
High Diversity of Long Terminal Repeat Retrotransposons in Compact Vertebrate Genomes: Insights from Genomes of Tetraodontiformes
 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
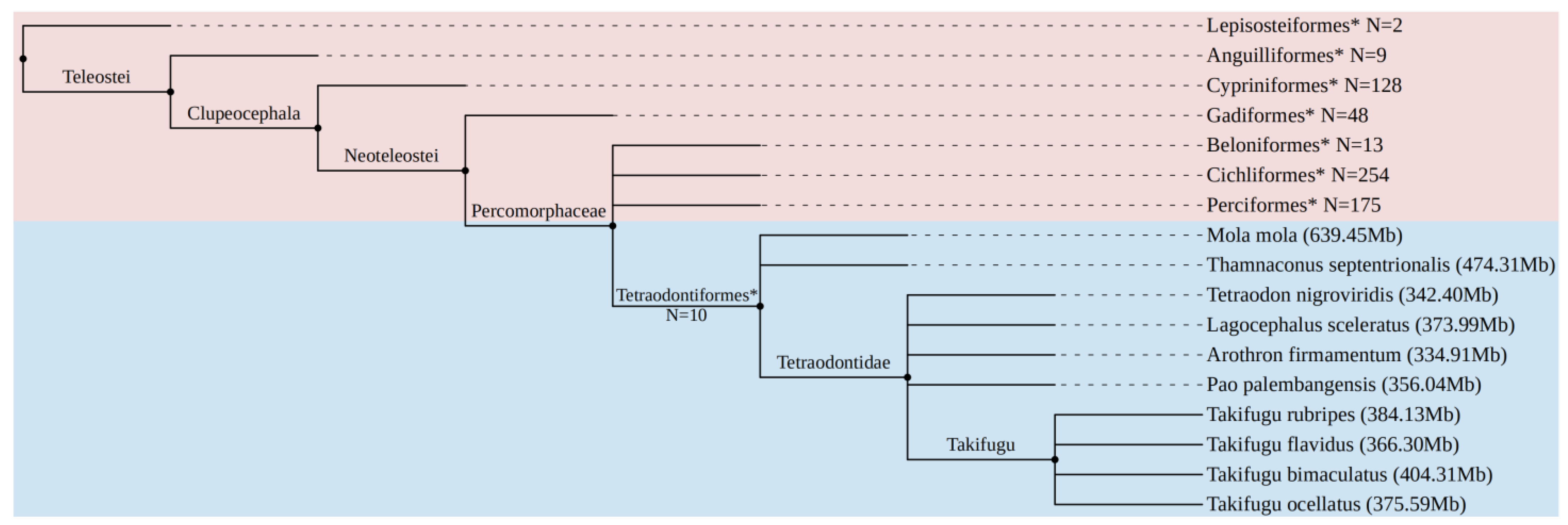
2.1. Genomes Used and LTR Retrotransposon Mining
2.2. Structure and Sequence Analysis of Retrotransposons and Proteins
2.3. Construction of Phylogenetic Tree
2.4. Evolution Activity of Retrotransposon
2.5. Theory/Calculation
3. Results
3.1. LTR-RTN Mining in Tetraodontiformes
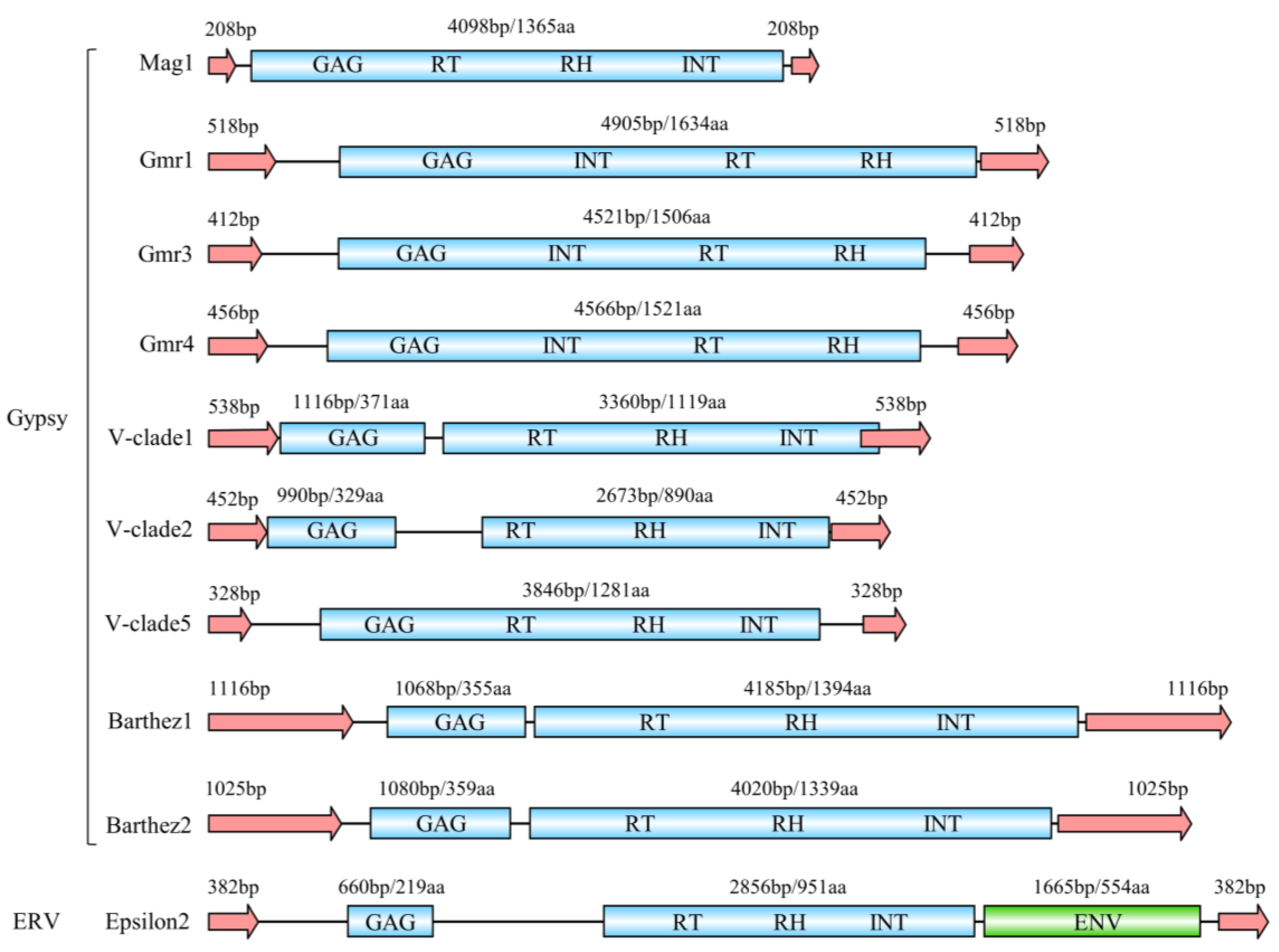
3.2. Classification and Structure Organization of LTR-RTNs in Tetraodontiformes
3.3. Distribution of LTR Families in Compact Genomes of Vertebrates
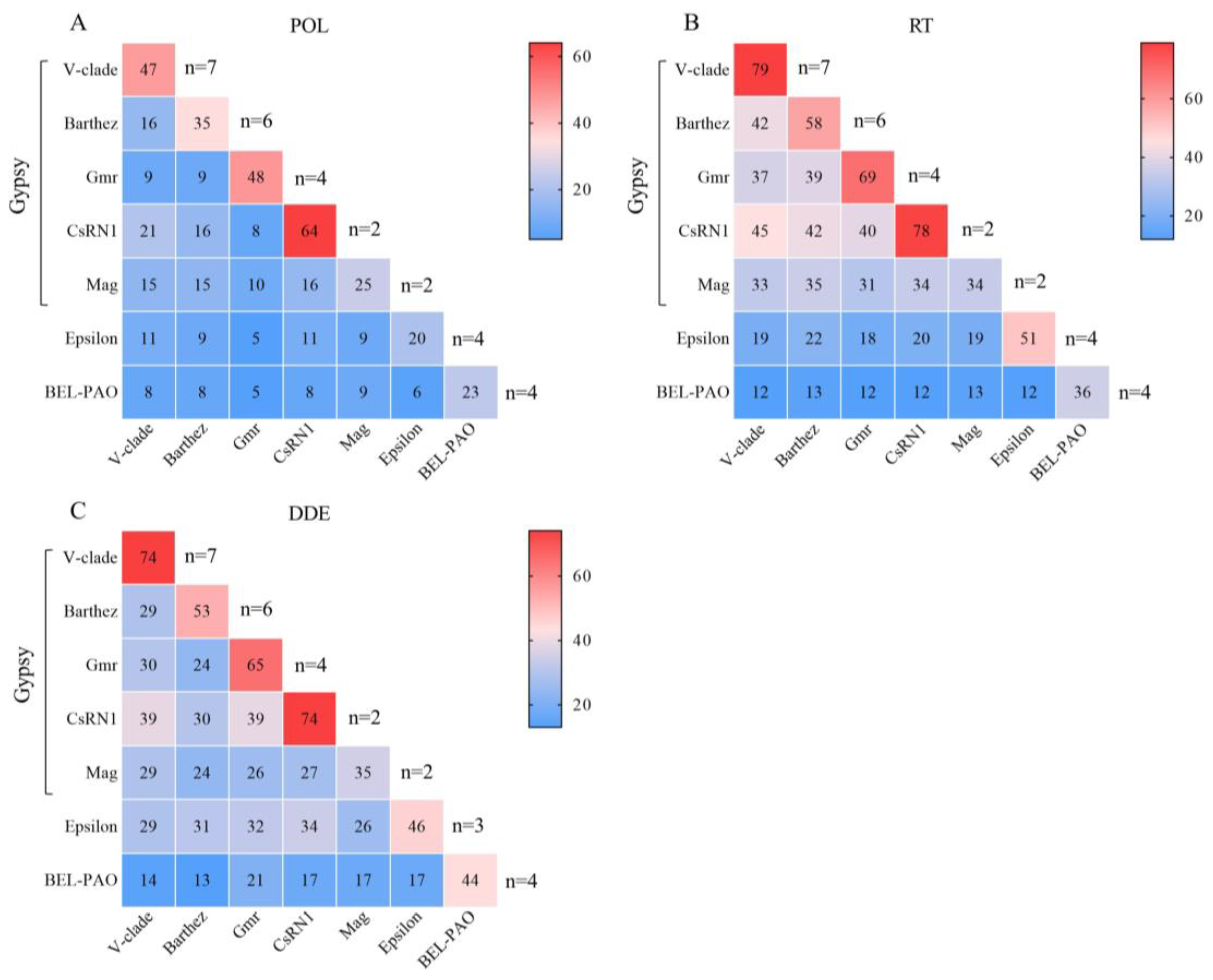
3.4. Protein Sequence Analysis
3.5. Evolution Dynamics of LTR in Compact Genomes of Vertebrates
4. Discussion
4.1. Diversity
4.2. Distribution and Abundances
4.3. Structure and Evolution Activity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Christian, M.L.; Dapp, M.J.; Scharffenberger, S.C.; Jones, H.; Song, C.; Frenkel, L.M.; Krumm, A.; Mullins, J.I.; Rawlings, D.J. CRISPR/Cas9-mediated insertion of HIV long terminal repeat within BACH2 promotes expansion of T regulatory–like cells. J. Immunol. 2022, 208, 1700–1710. [Google Scholar] [CrossRef]
- Benachenhou, F.; Sperber, G.O.; Bongcam-Rudloff, E.; Andersson, G.; Boeke, J.D.; Blomberg, J. Conserved structure and inferred evolutionary history of long terminal repeats (LTRs). Mob. DNA 2013, 4, 5. [Google Scholar] [CrossRef]
- Liu, H.-N.; Pei, M.-S.; Ampomah-Dwamena, C.; He, G.-Q.; Wei, T.-L.; Shi, Q.-F.; Yu, Y.-H.; Guo, D.-L. Genome-wide characterization of long terminal repeat retrotransposons provides insights into trait evolution of four cucurbit species. Funct. Integr. Genom. 2023, 23, 218. [Google Scholar] [CrossRef] [PubMed]
- Jedlicka, P.; Lexa, M.; Kejnovsky, E. What can long terminal repeats tell us about the age of LTR retrotransposons, gene conversion and ectopic recombination? Front. Plant Sci. 2020, 11, 644. [Google Scholar] [CrossRef] [PubMed]
