcite, and 2.2% sphene. The given calcite content was calculated based on the sample’s LOI. During SEM studies of the samples, we additionally found minor amounts of a Ba-containing silicate that was most likely Ca-bearing banalsite ((Ba,Ca)Na2Al4Si4O16).
The third sample investigated was an ilmenite norite-based waste material produced by the titanium mining operations in Tellnes. Based on our XRD and bulk chemical analyses, and mineral chemical analyses provided by the provider of the sample, Titania AS, the powder sample consisted of 65.3% plagioclase (Na0.54Ca0.46Al1.46Si2.54O8), 14.4% pyroxene, 11.4% ilmenite (containing some Mg), 7.3% biotite, 1.0% magnetite, and 0.7% periclase (MgO, although this Mg was more likely present as component in other phases). Many ilmenite grains contained sub-micron scale lamellae with a regular spacing in the order of a few µm, formed by the exsolution of a phase enriched in Fe. Many pyroxene grains were also observed to have non-continuous, thin (sub-micron), irregularly spaced lamellae of ilmenite. Note that while our nepheline syenite and ilmenite norite samples are actually residues from the mineral processing that was performed on these rocks, here they will be referred to by those names. Furthermore, it should be noted that both of these products were removed from their processing before flotation or similar processes involving chemical agents were performed.
The acid solutions that were used in our experiments were all prepared from pro analysi quality chemicals, and using Milli-Q water. First, a 1000× concentrated stock solution was prepared from the as-received acid (65% solution nitric acid, 35% solution hydrochloric acid, or solid powder in the case citric and oxalic acid). Next, 10-L batches of the intended acid solution were prepared by diluting 10.0 g of this stock solution to 10.0 kg of total mass using Milli-Q water. All of the experiments were performed at a starting pH of 4.0. Blank samples were taken from each acid batch that was prepared, and were analyzed using ICP-MS.
3.2. Dissolution Cell and Procedures
Dissolution experiments were carried out in purpose-built flow-through cells. These cells (see Figure 1) consisted of Polyether ether ketone (PEEK) top and bottom pieces, with a cylindrical Perspex body. The cell chamber had a height of 10 mm and a diameter of 20 mm. The top and bottom pieces had central bores as fluid inlets and outlets, and Whatman QMA grade quartz fiber filters were used as flow distributors at both inlet and outlet to promote a more evenly distributed flow. Whatman membrane filters with 0.2-µm (inlet) and 1.2-µm (outlet) pore sizes were used to ensure that only fluid was transported into and out of the cells.
Eight individual cells were operated simultaneously: two for each silicate powder (labeled 1ÅD and 8ÅD for Åheim dunite; 4NS and 6NS for nepheline syenite; and 3IN and 5IN for ilmenite norite), plus two empty blank cells. An eight-channel Gilson Minipuls3 peristaltic pump was used to pump the acid solution through these cells, with average flow rates in the range of 0.80–0.87 mL/min. This range of flow rates was chosen to ensure that variations in fluid pH would remain minimal, and would not affect the reaction rates. Minor variations in flow rate existed between the various cells, and over time. The outlet fluids were collected in large individual vessels, and the collected fluid mass was used to calculate the average flow rates between samplings. Fluid samples for analysis were tapped directly from the cell outlet tubes into 13-mL fluid sampling tubes, and the collected fluid mass was likewise used to determine the flow rates during samplings.
The dissolution cells were unstirred to more closely represent field weathering conditions. While stirring is commonly used in dissolution kinetics studies to ensure the full exposure of the particle surface area to the fluid and prevent the build-up of high concentrations of elements due to transport (diffusion) kinetics limitations, particle collisions can lead to abrasion and the removal of leached or precipitated layers from particle surfaces. Furthermore, as stirring eliminates concentration gradients in the solution, it may also prevent layer precipitation. As the effects of such layers on sample reactivity can be significant [33,38,40], and we were also interested in studying microstructural changes resulting from dissolution, no stirring was used. Considering the average flow rate (~0.83 mL/min) and the inner cell volume minus the volume of powder (2.8 mL), the average residence time of the fluid in the cells was about 3.4 min.
At the beginning of the experiment, 1.005 ± 0.0004 g of cleaned silicate powder (grain size 200–300 µm) was weighed to a precision of 0.0001 g, and then added to each cell. Next, the cells were closed and connected to the pump. Once the pump inlet leads were inserted into a 10-L barrel of acid solution, and the cell outlet leads were inserted into closed 2-L barrels, the pump was started, and the cells started filling with acid solution flowing in from below. Samples were taken several times per week, with a higher daily sampling density in the days immediately after switching to a new acid. These samples were tapped directly from the outlets by inserting them into 13-mL sampling tubes, without halting the flow. When new inlet solution was required, the pump inlet leads were quickly switched from one 10-L barrel to another, stopping the flow for only a few seconds.
When switching from one acid to another, the experiment was switched to Milli-Q water for at least 16 h to wash out the previous acid before switching to the new acid. After running for 51 days, the experiment was likewise switched to Milli-Q water, and then flow was stopped entirely for a period of 48 days. After that period, flow was re-initiated with Milli-Q water, and then switched back to acid.
During the experiment, blockage of the filters occasionally started to inhibit flow. When this occurred, the cells were carefully opened to replace the filters. During one such replacement on cell 3 (containing ilmenite norite), at a duration of 68 days, some powder was spilt, and it was decided to continue running the experiment in this cell with a fresh sample of ilmenite norite.
3.3. Analyses
A number of analysis methods were used both to characterize our starting materials and track dissolution reactions and determine the effects of dissolution on our materials. Here, we will describe the applied methods.
Fluid chemical compositions were determined using a Thermo Scientific ELEMENT XR™ high-resolution ICP-MS. The concentrations of a range of trace elements (Sc, Ti, V, Cr, Mn, Co, Ni, Cu, Zn, Rb, Sr, Y, Cs, Ba, Pb, Th, U, Na, Mg, Al, Si, Ca, Fe, and K) were measured using five-point calibration curves, with indium as the internal standard. Before analysis, the samples were diluted five times, or more if necessary, with 2% nitric acid. All of the elements were analyzed in a medium resolution except K, which was analyzed in high resolution. As a matrix-matched, certified reference material for quality control was not available, synthetic fresh water certified reference material (SPS-SW-2) was diluted with 2% nitric acid to two different levels and analyzed for quality control.
Fluid pH values were measured using a Metrohm 827 pH Lab pH meter with an LL aquatrode plus Pt1000. These measurements were performed to confirm that changes in the fluid pH were negligible. XRD analyses were performed on powdered samples using a Bruker D8 Advance. XRF analyses were carried out on a Phillips PV1404. LOI was measured as weight loss after 2 h at 1000 °C. The specific surface areas of the samples were measured by N2 adsorption measurements at 77.35 K, using a BELSORP-max instrument, and analyzing the data according to the BET method. SEM was performed on a Zeiss Supra 55VP with a Thermo Scientific Ultradry EDS detector to identify minerals.
4. Results and Discussion
To study the applicability of silicate rock-derived, powdered waste products from mining as alternative liming agents in agriculture, we performed dissolution experiments under conditions approximating natural weathering conditions (room temperature, pH 4, flow-through without stirring). The release of cations was measured and will be used to estimate the mineral dissolution rates and H+-uptake rates.
4.1. Elements Released
ICP-MS analyses were performed on the fluid samples that were collected at the cell outlets to determine the quantities of elements released from our samples due to dissolution or leaching. The results obtained were corrected with respect to Si and Al based on blank analyses performed on the inlet acid as well as outlet fluids from the control channels (empty cells). From these concentrations, we can calculate the most important major element release rates (in mol/m2/s) for each silicate powder, using the initial powder surface area (assuming no change due to dissolution). These major element release rates are presented for each sample in Figure 2a–f.
An important concern when applying silicate rock-derived powders to agricultural land is the potential release of heavy metals, as these rocks can contain such elements in relatively high concentrations. The quantities of minor elements, including (potentially toxic) heavy metals released during dissolution were also measured in our experiments. Åheim dunite dissolution resulted in the release of minor amounts of Mn, Ni, Zn, and Ba. The release of Co and Cu was negligible, while the release of Cr, Pb, Th, and U was below the detection limit in most of the individual fluid samples. Nepheline syenite dissolution resulted in the release of minor amounts of Mn, Ti, Zn, Sr, and Ba, while the release of Cu, Ni, Cr, and Co was negligible, and the release of Pb, Th, and U was below the detection limit in most of the fluid samples. Ilmenite norite dissolution resulted in the release of Ti, Ni, Cu, Zn, and Ba, while the release of Cu, Mn, and Co was negligible, and Cr, Pb, Th, and U were not detected in most of the fluid samples.
4.2. pH Measurements
Whereas pH measurements were performed on all of the fluid samples that were taken during our experiment, these measurements generally confirmed that pH changes were negligible (<0.1 units), which was a result of the flow rates chosen for our tests. Notably, the pH measurements performed on oxalic acid samples suggested much larger changes pH changes, but these measurements were likely incorrect, due to either the unidentified aging effects that took place during storage, or some unidentified electrode effect, and were therefore discarded.
4.3. SEM Imaging
We performed SEM imaging on our silicate powders before and after dissolution. This was done on loose grains, as well as on grains that were cast in resin and then polished to obtain cross-sections through these grains. Selected images are presented in Figure 3, Figure 4 and Figure 5.
