Diversity and Co-Occurrence Patterns of Soil Bacterial and Fungal Communities of Chinese Cordyceps Habitats at Shergyla Mountain, Tibet: Implications for the Occurrence

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
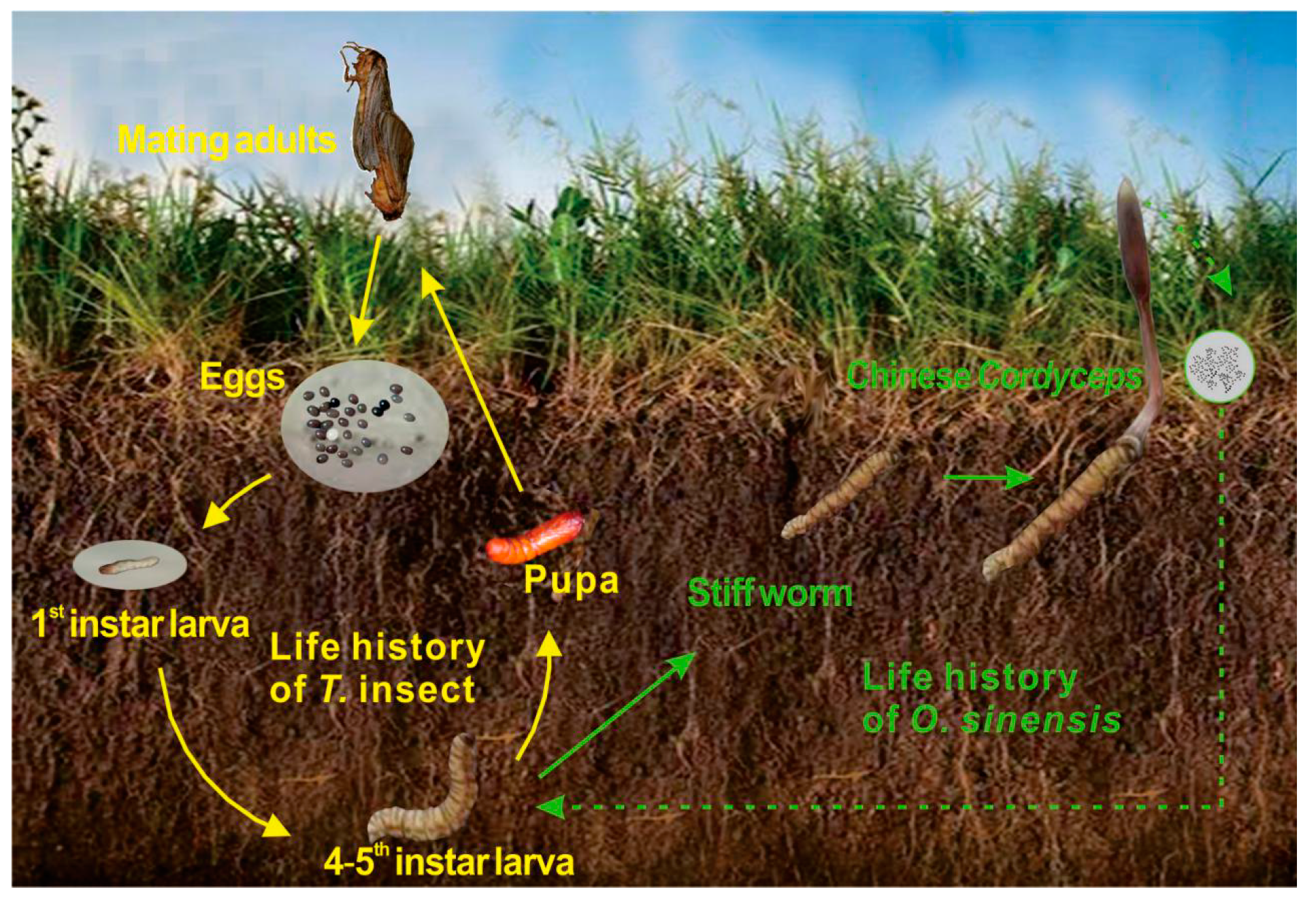
2.1. Field Site Description and Sample Collection