- Havecker, E.R.; Gao, X.; Voytas, D.F. The diversity of LTR retrotransposons. Genome Biol. 2004, 5, 225. [Google Scholar] [CrossRef] [PubMed]
- Aroh, O.; Halanych, K.M. Genome-wide characterization of LTR retrotransposons in the non-model deep-sea annelid Lamellibrachia luymesi. BMC Genom. 2021, 22, 466. [Google Scholar] [CrossRef]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.-N. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef]
- Chang, N.C.; Rovira, Q.; Wells, J.; Feschotte, C.; Vaquerizas, J.M. Zebrafish transposable elements show extensive diversification in age, genomic distribution, and developmental expression. Genome Res. 2022, 32, 1408–1423. [Google Scholar] [CrossRef]
- Shao, F.; Han, M.; Peng, Z. Evolution and diversity of transposable elements in fish genomes. Sci. Rep. 2019, 9, 15399. [Google Scholar] [CrossRef]
- Gebrie, A. Transposable elements as essential elements in the control of gene expression. Mob. DNA 2023, 14, 9. [Google Scholar] [CrossRef]
- Sotero-Caio, C.G.; Platt, R.N.; Suh, A.; Ray, D.A. Evolution and diversity of transposable elements in vertebrate genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Böhne, A.; Brunet, F.; Galiana-Arnoux, D.; Schultheis, C.; Volff, J.-N. Transposable elements as drivers of genomic and biological diversity in vertebrates. Chromosome Res. 2008, 16, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Neafsey, D.E.; Palumbi, S.R. Genome size evolution in pufferfish: A comparative analysis of diodontid and tetraodontid pufferfish genomes. Genome Res. 2003, 13, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Hinegardner, R.; Rosen, D.E. Cellular DNA content and the evolution of teleostean fishes. Am. Nat. 1972, 106, 621–644. [Google Scholar] [CrossRef]
- Brenner, S.; Elgar, G.; Sanford, R.; Macrae, A.; Venkatesh, B.; Aparicio, S. Characterization of the pufferfish (Fugu) genome as a compact model vertebrate genome. Nature 1993, 366, 265–268. [Google Scholar] [CrossRef]