In Figure 3, we present images showing the effects of dissolution on the Åheim dunite. Here, dissolution resulted in the formation of deep channels into the grains, which were possibly located along the boundaries between the individual olivine crystals that our grains were composed of, or along crystallographic defects (Figure 3d,e). Crystallographically controlled etch pits were observed on some surfaces (Figure 3f). Furthermore, thin layers were observed on the surface of some olivine grains, which may represent leached layers, or, more likely in the case of a nesosilicate such as olivine, precipitation (Figure 3g,h). While these layers were too thin for accurate chemical analysis using EDS without including the underlying material, analysis on the layer that was shown in Figure 3g gave an elevated Si content compared to the underlying grain, suggesting that this layer may have been silica. No significant dissolution effects were observed on the chlorite grains in this sample, beyond rather minor surface damage.
In the nepheline syenite (Figure 4), dissolution resulted in various surface effects. Alkali feldspar grains typically showed increased dissolution alongside exsolution features such as lamellae, where (presumably) the Na-rich phase dissolved preferentially (Figure 4d,e). Furthermore, dissolution resulted in crystallographically controlled roughening at mineral surfaces (Figure 4f). Thin precipitated or leached layers were observed on some grains (Figure 4g), but could not be analyzed accurately. In cross-section imaging, many grains likewise showed the effects of dissolution at their surfaces. Note that the apparent holes in the middle of a grain, or “islands” that can be seen in these figures (cf. Figure 4h), resulted from the intersection of the polishing surface through grain surface roughness. In addition, large cavities were observed on many grains, which implied the complete removal of a mineral phase or phases (Figure 4i,j). Since no nepheline was found in the reacted grains (while it was observed in the unreacted grains), we conclude that these cavities were most likely formed as a result of the preferential dissolution of nepheline.
The ilmenite norite grains (Figure 5) showed fewer and less intense signs of dissolution than the other two silicate powders. Still, we observed etching along exsolution lamellae on ilmenite grains (identified from the continuous, narrowly spaced lamellae), where the Fe-enriched phase was dissolved preferentially (Figure 5c,d), and surface effects on biotite (Figure 5e), where leaching or precipitation resulted in the formation of a thin layer that was either elevated in Si or consisted of pure silica. In the cross-section (Figure 5f–h), we observed cavities where grains were removed, which was presumably due to dissolution (Figure 5g), and surface roughening effects due to preferential dissolution (Figure 5h).
4.4. Effects of Fluid Composition and Flow
The main acid that was used in our experiments was hydrochloric acid. However, during the ongoing test, the solution used was switched from hydrochloric acid to a selected other acid and back again, in order to be able to directly compare the effects of various acid types on the cation release rates and dissolution rates. The other acids used were oxalic acid, citric acid, and nitric acid.
Switching to oxalic acid immediately resulted in a 6.3 times faster dissolution rate for Åheim dunite (see Figure 2a,b). Notably, though, the fluid Fe-content was below the detection limit for several days following the switch, after which Fe was measured in the fluid again, and at concentrations congruent with its content in the olivine. Switching back to hydrochloric acid resulted in a dissolution rate that was 6.3 times slower, which was similar to the dissolution rate measured in hydrochloric acid prior to the switch. For nepheline syenite (Figure 2c,d), switching to oxalic acid resulted in approximately doubling the dissolution rate relative to the rate measured using hydrochloric acid. However, for one of the two samples (4NS), the rate of release of Al increased significantly less. For the other sample (6NS), the Al-release rate did increase, but only after an initial delay of about one week, during which Al release was suppressed. After this week, the Al-release rate jumped up to a similar level as the Si release rate again. After switching back to hydrochloric acid, the nepheline syenite dissolution rate slowed again, but the various element release rates became less close to stoichiometric for nepheline dissolution. This may have been a result of nepheline having been dissolved fully, as suggested by the SEM observations. For ilmenite norite (Figure 2e,f), switching to oxalic acid did not result in a change in the dissolution rate for roughly three days, and then caused an increase to roughly four times the prior rate for most elements. The dissolution rate then gradually decreased over the remaining 23 days that oxalic acid was used. After switching back to hydrochloric acid, on average, the dissolution rate was roughly halved, to rates that were still somewhat faster than before the switch. Dissolution rates kept decreasing over the next days.
Switching to citric acid resulted in a dissolution rate that was four times faster for the Åheim dunite, and this enhancement fully disappeared as soon as hydrochloric acid was used again, with no transient effects. In the case of nepheline syenite, the effect of switching to citric acid was more pronounced in one sample (4NS), where it resulted in increases in individual element release rates that were two to 10 times higher than in the other sample (6NS), where the effect was only increased up to three times. The strength of the effect ranged from Na > Al > Si > K for 4NS and Al > Na > Si > K for 6NS. For 4NS, the average increase in the dissolution rate was about four times. For 6NS, the average increase in the dissolution rate was about two times. The difference observed between these two samples may be related to differences in flow distribution, and (resulting) differences in mineral content of the samples. For ilmenite norite, a clear indication of the effect of citric acid could not be derived for one sample (3IN), because the silicate powder had to be replaced due to an experimental problem. For the other sample (5IN), switching to citric acid resulted in an increase in element release rate (Al > Fe > Mg) by a factor of between four and 10, although the increase in the rate of Si-release was only ~1.3 times. The average increase was about 2.5 times. After switching back to hydrochloric acid, the average dissolution rate decreased by a similar amount.
Finally, switching to nitric acid and back to hydrochloric acid had no discernible effect on the dissolution rates that were measured on any of the samples.
There is a clear two to 10 times increase in the dissolution rate for all three rocks as a result of changing to organic acids, while changing between hydrochloric acid and nitric acid did not result in any significant change. In general, these observations agree well with the observed effects of organic ligands (oxalate and citrate) on the single mineral phase dissolution that were reported in the literature [11,14,50,51]. Individual ligands have different effects on different minerals, as was observed in our results. Even though our experiments were too simplified to accurately reflect in situ conditions when it comes to dissolution in soils, our results do show that organic acids, such as those found in soil–plant systems, may enhance silicate mineral dissolution rates. We suggest that the most likely explanation for the significant differences observed between the duplicate tests on nepheline syenite (4NS and 6NS), in particular when switching to citric acid, may be that certain mineral phases had disappeared from one duplicate, but were still exposed in the other. Alternatively, differences in the presence of non-reactive layers of leached or precipitated material may have played a part in causing these differences.
After running for 51 days (before testing with citric acid, but after having tested with oxalic acid), our experiment was paused for 48 days, during which period the cells were filled with stagnant Milli-Q water. After this pause, the experiment was re-started by pumping Milli-Q water through the cells for about 24 h, followed by hydrochloric acid solution. As a result of this pause, some effects were observed on the dissolution rates. The initial Åheim dunite dissolution rate after the pause was about half the rate measured before the pause. Over the next two days, the dissolution rate recovered to the original value. The nepheline syenite dissolution rate after the pause was likewise about half the rate that was measured before the pause. However, the nepheline syenite dissolution rate did not recover over the following 12 days. For the ilmenite norite, no change in the rate of release of Si or Mg was observed, but the release of Al and Fe were, respectively, ~4 times and ~1.6 times slower, recovering slowly to prior values over two to three days.
4.5. Mineral Dissolution Rates
4.5.1. Olivine Dissolution in Åheim Dunite
Based on the element release rates reported above, it is possible to estimate apparent mineral dissolution rates. For Åheim dunite, based on literature dissolution rates, olivine should by far be the fastest dissolving phase [24,27,28,30,31]. Microstructural observations (Figure 3) further support that olivine was the main dissolving phase. Hence, using the sample olivine content (95%) and BET surface area, we can estimate an apparent olivine surface area (assuming 95% of the total BET surface area to be representative for the olivine surface area, and assuming the full exposure of all of the surfaces to fluid flow). Using this apparent olivine surface area, we can then estimate dissolution rates from the fluid Mg, Si, and Fe contents, further assuming stoichiometric dissolution [27]. The apparent olivine dissolution rates from the two cells, which were based on the release of Mg, Fe, and Si, are plotted against time in Figure 6a. The averaged rates per cell are plotted in Figure 6b. This shows that initially, the apparent dissolution rates start relatively high, with the average apparent olivine dissolution rate (Figure 6b) decreasing by 10–20% over the first two days, and a total 40% over the first two weeks. Furthermore, it shows that during the first two days, the Mg/Si ratio is somewhat higher than stoichiometric (2.9–3.9 versus 1.86), while the Fe/Si ratio is too low (0.10–0.11 versus 0.14).
The initially observed rapid dissolution can be ascribed to fine grains and sharp edges on the dunite grains (Figure 3a), which resulted in an increased specific surface area, as well as enhanced dissolution at high-energy surface sites, such as at defects and dislocations. The high initial Mg/Si ratio suggests minor leaching, and is in accordance with some of the literature observations [27]. The relatively low initial Fe release rates may indicate the precipitation of some Fe oxides or Fe hydroxides. After about 30 days, the apparent olivine dissolution rate stabilized at ~1.2 × 10−10 mol/m2/s. This is roughly 1–1.5 orders of magnitude slower than the olivine dissolution rates that have been reported in the literature based on single mineral dissolution experiments ([55]; and references therein). We suggest that this discrepancy resulted from our assumptions regarding the surface area exposed to flowing fluid. The BET surface area measurements on the starting material included the very fine grains and sharp edges that were rapidly removed during the initial phase of dissolution (resulting in a ~50% decrease in the apparent dissolution rate, suggesting a similar decrease in surface area). However, more significantly, our assumption that all of the powder surface was fully exposed to the flow of acid solution was likely incorrect. Therefore, the effective surface area of our sample was likely lower than the total initial surface area that was used here to calculate the apparent dissolution rate (i.e., similar to the exposure effects discussed with regards to natural weathering; cf. [33]). For each dissolution step (i.e., between each measurement), the total amount of olivine dissolved can be estimated by multiplying the fluid olivine content (based on its Mg, Si, and Fe contents) with the total fluid flow volume during that step (average flow rate times duration). Doing so, it is estimated that 16–17% of the olivine that was present dissolved during the experiment.