2.2. Analysis of Soil Physicochemical Properties
2.3. DNA Extraction, PCR, MiSeq Sequencing, and Sequence Data Analysis
2.4. Statistical Analysis
3. Results
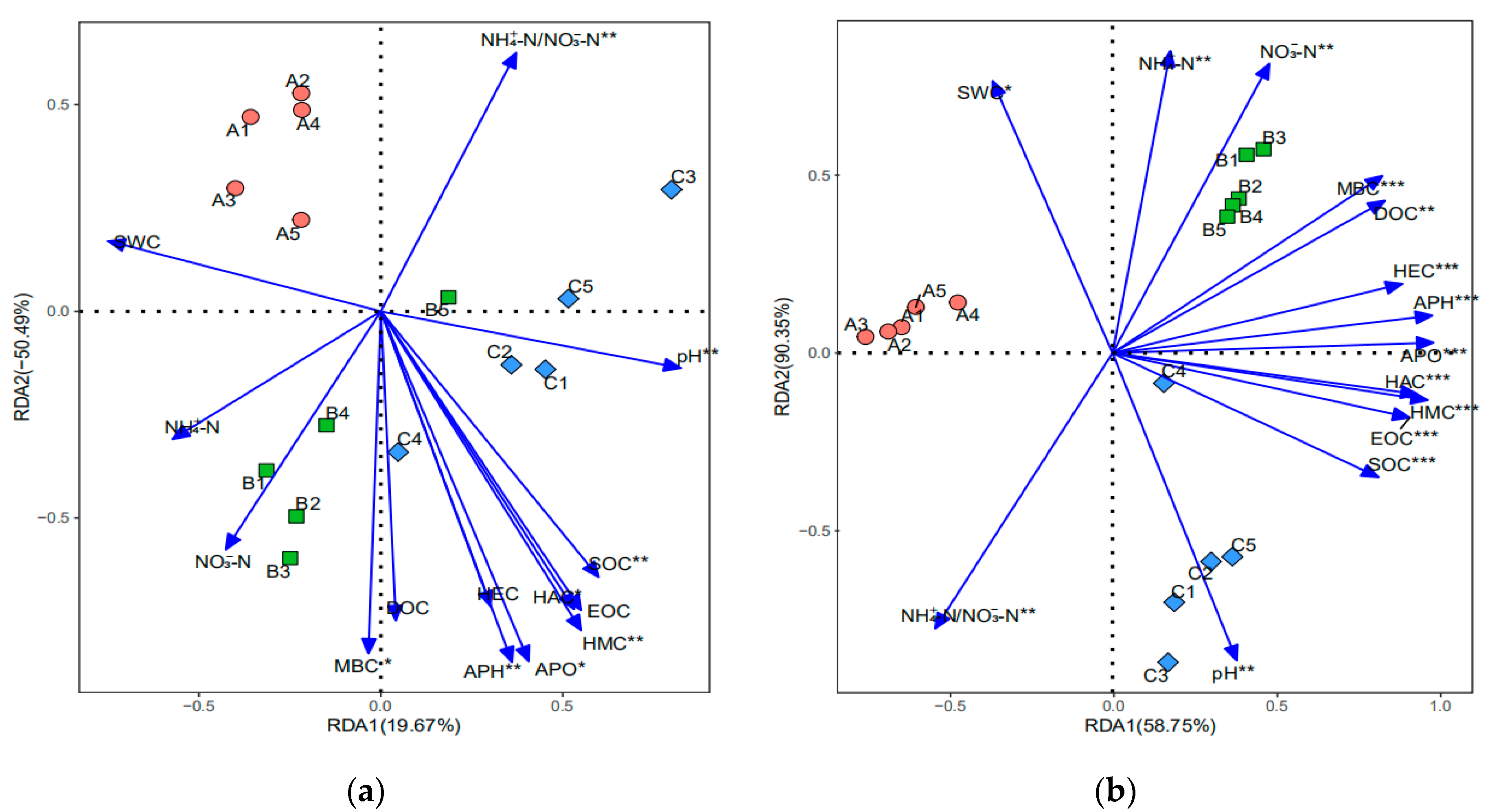
3.1. Soil Physicochemical Properties