- Lamatsch, D.; Steinlein, C.; Schmid, M.; Schartl, M. Noninvasive determination of genome size and ploidy level in fishes by flow cytometry: Detection of triploid Poecilia formosa. J. Int. Soc. Anal. Cytol. 2000, 39, 91–95. [Google Scholar] [CrossRef]
- Eryılmaz, L.; Özuluğ, M.; Meriç, N. The smooth pufferfish, Sphoeroides pachygaster (müller & troschel, 1848) (Teleostei: Tetraodontidae), new to the northern Aegean Sea. Zool. Middle East 2003, 28, 125–126. [Google Scholar]
- Volff, J.-N.; Bouneau, L.; Ozouf-Costaz, C.; Fischer, C. Diversity of retrotransposable elements in compact pufferfish genomes. Trends Genet. 2003, 19, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Kosker, A.R.; Özogul, F.; Ayas, D.; Durmus, M.; Ucar, Y.; Regenstein, J.M.; Özogul, Y. Tetrodotoxin levels of three pufferfish species (Lagocephalus sp.) caught in the North-Eastern Mediterranean sea. Chemosphere 2019, 219, 95–99. [Google Scholar] [CrossRef]
- Basu, S.; Hadzhiev, Y.; Petrosino, G.; Nepal, C.; Gehrig, J.; Armant, O.; Ferg, M.; Strahle, U.; Sanges, R.; Müller, F. The Tetraodon nigroviridis reference transcriptome: Developmental transition, length retention and microsynteny of long non-coding RNAs in a compact vertebrate genome. Sci. Rep. 2016, 6, 33210. [Google Scholar] [CrossRef]
- Gao, B.; Shen, D.; Xue, S.; Chen, C.; Cui, H.; Song, C. The contribution of transposable elements to size variations between four teleost genomes. Mob. DNA 2016, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Chernyavskaya, Y.; Zhang, X.; Liu, J.; Blackburn, J. Long-read sequencing of the zebrafish genome reorganizes genomic architecture. BMC Genom. 2022, 23, 116. [Google Scholar] [CrossRef] [PubMed]
- Carducci, F.; null, n.; Biscotti, M.A.; Barucca, M.; null, n.; Canapa, A.; null, n. Transposable elements in vertebrates: Species evolution and environmental adaptation. Eur. Zool. J. 2019, 86, 497–503. [Google Scholar] [CrossRef]
- Zhou, S.-S.; Yan, X.-M.; Zhang, K.-F.; Liu, H.; Xu, J.; Nie, S.; Jia, K.-H.; Jiao, S.-Q.; Zhao, W.; Zhao, Y.-J.; et al. A comprehensive annotation dataset of intact LTR retrotransposons of 300 plant genomes. Sci. Data 2021, 8, 174. [Google Scholar] [CrossRef]