Using oxalic or citric acid resulted, respectively, in a 6.3 times and a four times increase of the olivine dissolution rate relative to the rate measured in hydrochloric acid. This is comparable to the 600% increase in the forsterite dissolution rate that was reported for a 0.001-m oxalate concentration at pH > 4.2 [51], although at pH < 3.6, Declerq et al. [13] and Wolff-Boenisch et al. [8] reported a negligible influence of oxalate or citrate on olivine dissolution. These pH-dependent differences are related to the pH-dependent dissociation of the acid. A mechanism whereby oxalate attaches to Mg and thereby helps release H4SiO4, promoting dissolution, has been proposed by Olsen and Rimstidt [51]. Citrate may enhance dissolution via a similar effect.
4.5.2. Nepheline Dissolution in Nepheline Syenite
In the nepheline syenite experiments, based on the inferred mineral compositions and relative mineral dissolution rates reported in the literature [18,19,23,29,56,57], the observed cation release likely resulted mainly from the dissolution of nepheline, while, at least initially, contributions from other mineral phases (through dissolution and/or leaching) were relatively minor. This is supported by the disappearance of nepheline from the powder after the experiments. Figure 7a shows the apparent dissolution rate of nepheline, which was calculated from the initial sample surface area and nepheline content (again, assuming full exposure of the powder to flowing acid), based on the fluid Na, Al, Si, and K contents. The plot shows some spread in the calculated apparent dissolution rates based on each cation, indicating that dissolution may not have been stoichiometric, our nepheline composition ((Na0.78K0.22)AlSiO4) was not representative of the nepheline in our sample, and/or that certain elements, in particular K and Si, were also released from other minerals, through dissolution or leaching. The precipitation of Al cannot be excluded, although Al concentrations in our fluids were typically below 0.7 ppm. The average apparent nepheline dissolution rates were calculated from the individual rates per element, and are given in Figure 7b. This shows that a constant rate was not reached during our experiments, as after about 20 days, the average apparent dissolution rate starts to decline, until it reaches a value of about 1/40 of the initial rate. Using the total amounts of each element released, we can estimate the amount of nepheline dissolved. This confirms that all of the nepheline would have been dissolved before the end of the experiment. The spread in element-based calculated dissolution rates (see Figure 7a) increases with increasing time (and as the apparent, calculated rates decrease), which suggests that the impact of additional element release from other mineral phases increased as nepheline disappeared from the mineral assembly. SEM with EDS was used to confirm that no detectable nepheline remained by the end of our experiment. Comparing the range of apparent nepheline dissolution rates determined here, we see that our rates are about 1–2.5 orders of magnitude slower than those reported by Tole et al. [56], but 0–1.5 orders of magnitude faster than the rates reported by Hamilton et al. [29]. Finally, as can be seen in Figure 2c,d, during the first two days of the experiment, Ca was the main cation that was released into solution. While the rate of Ca release slowed down rapidly, Ca was measured in the fluid for more than 40 days. The initially rapid release of Ca2+, likely resulting from the dissolution of calcite from the sample, caused a significant increase in pH, and resulted in a suppressed initial nepheline dissolution rate (which can be observed in Figure 7).
Using oxalate caused only minor increases in the dissolution of the nepheline syenite. Stronger, element-dependent effects were observed with citrate. However, literature data on the effect of organic ligands on nepheline dissolution are not available. Furthermore, since nepheline likely fully dissolved during our experiments, the observed effects may instead represent increases in the dissolution rate of other minerals, such as plagioclase, for which increases in the dissolution rate with organic ligands of up to an order of magnitude have been reported [11,52].
4.5.3. Mineral Dissolution in Ilmenite Norite
Based on the dissolution rates in the literature, and the mineral composition of the ilmenite norite, no single phase could be identified that would have controlled dissolution and cation release, as for the other silicate powders. Figure 2e,f give the element release rates for the main elements released. Comparing these element release rates to the literature dissolution rates for biotite [14,21] shows that biotite dissolution would be up to more than an order of magnitude too slow to account for the elemental release rates that were measured. We infer that during dissolution, multiple minerals released elements at comparable rates. Alternatively, the exposed biotite surface area may have been larger than assumed based on multiplying the BET surface area with the biotite fraction in the sample.
Bray et al. [14] report non-stoichiometric biotite dissolution at pH in the range of 2–6, with increased release of Fe, Mg, and Al relative to Si. In our experiments, the ratios of released Fe, Mg, and Al to Si were similarly non-stoichiometric for biotite dissolution. Initially, Mg/Si and Fe/Si ratios are elevated. Changing from hydrochloric acid to oxalic acid does not influence these ratios, but changing back from oxalic acid to hydrochloric acid caused these ratios to drop. The ratio Al/Si was initially stoichiometric for biotite, and increased when changing to oxalic acid. After switching back to hydrochloric acid, the ratio dropped to below stoichiometric. Switching to citric acid caused an increase in the release of all of the elements relative to Si, to about double the stoichiometric value, which then transiently disappeared as dissolution in citric acid continued. Finally, nitric acid caused a minor increase in the Al/Si ratio. In considering these ratios, note that Si (and other elements) were likely also released through the dissolution of other minerals, such as plagioclase, hornblende, and pyroxene. Switching to oxalic acid resulted in a rough increase in the dissolution rate by a factor of about four, but this enhancement did not occur until approximately three days after the switch. Switching to citric acid resulted in a four to 10 times increase in the element release rate, with the strongest effects observed for Al > Fe > Mg. Bray et al. [14] similarly reported the enhanced release of selected cations (in particular Al and Fe) from biotite in the presence of low concentrations of organic ligands, including oxalate and citrate.
4.6. H+-Uptake Rates and Potential
As our experiments were designed to minimize pH changes, pH measurements were not precise enough to determine H+ uptake rates. Instead, such rates were calculated based on the elements that were released. The calculated H+ uptake rates per sample are given in Figure 8a–c. These plots show that for Åheim dunite, after the first three to four weeks, the H+ uptake rate stabilizes at about 4–5 × 10−10 mol/m2/s. For the nepheline syenite, during the first three weeks, the H+-uptake rate is quite stable at 5 × 10−9 mol/m2/s. During the next ~10 weeks, this rate gradually declines, until it becomes stable again at a rate of about 1 × 10−10 mol/m2/s. For the ilmenite norite, after the initial few days, the rate stabilized at about 1.6 × 10−10 mol/m2/s, and then gradually decreased down to 0.5 × 10−10 mol/m2/s.
As a comparison, H+ uptake rates can be calculated for calcite or dolomite, which are the most commonly used liming products, based on the literature dissolution rates for these minerals [58] and neglecting any subsequent release of H+ through the dissociation of H2CO3. For calcite, a dissolution rate of 2.2 × 10−5 mol/m2/s then gives a neutralization (H+ uptake) rate of 4.4 × 10−5 mol/m2/s. For dolomite, a dissolution rate of 0.6–2.3 × 10−7 mol/m2/s gives a neutralization rate of 1.2–2.6 × 10−7 mol/m2/s. These carbonate neutralization rates are 200 to 400,000 times faster than the rates estimated for the silicate powders in our experiments. However, due to experimental differences, such as our experiments being unstirred, and the use of multi-mineral powders rather than single minerals, the mineral dissolution rates that were determined in our experiments were slower than those measured under ideal (for dissolution) circumstances. Based on comparing our apparent olivine dissolution rates to literature values, it was estimated that this can account for differences of a factor of up to 300. Furthermore, Pokrovsky et al. [12] reported the negligible effects of high concentrations of organic ligands such as oxalate and citrate on carbonate dissolution rates. Even then, pH neutralization by liming with a carbonate can be expected to be considerably faster compared to using one of the silicates considered here. In part, this difference could be compensated for by increasing the surface area of the silicate powder, either by using a finer fraction (which may require additional grinding), or by increasing the amount of silicate powder that was used. A popular dolomitic liming product in Norway, Agri Dol, is reported to have a particle size distribution in which 64% of its mass is coarser than 130 µm (52% coarser than 250 µm [59]). Hence, a sufficiently fine silicate powder (for example, finer than 100 µm) may react at a rate that is comparable enough to such a dolomitic liming product. Furthermore, when a sufficiently large amount of silicate powder is applied, the slower pH neutralization reaction of the silicate powder may also be an advantage, as it could potentially help maintain a more stable soil pH over a longer period, whereas a highly reactive lime gives rise to a rapid pH increase followed by a drop in pH once all of the lime has reacted. Therefore, the use of silicate powders may have the potential to reduce the required frequency of liming.
In addition to the rate with which H+ is taken up through the dissolution of any rock powder, we should also consider the potential total amount of H+ that is taken up per kg of rock powder, assuming full dissolution. Table 4 presents the total H+ uptake capacity per kg for each silicate powder that is used. The maximum potential uptake by the silicate rock-derived powders is somewhat higher than the maximum uptake by carbonates, meaning that if such a silicate powder were to dissolve fully, this would potentially take up more H+ than would be taken up by the full dissolution of a carbonate. Typically, though, the weathering of silicates such as plagioclase results in the formation of other, stable silicates, such as clays, which may considerably lessen the H+ uptake potential of silicate dissolution.
4.7. Trace Elements
Besides pH neutralization while avoiding the release of CO2, the use of silicate rock-derived powders can have the additional benefit of releasing elements that are important nutrients to the crops that are grown, such as Mg, K, Zn, and Mn. However, these silicates may also contain heavy metals that are potentially toxic. Åheim dunite contains relatively high concentrations of Cr (0.14%) and Ni (0.25%). Ilmenite norite contains 0.02% Cr and 0.02% Ni.