3.2. Soil Microbial Diversity
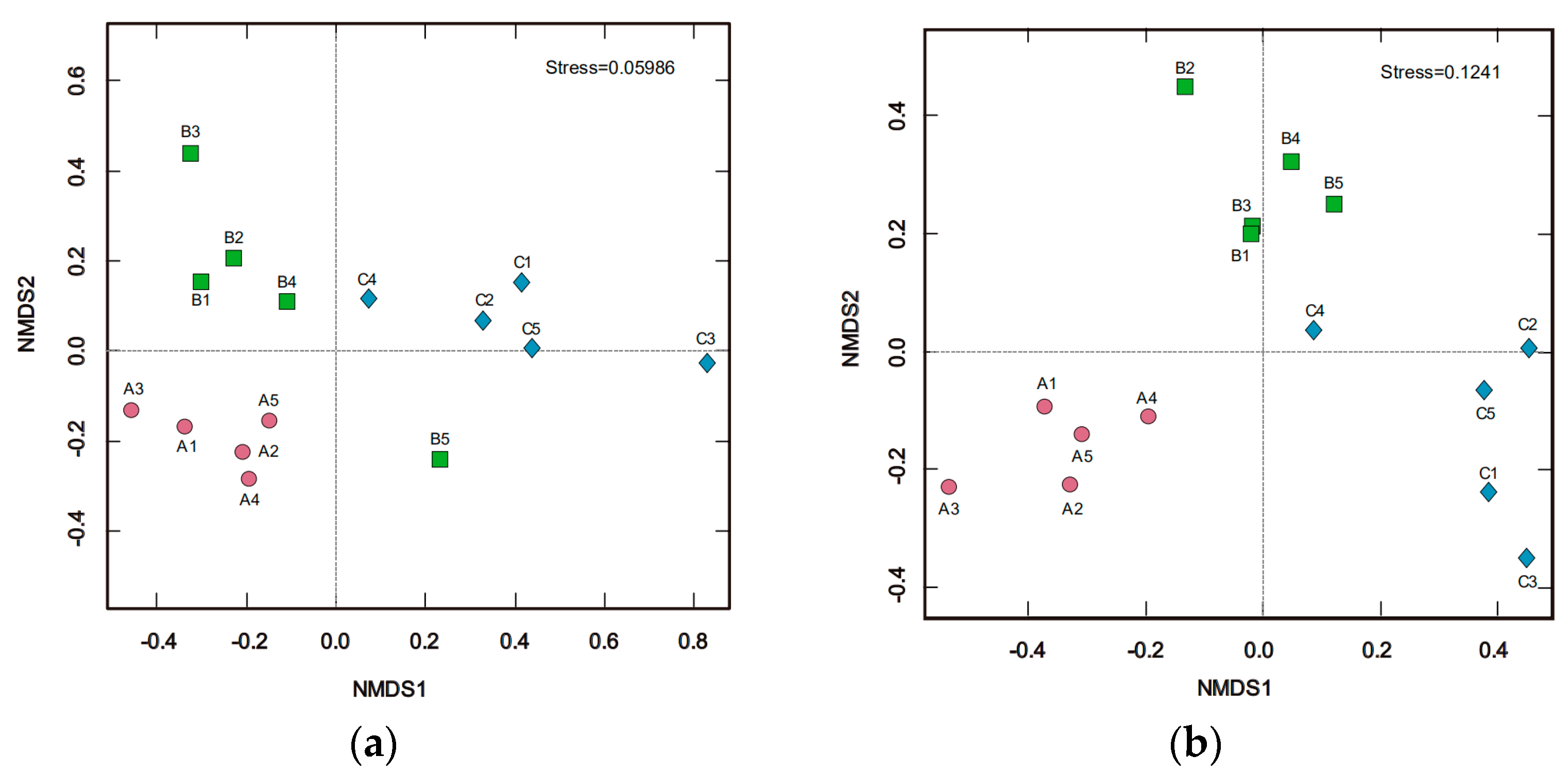
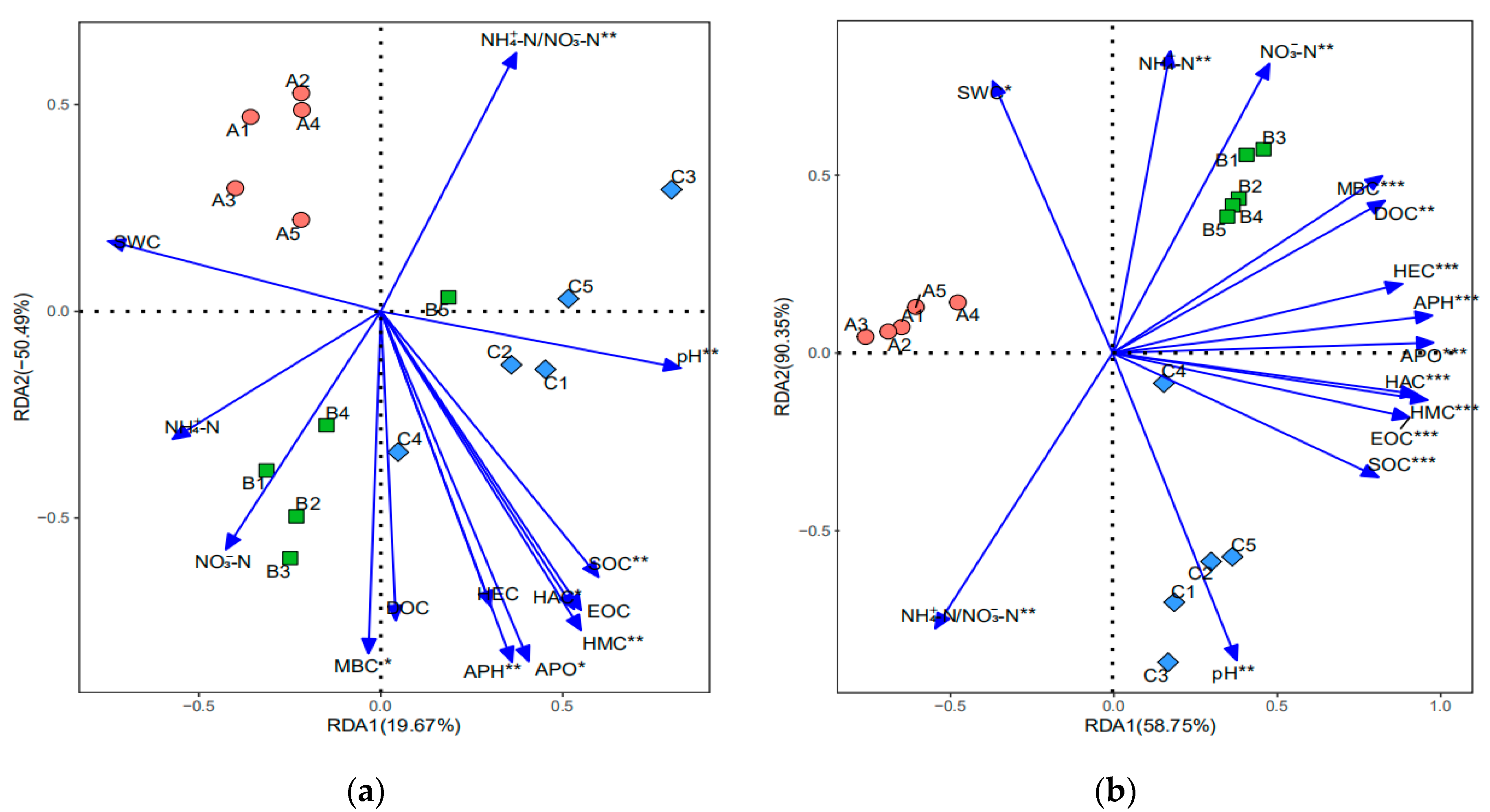