- Brainerd, E.L.; Slutz, S.S.; Hall, E.K.; Phillis, R.W. Patterns of genome size evolution in tetraodontiform fishes. Evolution 2001, 55, 2363–2368. [Google Scholar]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 1–14. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Schemberger, M.; Nascimento, V.; Coan, R.; Ramos, É.; Nogaroto, V.; Ziemniczak, K.; Valente, G.; Moreira-Filho, O.; Martins, C.; Vicari, M. DNA transposon invasion and microsatellite accumulation guide W chromosome differentiation in a Neotropical fish genome. Chromosoma 2019, 128, 547–560. [Google Scholar] [CrossRef]
- Roest Crollius, H.; Jaillon, O.; Dasilva, C.; Ozouf-Costaz, C.; Fizames, C.; Fischer, C.; Bouneau, L.; Billault, A.; Quetier, F.; Saurin, W.; et al. Characterization and repeat analysis of the compact genome of the freshwater pufferfish Tetraodon nigroviridis. Genome Res. 2000, 10, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Elgar, G. Quality not quantity: The pufferfish genome. Hum. Mol. Genet. 1996, 5, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Holcroft, N.I. A molecular test of alternative hypotheses of tetraodontiform (Acanthomorpha: Tetraodontiformes) sister group relationships using data from the RAG1 gene. Mol. Phylogenet. Evol. 2004, 32, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.C.; Orti, G.; Huang, Y.; Sun, Y.; Baldwin, C.C.; Thompson, A.W.; Arcila, D.; Betancur, R.R.; Li, C.; Becker, L.; et al. Comprehensive phylogeny of ray-finned fishes (Actinopterygii) based on transcriptomic and genomic data. Proc. Natl. Acad. Sci. USA 2018, 115, 6249–6254. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-López, M.; García-Pérez, J.L. DNA transposons: Nature and applications in genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Namgoong, S.-Y.; Kim, K.; Saxena, P.; Yang, J.-Y.; Jayaram, M.; Giedroc, D.P.; Harshey, R.M. Mutational analysis of domain IIβ of bacteriophage Mu transposase: Domains IIα and IIβ belongs to different catalytic complementation groups. J. Mol. Biol. 1998, 275, 221–232. [Google Scholar] [CrossRef]