During the dissolution of Åheim dunite, the mass of Ni that was released was typically less than 1.0% of the mass of Mg plus Fe that was released, although Ni release was somewhat enhanced in the presence of oxalic, citric, or nitric acid (0.8–1.0% versus 0.6–1.0%). During the first 10 days of ilmenite norite dissolution, Ni made up about 1% of the total mass of cations released, after which this ratio decreased. Over the full experiment, Ni accounted for about 0.3% of the mass of cations that was released. For both Åheim dunite and ilmenite norite, the Cr content in the fluid samples after dissolution was consistently below the detection limit (0.025 ppb).
In our study, we have not evaluated the agronomic effect of this release of heavy metals, as this involves many factors that are not assessed in our experiments. Before these silicate powders are applied in a large-scale agronomic setting, such effects should be assessed. Note that Ten Berge et al. [9] reported no negative effects to plant growth caused by Ni release due to olivine dissolution.
4.8. Comparison to Natural Weathering
The SEM observations on grains that underwent dissolution in this study show similarities to naturally weathered grains. In all of our samples, we observed the formation of etch pits and surface roughening on some grain surfaces, while other surfaces (or parts of surfaces) appeared unaffected. We also observed dissolution and etch pit formation focused along exsolution lamellae, which was similar to the structures that were seen on naturally weathered mineral grains. Finally, we observed thin layers that were Si-enriched or consisted of pure silica, and which had formed either due to leaching or by the precipitation of a secondary phase.
In the Åheim dunite sample, dissolution resulted in the formation of deep channels through the olivine grains. These channels may have formed along crystal boundaries, or along defects such as dislocations. However, the channels that were observed here were about three orders of magnitude wider than those reported on naturally weathered olivine [38]. We cannot exclude the possibility that fluid flow patterns in our cells influenced the formation of these channels. On the nepheline syenite, the main observed dissolution features were the complete removal of the nepheline, along with preferred dissolution at exsolution lamellae in plagioclase, and crystallographically controlled surface roughening. On the ilmenite norite, preferential dissolution at exsolution lamellae and the selective dissolution of specific phases were observed.
Our observation of lower apparent mineral dissolution rates than the typical literature values was most likely due to uneven flow around particle surfaces, and thus demonstrates how soil structure and fluid flow patterns can limit dissolution in the field. Since there is high variability in the concentration and type of organic acids between soils and different plants rhizospheres, and the lifetime of aliphatic organic acids in the rhizosphere is uncertain (cf. [42,45]), this limits how representatively lab dissolution rates can be extrapolated to field rates, especially noting that different (organic) acids have different effects on different minerals. The observed similarities between naturally weathered grains and the grains that were dissolved in our experiments support the assumption that the processes by which natural weathering occurs were at least partly reproduced in our relatively long-term, unstirred flow-through experiments. Our results also showed that, even for a relatively rapidly dissolving mineral such as olivine, it can take days (more than 10 days for olivine) for the dissolution rate to become (apparently) stable, and thus more representative of long-term, natural dissolution. This means that experiments with a very long duration that are performed on sufficiently large grains (to limit changes in surface area) are required in order to properly measure representative dissolution rates.
White and Brantley [34] reported experimental weathering rates that decreased constantly with time over at least six years. Furthermore, they showed even slower weathering rates when using weathered grains compared to fresh grains. Their observations thus showed that ongoing weathering will lead to a slowing down of dissolution. However, in the application of silicate powders from mining as liming agents in agriculture, the mineral powders that will be distributed on the field will be freshly crushed and not yet affected by weathering. Therefore, these powders will have relatively high reactivity, which is similar to the minerals studied in the dissolution experiments. As liming should typically be repeated every 10 years or so, this means that the negative effects of surface ageing on weathering rate, such as the intrinsic effects addressed by White and Brantley [34], will not have a significant impact on the agricultural application discussed here. As discussed above, extrinsic effects limiting the dissolution rates during weathering (related to the mineral–fluid ratio, fluid composition and fluid flow) will still apply.
5. Implications
While the increased dissolution rates measured when using organic acids suggest that mineral dissolution in agricultural soils can be faster than the rates that are normally measured using mineral acids, we also observed that our unstirred experiments yielded lower rates than the other experiments that were performed using stirred reactors. Furthermore, the observed preferential dissolution patterns on rock grains suggest that even in our relatively homogenous reactors, spatially uneven dissolution reduced the average rates that were measured. Similar effects are expected to be even more pronounced in soils, and therefore, real dissolution rates in the field could be still lower than those measured here. However, as liming is performed with freshly crushed, fine rock powders, and should typically be repeated every 10 or so years, other rate-limiting effects that control the natural weathering of silicates as these silicates age will not impact their use as liming agents. It should also be noted that Pokrovsky et al. [12] reported negligible effects of organic ligands, such as oxalate and citrate, on calcite and magnesite dissolution, for concentrations in excess of those expected in agricultural soils.
The spreading of rock-derived silicate powders produced as waste products in mining to neutralize soil acidification on agricultural fields, as a replacement for lime, can have many benefits. It prevents the release of CO2 that would otherwise be released during the quarrying, grinding, and dissolution or calcination of carbonates, and may have additional beneficial CO2-uptake effects [6,7,8,9]. Furthermore, using these waste silicate powders decreases the volume of materials that are currently stored in stockpiles. Additional nutrients, such as Mg and K, may also be released as the powders dissolve. However, even with the increase in dissolution rates observed with organic acids, the relatively low dissolution rates that can be expected from these silicates relative to the normal liming materials mean that the application of silicate powders as liming agents may not be feasible unless larger quantities of material with a finer particle size than would normally be used when liming with (calcined) carbonates.
6. Summary and Conclusions
In order to investigate whether silicate rock-derived powders that were produced during mining and mineral processing can be used to neutralize soil pH in agricultural soils, we have performed unstirred dissolution experiments on selected silicate powders, and under conditions that were representative of dissolution in acidified soils (pH 4, RT). During these experiments, we have addressed the effects of different (organic) acids, and we have used an SEM to directly study the effects of dissolution on our mineral grains. We observed the following:
- The dissolution rates of dunite at pH 4.0 increased six times with oxalic acid and four times with citric acid compared to hydrochloric acid and nitric acid at the same pH. The increase that was observed with oxalic acid is comparable to that reported by Olsen and Rimstidt [51].
- The dissolution rates of nepheline syenite at pH 4.0 increased two times with oxalic acid and two to four times with citric acid compared to hydrochloric acid and nitric acid at the same pH.
- The dissolution rates of ilmenite norite at pH 4.0 increased four times with oxalic acid, after a delay of about three days, and on average 2.5 times with citric acid compared to hydrochloric acid and nitric acid at the same pH.
- For nephline syenite and ilmenite norite, the effects of changing to organic acids was different for different elements, which suggests that a ligand effect is the main reason for the change in the dissolution rate (rather than increased H+ ion availability through buffering effects). Åheim dunite dissolution rates were likely enhanced by ligand effects (cf. [51]).
Based on our observations and the calculated H+ uptake rates, we concluded that while the application of silicate powders as liming agents would have several benefits over traditional liming agents, it may not be feasible due to the relatively slow neutralization kinetics. To overcome these slow kinetics, larger quantities of finer powders would have to be used. The application of larger volumes of slower-reacting silicates may have the benefit of reducing the required frequency of liming.
Author Contributions
Conceptualization, P.T.M.; Methodology, R.v.N.; Validation, R.v.N.; Formal Analysis, R.v.N.; Investigation, R.v.N. and S.H.D.; Resources, P.T.M. and S.H.D.; Data Curation, R.v.N.; Writing-Original Draft Preparation, R.v.N.; Writing-Review & Editing, R.v.N. and P.T.M.; Visualization, R.v.N.; Funding Acquisition, P.T.M.
Funding
This research was funded by the Research Council of Norway, under grant 234382: Mitigation of greenhouse gas emission from cropped soils by mafic mineral applications.