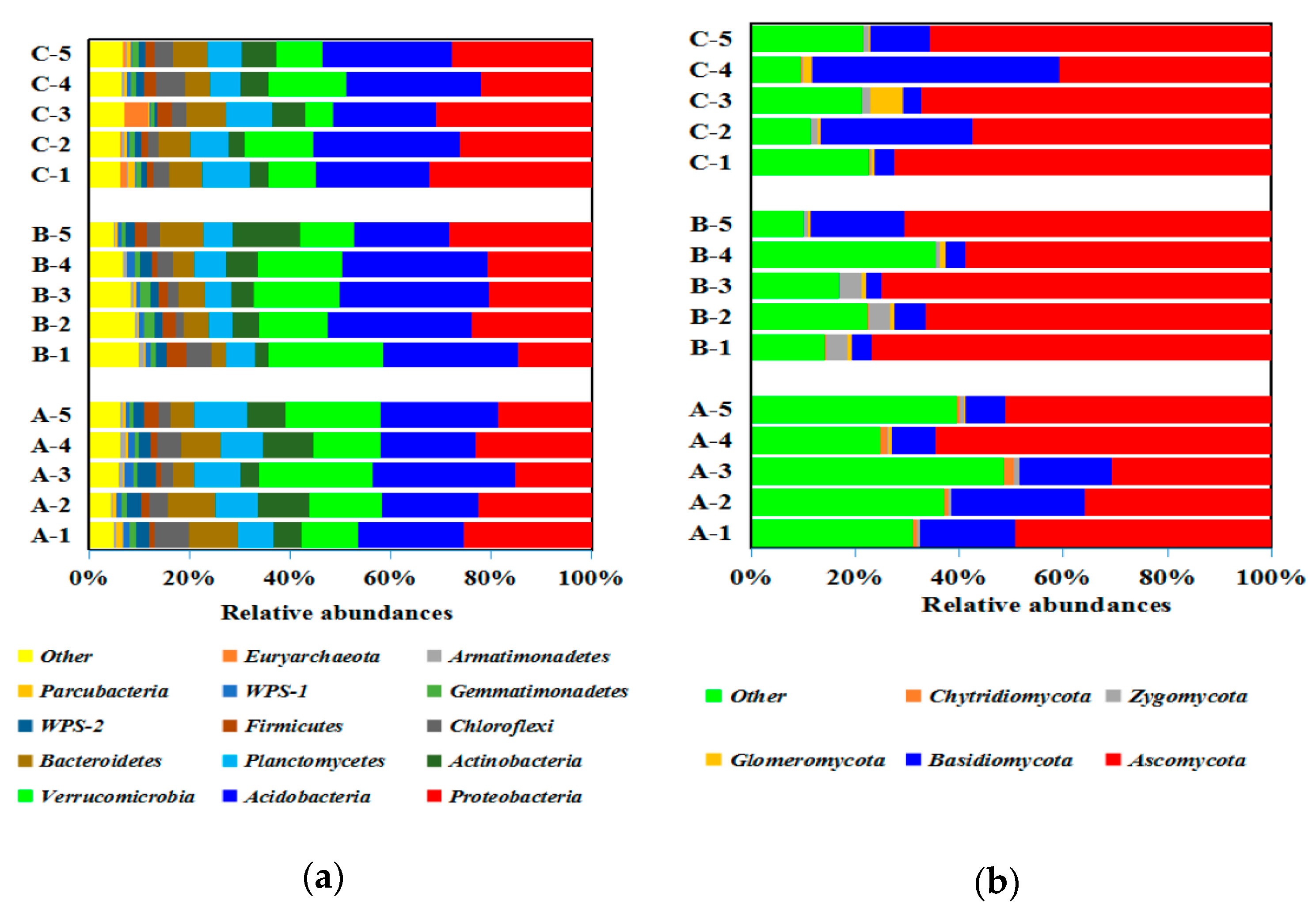
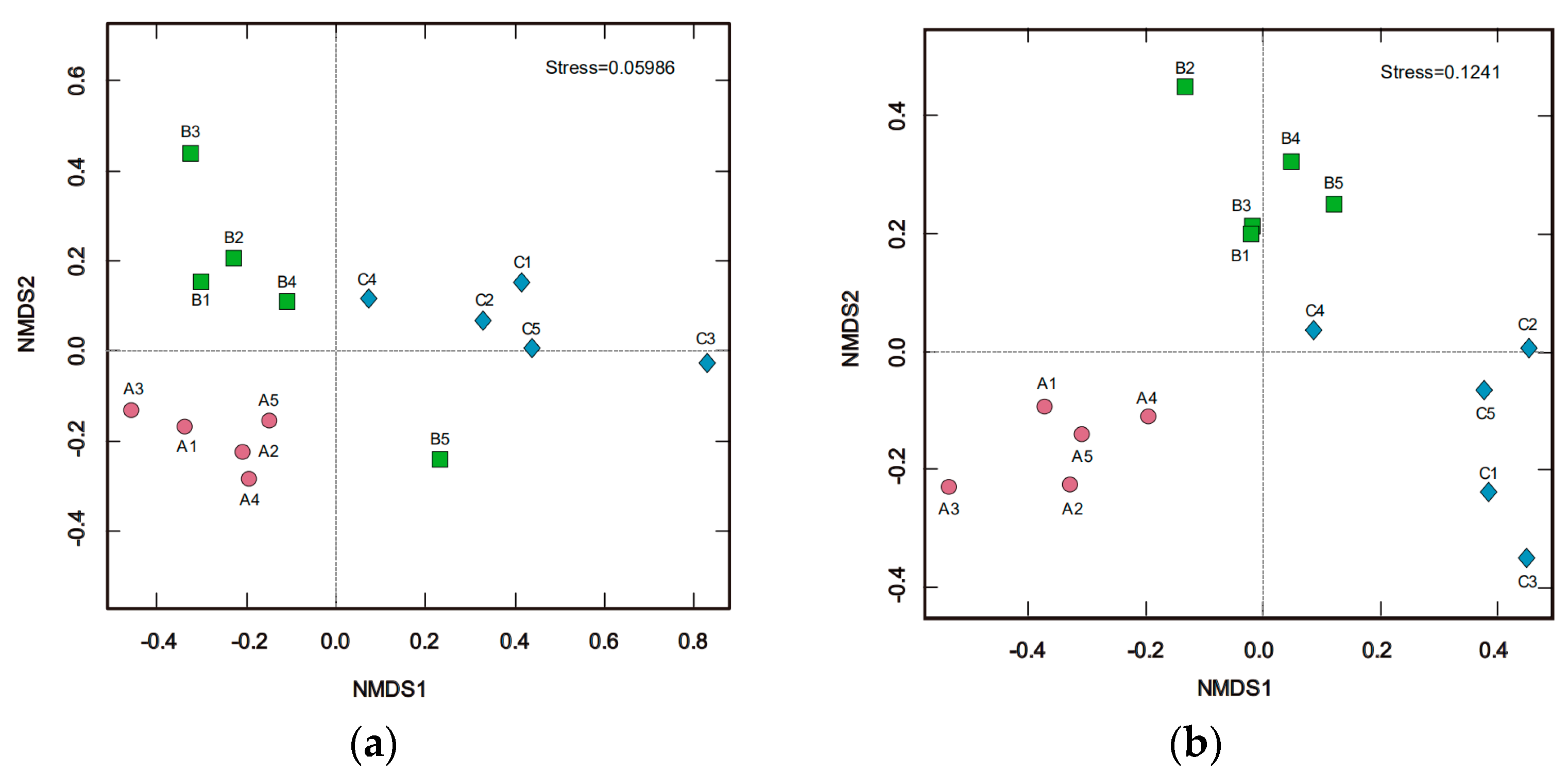