- Sandoval-Villegas, N.; Nurieva, W.; Amberger, M.; Ivics, Z. Contemporary transposon tools: A review and guide through mechanisms and applications of Sleeping Beauty, piggyBac and Tol2 for genome engineering. Int. J. Mol. Sci. 2021, 22, 5084. [Google Scholar] [CrossRef]
- Fischer, C.; Bouneau, L.; Coutanceau, J.P.; Weissenbach, J.; Ozouf-Costaz, C.; Volff, J.N. Diversity and clustered distribution of retrotransposable elements in the compact genome of the pufferfish Tetraodon nigroviridis. Cytogenet. Genome Res. 2005, 110, 522–536. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Al-Shomrani, B.M.; Simenc, M.; Alharbi, S.N.; Alqahtani, F.H.; Al-Fageeh, M.B.; Manee, M.M. Comparative analysis of transposable elements provides insights into genome evolution in the genus Camelus. BMC Genom. 2021, 22, 842. [Google Scholar] [CrossRef] [PubMed]
- Li, S.F.; She, H.B.; Yang, L.L.; Lan, L.N.; Zhang, X.Y.; Wang, L.Y.; Zhang, Y.L.; Li, N.; Deng, C.L.; Qian, W.; et al. Impact of LTR-retrotransposons on genome structure, evolution, and function in Curcurbitaceae species. Int. J. Mol. Sci. 2022, 23, 10158. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Stöck, M.; Kneitz, S.; Klopp, C.; Woltering, J.M.; Adolfi, M.C.; Feron, R.; Prokopov, D.; Makunin, A.; Kichigin, I.; et al. The sterlet sturgeon genome sequence and the mechanisms of segmental rediploidization. Nat. Ecol. Evol. 2020, 4, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Tafalla, C.; Estepa, A.; Coll, J.M. Fish transposons and their potential use in aquaculture. J. Biotechnol. 2006, 123, 397–412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yan, L.; Jiang, J.; Wang, Y.; Jiang, Y.; Yan, T.; Cao, Y. The structure and retrotransposition mechanism of LTR-retrotransposons in the asexual yeast Candida albicans. Virulence 2014, 5, 655–664. [Google Scholar] [CrossRef]