Acknowledgments
The authors would like to thank the three anonymous reviewers for their thorough reviews, that helped improve our final paper.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Bergaust, L.; Mao, Y.; Bakken, L.R.; Frostegard, A. Denitrification Response Patterns during the Transition to Anoxic Respiration and Posttranscriptional Effects of Suboptimal pH on Nitrogen Oxide Reductase in Paracoccus denitrificans. Appl. Environ. Microbiol. 2010, 76, 6387–6396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuhel, J.; Simek, M.; Laughlin, R.J.; Bru, D.; Cheneby, D.; Watson, C.J.; Philippot, L. Insights into the Effect of Soil pH on N2O and N2 Emissions and Denitrifier Community Size and Activity. Appl. Environ. Microbiol. 2010, 76, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Mørkved, P.T.; Frostegård, A.; Bakken, L.R. Denitrification gene pools, transcription and kinetics of NO, N2O and N2 production as affected by soil pH. FEMS Microbiol. Ecol. 2010, 72, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Bakken, L.R.; Bergaust, L.; Liu, B.; Frostegard, A. Regulation of denitrification at the cellular level: A clue to the understanding of N2O emissions from soils. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- West, T.O.; McBride, A.C. The contribution of agricultural lime to carbon dioxide emissions in the United States: Dissolution, transport, and net emissions. Agric. Ecosyst. Environ. 2005, 108, 145–154. [Google Scholar] [CrossRef]
- Schuiling, R.D.; Krijgsman, P. Enhanced Weathering: An Effective and Cheap Tool to Sequester CO2. Clim. Chang. 2006, 74, 349–354. [Google Scholar] [CrossRef]
- Koehler, P.; Hartmann, J.; Wolf-Gladrow, D.A. Geoengineering potential of artificially enhanced silicate weathering of olivine. Proc. Natl. Acad. Sci. USA 2010, 107, 20228–20233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff-Boenisch, D.; Wenau, S.; Gislason, S.R.; Oelkers, E.H. Dissolution of basalts and peridotite in seawater, in the presence of ligands, and CO2: Implications for mineral sequestration of carbon dioxide. Geochim. Cosmochim. Acta 2011, 75, 5510–5525. [Google Scholar] [CrossRef]
- Ten Berge, H.F.M.; van der Meer, H.G.; Steenhuizen, J.W.; Goedhart, P.W.; Knops, P.; Verhagen, J. Olivine Weathering in Soil, and Its Effects on Growth and Nutrient Uptake in Ryegrass (Lolium perenne L.): A Pot Experiment. PLoS ONE 2012, 7, e42098. [Google Scholar] [CrossRef] [PubMed]
- Welch, S.A.; Ullman, W.J. The effect of organic acids on plagioclase dissolution rates and stoichiometry. Geochim. Cosmochim. Acta 1993, 57, 2725–2736. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Gislason, S.R. The mechanism, rates and consequences of basaltic glass dissolution: I. An experimental study of the dissolution rates of basaltic glass as a function of aqueous Al, Si and oxalic acid concentration at 25 °C and pH = 3 and 11. Geochim. Cosmochim. Acta 2001, 65, 3671–3681. [Google Scholar] [CrossRef]
- Pokrovsky, O.S.; Golubev, S.V.; Jordan, G. Effect of organic and inorganic ligands on calcite and magnesite dissolution rates at 60 °C and 30 atm pCO2. Chem. Geol. 2009, 265, 33–43. [Google Scholar] [CrossRef]
- Declercq, J.; Bosc, O.; Oelkers, E.H. Do organic ligands affect forsterite dissolution rates? Appl. Geochem. 2013, 39, 69–77. [Google Scholar] [CrossRef]
- Bray, A.W.; Oelkers, E.H.; Bonneville, S.; Wolff-Boenisch, D.; Potts, N.J.; Fones, G.; Benning, L.G. The effect of pH, grain size, and organic ligands on biotite weathering rates. Geochim. Cosmochim. Acta 2015, 164, 127–145. [Google Scholar] [CrossRef] [Green Version]
- Oelkers, E.H.; Benning, L.G.; Lutz, S.; Mavromatis, V.; Pearce, C.R.; Plümper, O. The efficient long-term inhibition of forsterite dissolution by common soil bacteria and fungi at Earth surface conditions. Geochim. Cosmochim. Acta 2015, 168, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Dove, P.M.; Crerar, D.A. Kinetics of quartz dissolution in electrolyte solutions using a hydrothermal mixed flow reactor. Geochim. Cosmochim. Acta 1990, 54, 955–969. [Google Scholar] [CrossRef]
- Dove, P.M. The dissolution kinetics of quartz in sodium chloride solutions at 25 degrees to 300 degrees C. Am. J. Sci. 1994, 294, 665–712. [Google Scholar] [CrossRef]
- Gautier, J.-M.; Oelkers, E.H.; Schott, J. Experimental study of K-feldspar dissolution rates as a function of chemical affinity at 150 °C and pH 9. Geochim. Cosmochim. Acta 1994, 58, 4549–4560. [Google Scholar] [CrossRef]
- Chen, Y.; Brantley, S.L. Temperature- and pH-dependence of albite dissolution rate at acid pH. Chem. Geol. 1997, 135, 275–290. [Google Scholar] [CrossRef]
- Dove, P.M.; Nix, C.J. The influence of the alkaline earth cations, magnesium, calcium, and barium on the dissolution kinetics of quartz. Geochim. Cosmochim. Acta 1997, 61, 3329–3340. [Google Scholar] [CrossRef]
- Malmström, M.; Banwart, S. Biotite dissolution at 25 °C: The pH dependence of dissolution rate and stoichiometry. Geochim. Cosmochim. Acta 1997, 61, 2779–2799. [Google Scholar] [CrossRef]
- Dove, P.M. The dissolution kinetics of quartz in aqueous mixed cation solutions. Geochim. Cosmochim. Acta 1999, 63, 3715–3727. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Schott, J. Experimental study of anorthite dissolution and the relative mechanism of feldspar hydrolysis. Geochim. Cosmochim. Acta 1995, 59, 5039–5053. [Google Scholar] [CrossRef]
- Oelkers, E.H.; Schott, J. An experimental study of enstatite dissolution rates as a function of pH, temperature, and aqueous Mg and Si concentration, and the mechanism of pyroxene/pyroxenoid dissolution. Geochim. Cosmochim. Acta 2001, 65, 1219–1231. [Google Scholar] [CrossRef]
- Icenhower, J.P.; Dove, P.M. The dissolution kinetics of amorphous silica into sodium chloride solutions: Effects of temperature and ionic strength. Geochim. Cosmochim. Acta 2000, 64, 4193–4203. [Google Scholar] [CrossRef]
- Pokrovsky, O.S.; Schott, J. Forsterite surface composition in aqueous solutions: A combined potentiometric, electrokinetic, and spectroscopic approach. Geochim. Cosmochim. Acta 2000, 64, 3299–3312. [Google Scholar] [CrossRef]
- Pokrovsky, O.S.; Schott, J. Kinetics and mechanism of forsterite dissolution at 25 °C and pH from 1 to 12. Geochim. Cosmochim. Acta 2000, 64, 3313–3325. [Google Scholar] [CrossRef]
- Oelkers, E.H. An experimental study of forsterite dissolution rates as a function of temperature and aqueous Mg and Si concentrations. Chem. Geol. 2001, 175, 485–494. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Brantley, S.L.; Pantano, C.G.; Criscenti, L.J.; Kubicki, J.D. Dissolution of nepheline, jadeite and albite glasses: Toward better models for aluminosilicate dissolution. Geochim. Cosmochim. Acta 2001, 65, 3683–3702. [Google Scholar] [CrossRef]
- Golubev, S.V.; Pokrovsky, O.S.; Schott, J. Experimental determination of the effect of dissolved CO2 on the dissolution kinetics of Mg and Ca silicates at 25 °C. Chem. Geol. 2005, 217, 227–238. [Google Scholar] [CrossRef]
- Lowson, R.T.; Brown, P.L.; Josick Comarmond, M.-C.; Rajaratnam, G. The kinetics of chlorite dissolution. Geochim. Cosmochim. Acta 2007, 71, 1431–1447. [Google Scholar] [CrossRef]
- Marini, L. Geological Sequestration of Carbon Dioxide Thermodynamics, Kinetics and Reaction Path Modelling. Dev. Geochem. 2007, 11, 453. [Google Scholar]
- Sverdrup, H.; Warfvinge, P. Estimating field weathering rates using laboratory kinetics. Rev. Miner. Geochem. 1995, 31, 485–539. [Google Scholar]
- White, A.F.; Brantley, S.L. The effect of time on the weathering of silicate minerals: Why do weathering rates differ in the laboratory and field? Chem. Geol. 2003, 202, 479–506. [Google Scholar] [CrossRef]
- Béarat, H.; McKelvy, M.J.; Chizmeshya, A.V.G.; Gormley, D.; Nunez, R.; Carpenter, R.W.; Squires, K.; Wolf, G.H. Carbon Sequestration via Aqueous Olivine Mineral Carbonation: Role of Passivating Layer Formation. Environ. Sci. Technol. 2006, 40, 4802–4808. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Agudo, E.; Putnis, C.V.; Rodriguez-Navarro, C.; Putnis, A. Mechanism of leached layer formation during chemical weathering of silicate minerals. Geology 2012, 40, 947–950. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Putnis, C.V.; Putnis, A. Coupled dissolution and precipitation at mineral-fluid interfaces. Chem. Geol. 2014, 383, 132–146. [Google Scholar] [CrossRef]
- Hochella, M.F.; Banfield, J.F. Chemical weathering of silicates in nature; a microscopic perspective with theoretical considerations. Rev. Mineral. Geochem. 1995, 31, 353–406. [Google Scholar]
- Lee, M.R.; Parsons, I. Microtextural controls of weathering of perthitic alkali feldspars. Geochim. Cosmochim. Acta 1995, 59, 4465–4488. [Google Scholar] [CrossRef]
- White, A.F. Chemical weathering rates of silicate minerals in soils. Rev. Mineral. Geochem. 1995, 31, 407–458. [Google Scholar]
- Jones, D.L. Organic acids in the rhizosphere—A critical review. Plant Soil 1998, 205, 25–44. [Google Scholar] [CrossRef]
- Jones, D.L.; Hodge, A.; Kuzyakov, Y. Plant and mycorrhizal regulation of rhizodeposition. New Phytol. 2004, 163, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Cieslinski, G.; van Rees, K.C.J.; Szmigielska, A.M.; Huang, P.M. Low molecular weight organic acids released from roots of durum wheat and flax into sterile nutrient solutions. J. Plant Nutr. 2008, 20, 753–764. [Google Scholar] [CrossRef]
- Sandnes, A.; Eldhuset, T.D.; Wollebæk, G. Organic acids in root exudates and soil solution of Norway spruce and silver birch. Soil Biol. Biochem. 2005, 37, 259–269. [Google Scholar] [CrossRef]
- Vranová, V.; Rejšek, K.; Formánek, P. Aliphatic, Cyclic, and Aromatic Organic Acids, Vitamins, and Carbohydrates in Soil: A Review. Sci. World J. 2013, 2013, 524239. [Google Scholar] [CrossRef] [PubMed]
- Gadd, G.M. Fungal Production of Citric and Oxalic Acid: Importance in Metal Speciation, Physiology and Biogeochemical Processes. Adv. Microb. Physiol. 1999, 41, 47–92. [Google Scholar] [PubMed]
- Dakora, F.D.; Phillips, D.A. Root exudates as mediators of mineral acquisition in low-nutrient environments. Plant Soil 2002, 245, 35–47. [Google Scholar] [CrossRef]
- El Zahar Haichar, F.; Santaella, C.; Heulin, T.; Achouak, W. Root exudates mediated interactions belowground. Soil Biol. Biochem. 2014, 77, 69–80. [Google Scholar] [CrossRef]
- Hinsinger, P.; Barros, O.N.F.; Benedetti, M.F.; Noack, Y.; Callot, G. Plant-induced weathering of a basaltic rock: Experimental evidence. Geochim. Cosmochim. Acta 2001, 65, 137–152. [Google Scholar] [CrossRef]
- Golubev, S.V.; Pokrovsky, O.S. Experimental study of the effect of organic ligands on diopside dissolution kinetics. Chem. Geol. 2006, 235, 377–389. [Google Scholar] [CrossRef]
- Olsen, A.A.; Rimstidt, J.D. Oxalate-promoted forsterite dissolution at low pH. Geochim. Cosmochim. Acta 2008, 72, 1758–1766. [Google Scholar] [CrossRef]
- Huang, W.H.; Kiang, W.C. Laboratory dissolution of plagioclase feldspars in water and organic acids at room temperature. Am. Mineral. 1972, 57, 1849–1859. [Google Scholar]
- Strand, T. Lillebukt Alkaline Kompleks: Karbonatittens Mineralogi og Petrokjemi. Ph.D. Thesis, University of Bergen, Bergen, Norway, 1981; 287p. (In Norwegian). [Google Scholar]
- Mjelde, Ø. Geologi og Petrografi av Nabberen Nefelinsyenitt. Ph.D. Thesis, University of Bergen, Bergen, Norway, 1983; 315p. (In Norwegian). [Google Scholar]
- Olsen, A.A. Forsterite Dissolution Kinetics: Applications and Implications for Chemical Weathering. Ph.D. Thesis, Virginia Polytechnic Institute and State University, Blacksburg, VI, USA, 2007. [Google Scholar]
- Tole, M.P.; Lasaga, A.C.; Pantano, C.; White, W.B. The kinetics of dissolution of nepheline (NaAlSiO4). Geochim. Cosmochim. Acta 1986, 50, 379–392. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, X.; Guy, B.; Hu, S.; de Ligny, D.; Moutte, J. Experimental study of dissolution rates of hedenbergitic clinopyroxene at high temperatures: Dissolution in water from 25 °C to 374 °C. Eur. J. Mineral. 2013, 25, 353–372. [Google Scholar] [CrossRef]
- Pokrovsky, O.S.; Golubev, S.V.; Schott, J. Dissolution kinetics of calcite, dolomite and magnesite at 25 °C and 0 to 50 atm pCO2. Chem. Geol. 2005, 217, 239–255. [Google Scholar] [CrossRef]
- Franzefoss Miljøkalk as Agri Dol Product Sheet. Available online: https://debio.no/content/uploads/2017/02/AgriDol_L-2.pdf (accessed on 17 September 2018).
Figure 1.
Schematic drawing of the flow-through cells used in this study.

Figure 2.
Major element release rates per sample plotted against experimental duration, for Åheim dunite (a,b), nepheline syenite (c,d), and ilmenite norite (e,f). Colored bands illustrate when different acids were used (blue = hydrochloric; red = oxalic; green = citric; orange is nitric acid). The black line at 51 days indicates when the experiment was paused, and the cells were filled with stagnant Milli-Q water for 48 days. The red line in (e) indicates when the sample was replaced with fresh unreacted powder.
Figure 2.
Major element release rates per sample plotted against experimental duration, for Åheim dunite (a,b), nepheline syenite (c,d), and ilmenite norite (e,f). Colored bands illustrate when different acids were used (blue = hydrochloric; red = oxalic; green = citric; orange is nitric acid). The black line at 51 days indicates when the experiment was paused, and the cells were filled with stagnant Milli-Q water for 48 days. The red line in (e) indicates when the sample was replaced with fresh unreacted powder.

Figure 3.
SEM micrographs of unreacted (a–c) and reacted (d–h) Åheim dunite grains. Both loose grains (a,b,d,f–h) and polished sections (c,e) are shown. In (d,e), note the deep channels caused by dissolution. In (f), note the crystallographically controlled etch pits. Finally, (e,g) show thin layers on grain surfaces, which were most likely formed by precipitation.
Figure 3.
SEM micrographs of unreacted (a–c) and reacted (d–h) Åheim dunite grains. Both loose grains (a,b,d,f–h) and polished sections (c,e) are shown. In (d,e), note the deep channels caused by dissolution. In (f), note the crystallographically controlled etch pits. Finally, (e,g) show thin layers on grain surfaces, which were most likely formed by precipitation.

Figure 4.
SEM micrographs of unreacted (a–c) and reacted (d–j) nepheline syenite grains. Both loose grains (a,d–g) and polished sections (b,c,h–j) are shown. Preferential dissolution along exsolution lamellae is shown in (d,e). Images (d–f) show crystallographically controlled roughening at mineral surfaces. Thin layers formed by precipitation or leaching are shown in (g). In images (h–j), note the roughening of grain surfaces, and the removal of mineral phases from grains, resulting in cavities in these grains.
Figure 4.
SEM micrographs of unreacted (a–c) and reacted (d–j) nepheline syenite grains. Both loose grains (a,d–g) and polished sections (b,c,h–j) are shown. Preferential dissolution along exsolution lamellae is shown in (d,e). Images (d–f) show crystallographically controlled roughening at mineral surfaces. Thin layers formed by precipitation or leaching are shown in (g). In images (h–j), note the roughening of grain surfaces, and the removal of mineral phases from grains, resulting in cavities in these grains.


Figure 5.
SEM micrographs of unreacted (a,b) and reacted (c–h) ilmenite norite grains. Both loose grains (a,c–e) and polished sections (b,f–h) are shown. In (c,d), note the etching along Fe-enriched exsolution lamellae in ilmenite. Image (e) shows effects of dissolution on a biotite surface, where leaching or precipitation resulted in the formation of a thin layer. The cross-sections (f–h) show some cavities resulting from the removal of mineral phases. The light phase in (g) shows minor surface roughening due to preferential dissolution.
Figure 5.
SEM micrographs of unreacted (a,b) and reacted (c–h) ilmenite norite grains. Both loose grains (a,c–e) and polished sections (b,f–h) are shown. In (c,d), note the etching along Fe-enriched exsolution lamellae in ilmenite. Image (e) shows effects of dissolution on a biotite surface, where leaching or precipitation resulted in the formation of a thin layer. The cross-sections (f–h) show some cavities resulting from the removal of mineral phases. The light phase in (g) shows minor surface roughening due to preferential dissolution.

Figure 6.
Apparent mineral dissolution rates for olivine in Åheim dunite. Graph (a) shows the rates calculated per individual element released, while (b) shows the averaged rates per sample. Note the slight decrease in dissolution rate during the first ~30 days, after which the rate stabilized.
Figure 6.
Apparent mineral dissolution rates for olivine in Åheim dunite. Graph (a) shows the rates calculated per individual element released, while (b) shows the averaged rates per sample. Note the slight decrease in dissolution rate during the first ~30 days, after which the rate stabilized.

Figure 7.
Apparent mineral dissolution rates for nepheline in nepheline syenite. Graph (a) shows the rates calculated per individual element released, while (b) shows the averaged rates per sample. Note the decline in dissolution rate starting after ~20 days.
Figure 7.
Apparent mineral dissolution rates for nepheline in nepheline syenite. Graph (a) shows the rates calculated per individual element released, while (b) shows the averaged rates per sample. Note the decline in dissolution rate starting after ~20 days.

Figure 8.
H+-uptake rates calculated per sample, based on measured cation release rates, plotted against time, for (a) Åheim dunite, (b) nepheline syenite, and (c) ilmenite norite.
Figure 8.
H+-uptake rates calculated per sample, based on measured cation release rates, plotted against time, for (a) Åheim dunite, (b) nepheline syenite, and (c) ilmenite norite.