- de Assis, R.; Baba, V.Y.; Cintra, L.A.; Gonçalves, L.S.A.; Rodrigues, R.; Vanzela, A.L.L. Genome relationships and LTR-retrotransposon diversity in three cultivated Capsicum L. (Solanaceae) species. BMC Genom. 2020, 21, 237. [Google Scholar] [CrossRef] [PubMed]
- Papolu, P.K.; Ramakrishnan, M.; Mullasseri, S.; Kalendar, R.; Wei, Q.; Zou, L.H.; Ahmad, Z.; Vinod, K.K.; Yang, P.; Zhou, M. Retrotransposons: How the continuous evolutionary front shapes plant genomes for response to heat stress. Front. Plant Sci. 2022, 13, 1064847. [Google Scholar] [CrossRef]
- Jangam, D.; Feschotte, C.; Betrán, E. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 2017, 33, 817–831. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Common Name | LTR-RTNs Identified | LTR-RTNs <4 kb and <10 kb | Full-Length LTR-RTNs * | Ref. Genome | Genome Size |
|---|---|---|---|---|---|---|
| Arothron firmamentum | Starry Pufferfish | 1224 | 405 | 3 | GCA_016586285.1 | 334.905 |
| Lagocephalus sceleratus | Silver-Cheeked Toadfish | 3389 | 35 | 3 | GCA_911728415.1 | 373.990 |
| Mola mola | Ocean Sunfish | 1125 | 381 | 63 | GCA_001698575.1 | 639.452 |
| Pao palembangensis | South Sumatran Puffer | 1199 | 365 | 65 | GCA_015343265.1 | 356.042 |
| Takifugu bimaculatus | Two-Spot Pufferfish | 3420 | 1062 | 192 | GCA_004026145.2 | 404.312 |
| Takifugu flavidus | Yellow Pufferfish | 2783 | 937 | 124 | GCF_003711565.1 | 366.303 |
| Takifugu ocellatus | Ocellated Pufferfish | 2367 | 678 | 117 | GCA_027382335.1 | 375.589 |
| Takifugu rubripes | Tiger Puffer | 2757 | 861 | 186 | GCF_901000725.2 | 384.127 |
| Tetraodon nigroviridis | Green Spotted Pufferfish | 1192 | 460 | 24 | GCA_000180735.1 | 342.403 |
| Thamnaconus septentrionalis | Northern Round Herring | 2550 | 979 | 42 | GCA_009823395.1 | 474.310 |
| Total | 22,006 | 6163 | 819 |
| Family | Element | Number of Sequences | Length of Sequence | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Copy | Full LTR | Gag | Pol | Env | Consensus (bp) | LTR (bp) | Gag (aa) | Pol (aa) | Gag and Pol (aa) | Env (aa) | ||
| Gmr | 47 | 41 | 40 | 47 | 0 | |||||||
| Gmr1 | 20 | 16 | 19 | 20 | 0 | 6465 | 518 | - | - | 1634 | - | |
| Gmr2 | 6 | 6 | 4 | 6 | 0 | 6058 | 337 | - | - | 1564 | - | |
| Gmr3 | 11 | 10 | 9 | 11 | 0 | 6273 | 412 | - | - | 1506 | - | |
| Gmr4 | 10 | 9 | 8 | 10 | 0 | 6223 | 456 | - | - | 1521 | - | |
| Mag | 24 | 22 | 24 | 23 | 0 | |||||||
| Mag1 | 20 | 18 | 20 | 19 | 0 | 4696 | 208 | - | - | 1365 | - | |
| Mag2 | 4 | 4 | 4 | 4 | 0 | 4813 | 211 | - | - | 1330 | - | |
| V-clade | 112 | 94 | 90 | 111 | 0 | |||||||
| V-clade1 | 55 | 47 | 47 | 55 | 0 | 5558 | 538 | 371 | 1119 | - | - | |
| V-clade2 | 29 | 25 | 25 | 28 | 0 | 5226 | 443 | 329 | 890 | - | - | |
| V-clade3 | 3 | 3 | 2 | 3 | 0 | 5346 | 497 | 326 | 822 | - | - | |
| V-clade4 | 5 | 3 | 1 | 5 | 0 | 6444 | 454 | 309 | 1016 | - | - | |
| V-clade5 | 12 | 9 | 11 | 12 | 0 | 5369 | 328 | - | - | 1281 | - | |
| V-clade6 | 4 | 3 | 1 | 4 | 0 | 5430 | 367 | 306 | 801 | - | - | |
| V-clade7 | 4 | 4 | 3 | 4 | 0 | 5264 | 396 | 414 | 1119 | - | - | |
| CsRN1 | 39 | 38 | 0 | 39 | 0 | |||||||
| CsRN1-1 | 21 | 21 | 0 | 21 | 0 | 4337 | 174 | - | 1061 | - | - | |
| CsRN1-2 | 18 | 17 | 0 | 18 | 0 | 4552 | 302 | - | 1069 | - | - | |
| Barthez | 56 | 38 | 30 | 56 | 0 | |||||||
| Barthez1 | 13 | 11 | 11 | 13 | 0 | 7871 | 1116 | 355 | 1394 | - | - | |
| Barthez2 | 12 | 9 | 8 | 12 | 0 | 7566 | 1025 | 359 | 1339 | - | - | |
| Barthez3 | 4 | 2 | 0 | 4 | 0 | 8756 | 1114 | - | 1563 | - | - | |
| Barthez4 | 5 | 5 | 0 | 5 | 0 | 7513 | 442 | - | 1580 | - | - | |
| Barthez5 | 17 | 7 | 9 | 17 | 0 | 7544 | 259 | 526 | 1580 | - | - | |
| Barthez6 | 5 | 4 | 2 | 5 | 0 | 7310 | 329 | 879 | 1137 | - | - | |
| BEL-PAO | 24 | 20 | 0 | 22 | 0 | |||||||
| BEL1 | 3 | 3 | 0 | 3 | 0 | 7624 | 728 | - | 1434 | - | - | |
| BEL2 | 7 | 5 | 0 | 7 | 0 | 6587 | 536 | - | 1808 | - | - | |
| BEL3 | 10 | 9 | 0 | 9 | 0 | 7341 | 652 | - | 1970 | - | - | |
| PAO1 | 4 | 3 | 0 | 3 | 0 | 5939 | 456 | - | 1617 | - | - | |
| Copia | Copia1 | 4 | 4 | 2 | 4 | 0 | 4794 | 233 | 454 | 653 | - | - |
| Orthoretrovirinae | Orthoretrovirinae1 | 3 | 3 | 0 | 3 | 0 | 8626 | 433 | - | 1108 | - | - |