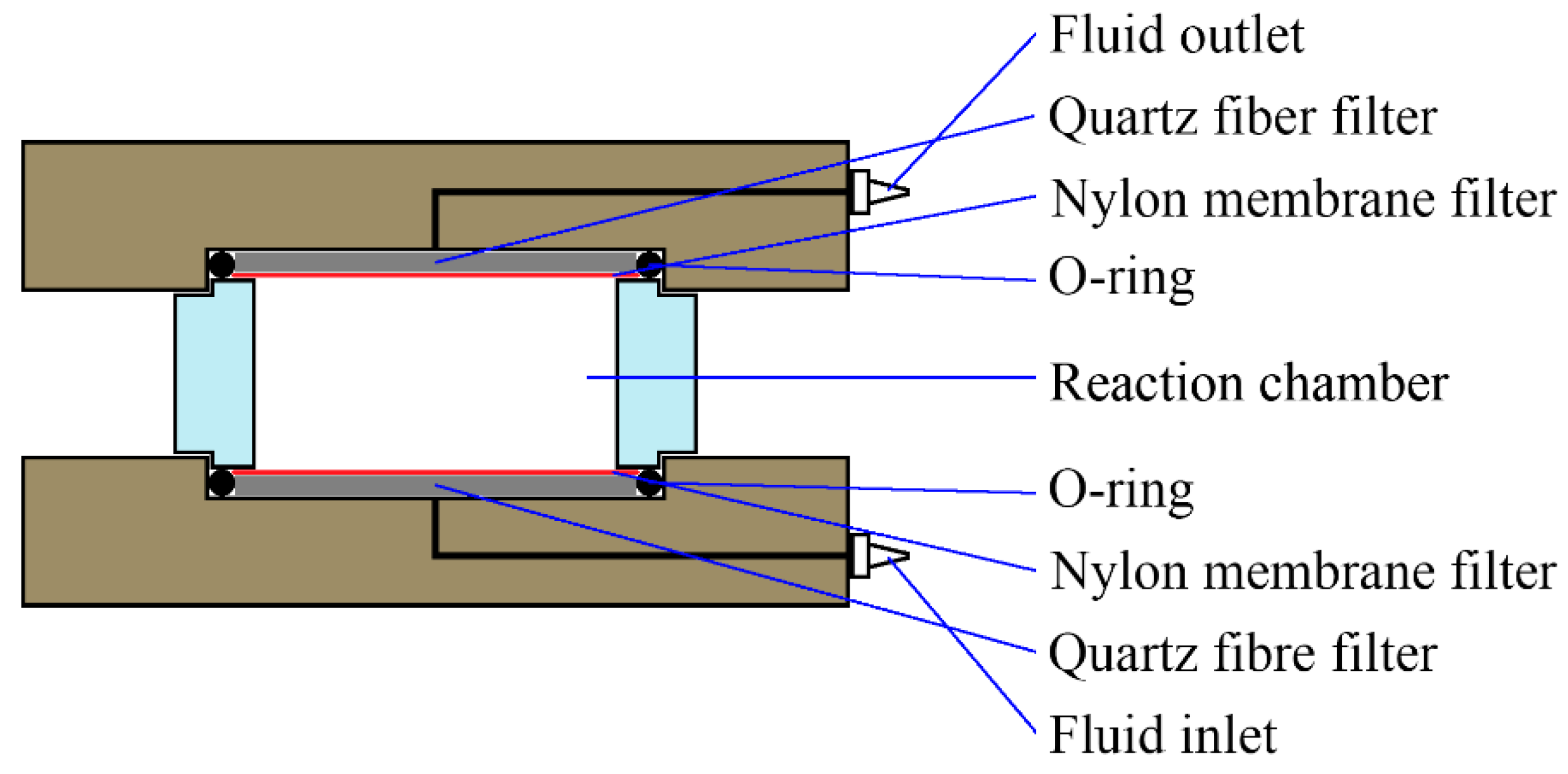{kind=link}
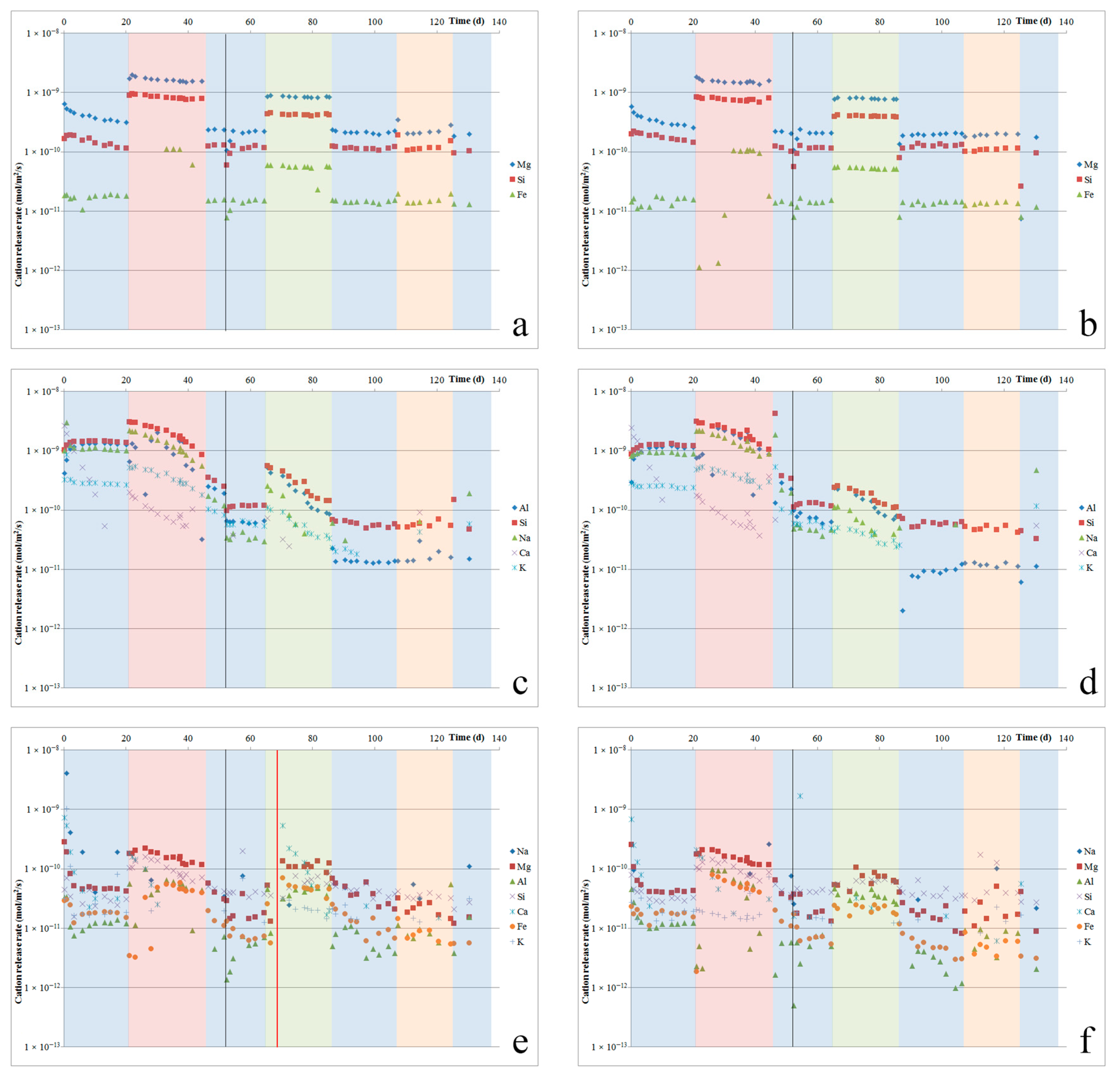{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
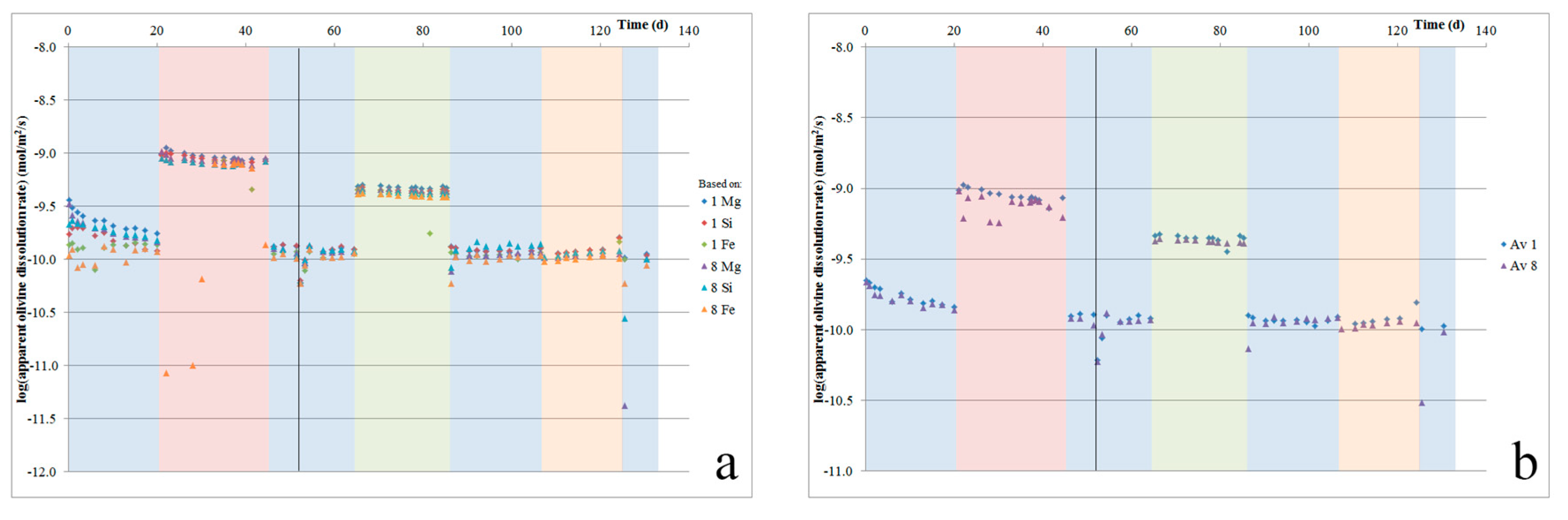{kind=link}
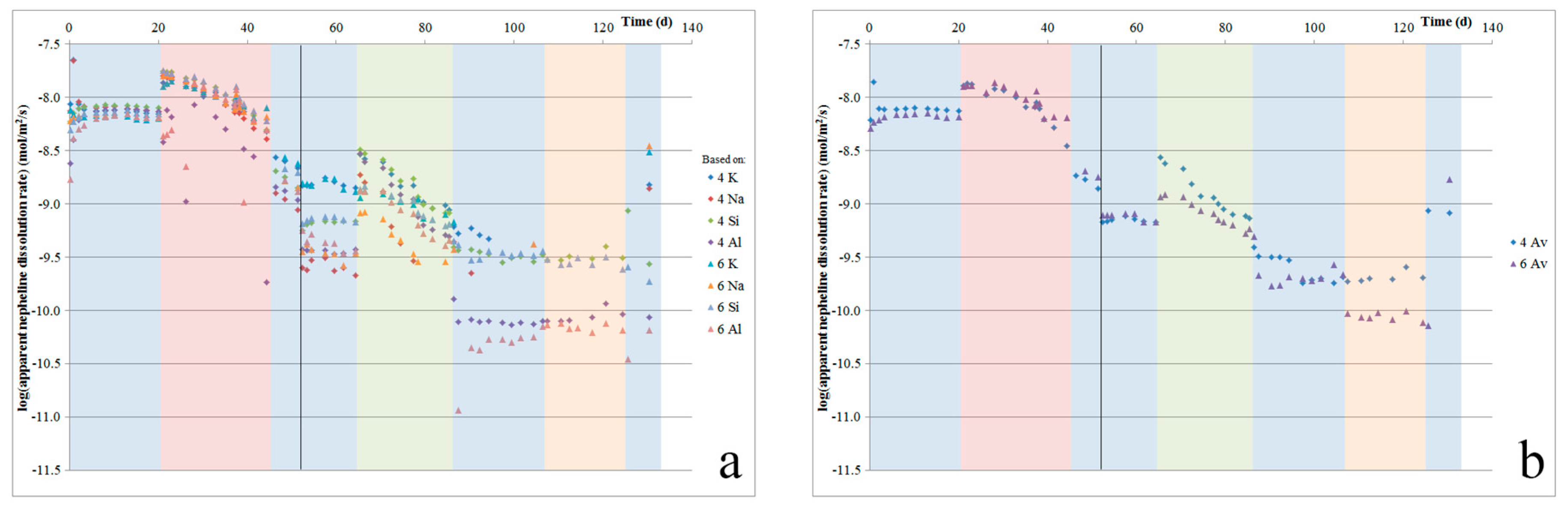{kind=link}
{kind=link}
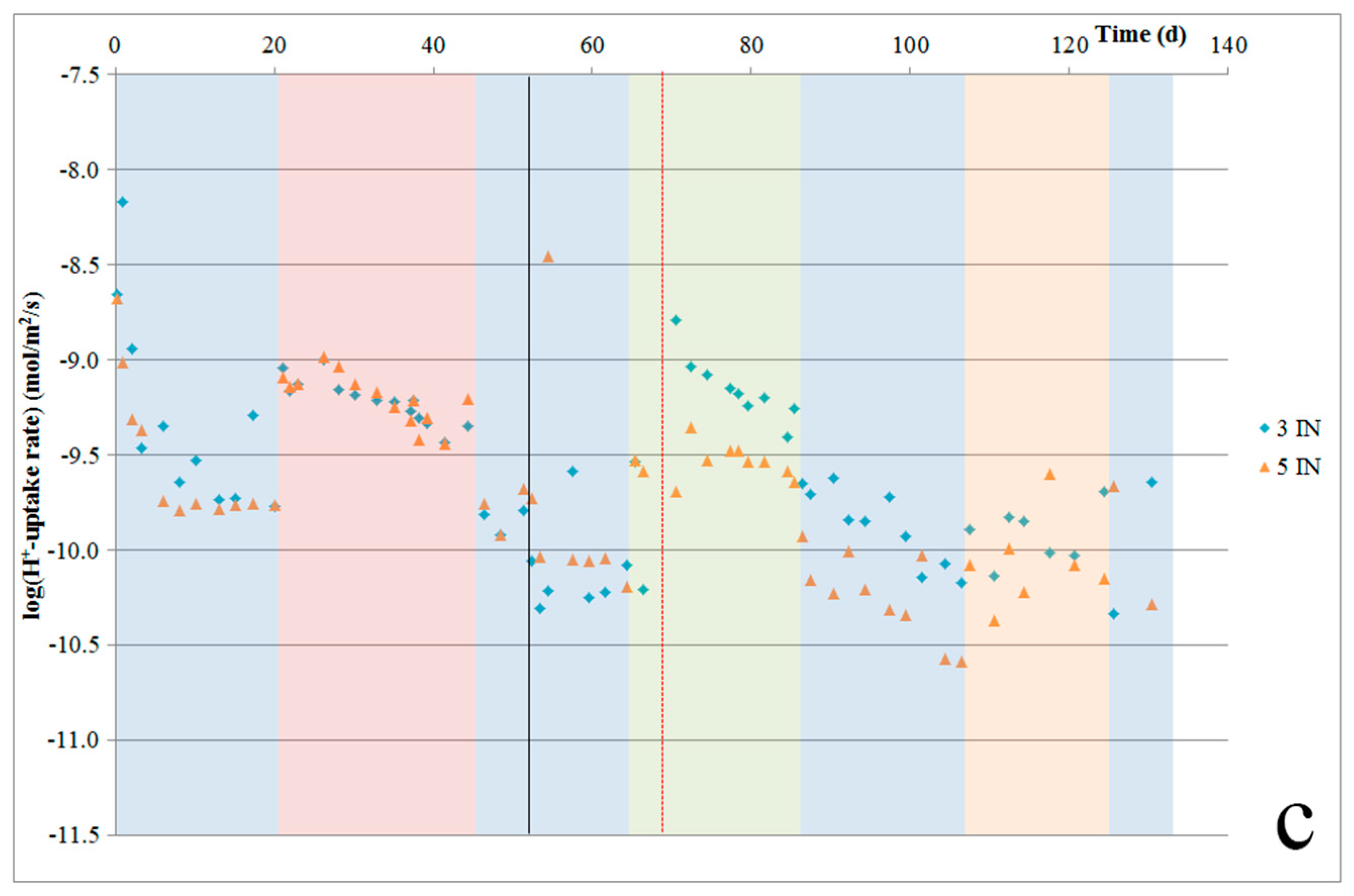{kind=link}
Table 1.
Major element bulk chemical compositions of the silicate powders.
| Sample | SiO2 | Al2O3 | Fe2O3 | MgO | CaO | Na2O | K2O | TiO2 | P2O5 | MnO | Cr2O3 | LOI | Sum |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| % | % | % | % | % | % | % | % | % | % | % | % | % | |
| Åheim dunite | 43.56 | 0.35 | 7.21 | 47.37 | 0.06 | <0.01 | 0.02 | <0.01 | <0.01 | 0.09 | 0.2 | 0 | 98.86 |
| Nepheline syenite | 46.76 | 18.94 | 8.78 | 3.41 | 6.34 | 6.08 | 5.31 | 1.89 | 0.13 | 0.22 | 0.01 | 1.6 | 99.47 |
| Ilmenite norite | 41.85 | 16.37 | 15.21 | 5.72 | 6.04 | 3.33 | 0.77 | 10.13 | 0.2 | 0.1 | 0.032 | −0.1 | 99.652 |
Table 2.
Trace element contents of the silicate powders in parts per million (ppm).
| Sample | Ba | Ni | Sc | Co | Cs | Rb | Sr | Th | U | V | Y | Cu | Pb | Zn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ppm | ppm | ppm | ppm | ppm | ppm | ppm | ppm | ppm | ppm | ppm | ppm | ppm | ppm | |
| Åheim dunite | 13 | 2507 | 5 | 112.1 | 0.2 | 0.9 | 7.1 | <0.2 | <0.1 | <8 | <0.1 | 6.4 | 0.3 | 28 |
| Nepheline syenite | 1301 | 32 | 8 | 16 | 0.5 | 92.9 | 1584.5 | 1.8 | 0.4 | 129 | 21.6 | 13.4 | 0.5 | 41 |