| Epsilon retrovirus | 134 | 116 | 82 | 125 | 51 | |||||||
| Epsilon1 | 21 | 19 | 11 | 14 | 9 | 8630 | 665 | 728 | 776 | - | 487 | |
| Epsilon2 | 105 | 93 | 65 | 103 | 40 | 8158 | 382 | 219 | 951 | - | 554 | |
| Epsilon3 | 3 | 2 | 2 | 3 | 0 | 7880 | 470 | 576 | 740 | - | - | |
| Epsilon4 | 5 | 2 | 4 | 5 | 2 | 9854 | 377 | 604 | 985 | - | 477 | |
| Superfamilies/Families | Arothron firmamentum | Lagocephalus sceleratus | Mola mola | Pao palembangensis | Takifugu bimaculatus | Takifugu flavidus | Takifugu ocellatus | Takifugu rubripes | Tetraodon nigroviridis | Thamnaconus septentrionalis |
|---|---|---|---|---|---|---|---|---|---|---|
| *Gypsy | 1 | 1 | 3 | 15 | 14 | 12 | 16 | 1 | 1 | |
| Gmr | 1 | 4 | 3 | 4 | 3 | |||||
| Mag | 1 | 1 | 1 | 2 | ||||||
| V-clade | 1 | 3 | 4 | 4 | 3 | 4 | ||||
| CsRN1 | 2 | 2 | 1 | 2 | 1 | |||||
| Barthez | 4 | 4 | 3 | 5 | 1 | |||||
| *ERV | 1 | 3 | 2 | 3 | 2 | 1 | ||||
| Epsilon retrovirus | 3 | 2 | 3 | 2 | 1 | |||||
| Orthoretrovirinae | 1 | |||||||||
| *BEL-PAO | 3 | 2 | 3 | 4 | ||||||
| *Copia | 1 | 1 | 1 | |||||||
| Total | 1 | 1 | 1 | 3 | 21 | 19 | 18 | 23 | 1 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Saleh, A.A.; Yang, N.; Asare, E.; Chen, H.; Wang, Q.; Chen, C.; Song, C.; Gao, B. High Diversity of Long Terminal Repeat Retrotransposons in Compact Vertebrate Genomes: Insights from Genomes of Tetraodontiformes. Animals 2024, 14, 1425. https://doi.org/10.3390/ani14101425
Wang B, Saleh AA, Yang N, Asare E, Chen H, Wang Q, Chen C, Song C, Gao B. High Diversity of Long Terminal Repeat Retrotransposons in Compact Vertebrate Genomes: Insights from Genomes of Tetraodontiformes. Animals. 2024; 14(10):1425. https://doi.org/10.3390/ani14101425
Chicago/Turabian StyleWang, Bingqing, Ahmed A. Saleh, Naisu Yang, Emmanuel Asare, Hong Chen, Quan Wang, Cai Chen, Chengyi Song, and Bo Gao. 2024. "High Diversity of Long Terminal Repeat Retrotransposons in Compact Vertebrate Genomes: Insights from Genomes of Tetraodontiformes" Animals 14, no. 10: 1425. https://doi.org/10.3390/ani14101425