| Ilmenite norite | 262 | 236 | 12 | 66.9 | <0.1 | 9.2 | 662.7 | 0.5 | 0.2 | 336 | 7.9 | 92.2 | 0.6 | 17 |
Table 3.
Specific surface areas (SSA) determined by BET.
| Sample | SSA |
|---|---|
| m2/g | |
| Åheim dunite | 0.31141 |
| Nepheline syenite | 0.1836 |
| Ilmenite norite | 0.28542 |
Table 4.
Total potential H+ uptake per silicate powder based on major elements compositions.
| mol/kg | % Rel. to Calcite | |
|---|---|---|
| Åheim dunite | 27.56 | 138 |
| Nepheline syenite | 22.17 | 111 |
| Ilmenite norite | 25.12 | 126 |
| Calcite | 19.98 | 100 |
| Dolomite ((Ca,Mg)CO3) | 21.69 | 109 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Van Noort, R.; Mørkved, P.T.; Dundas, S.H. Acid Neutralization by Mining Waste Dissolution under Conditions Relevant for Agricultural Applications. Geosciences 2018, 8, 380. https://doi.org/10.3390/geosciences8100380
AMA Style
Van Noort R, Mørkved PT, Dundas SH. Acid Neutralization by Mining Waste Dissolution under Conditions Relevant for Agricultural Applications. Geosciences. 2018; 8(10):380. https://doi.org/10.3390/geosciences8100380
Chicago/Turabian StyleVan Noort, Reinier, Pål Tore Mørkved, and Siv Hjorth Dundas. 2018. "Acid Neutralization by Mining Waste Dissolution under Conditions Relevant for Agricultural Applications" Geosciences 8, no. 10: 380. https://doi.org/10.3390/geosciences8100380
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.