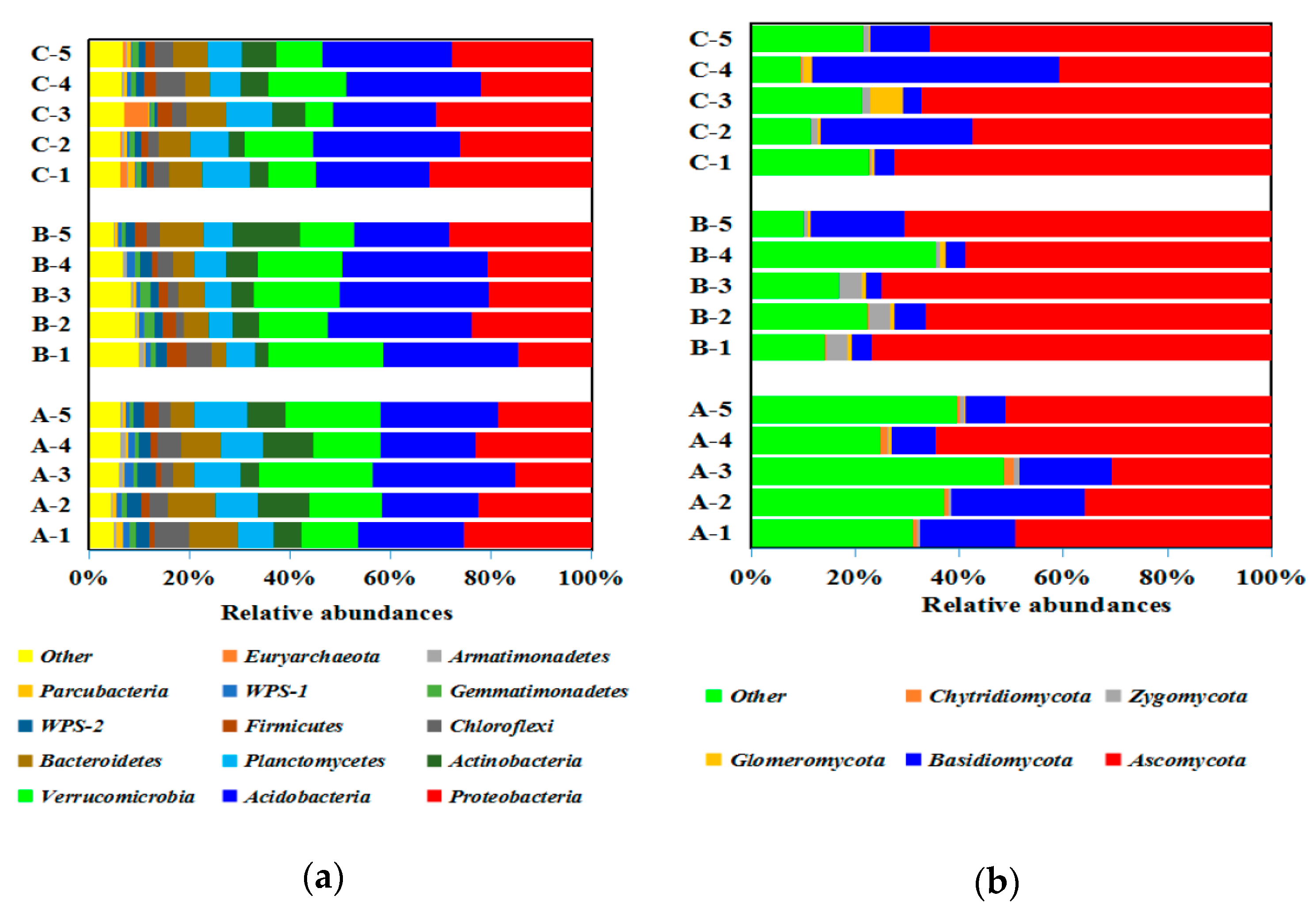
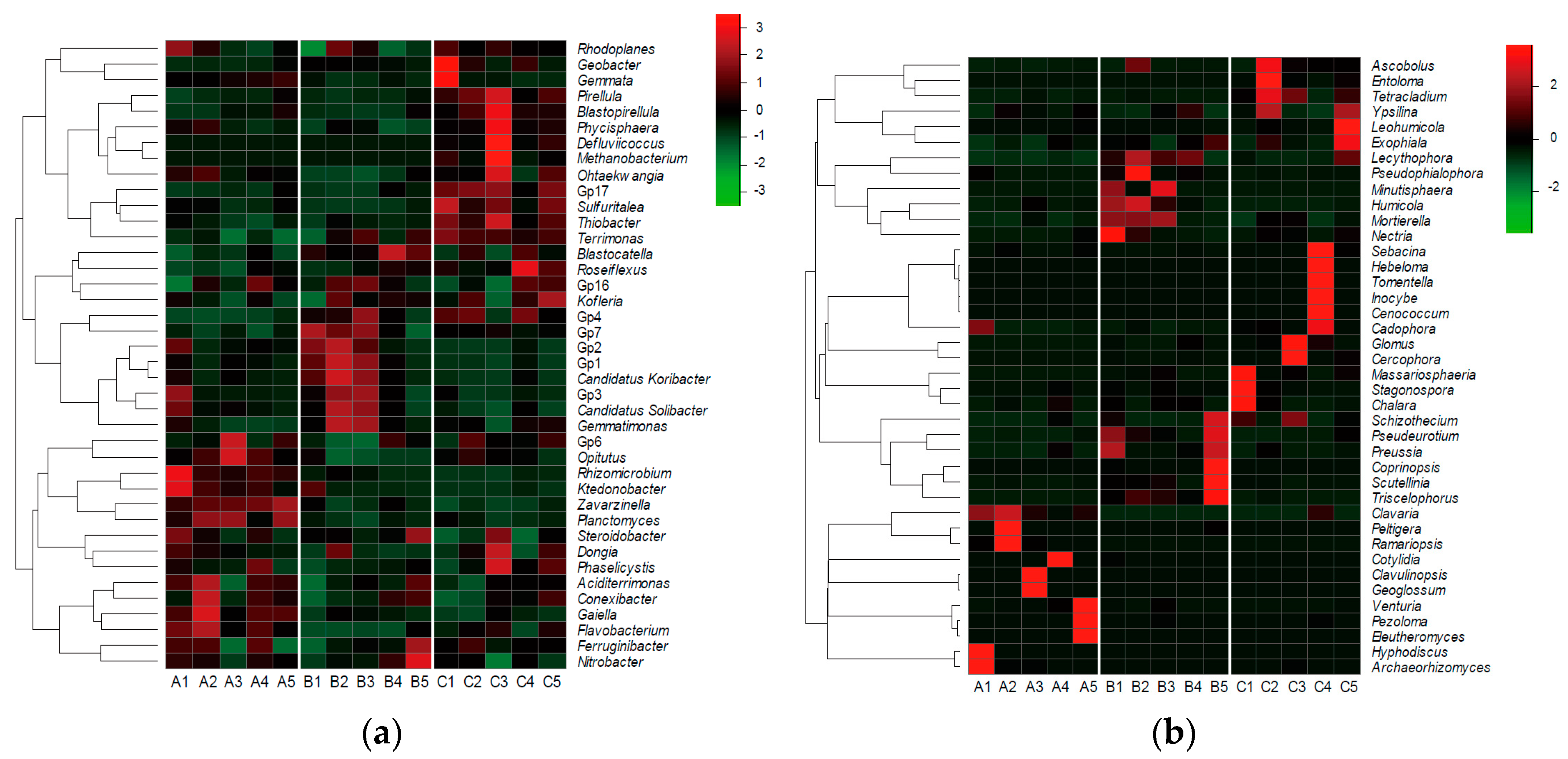
3.3. Soil Bacterial and Fungal Structure
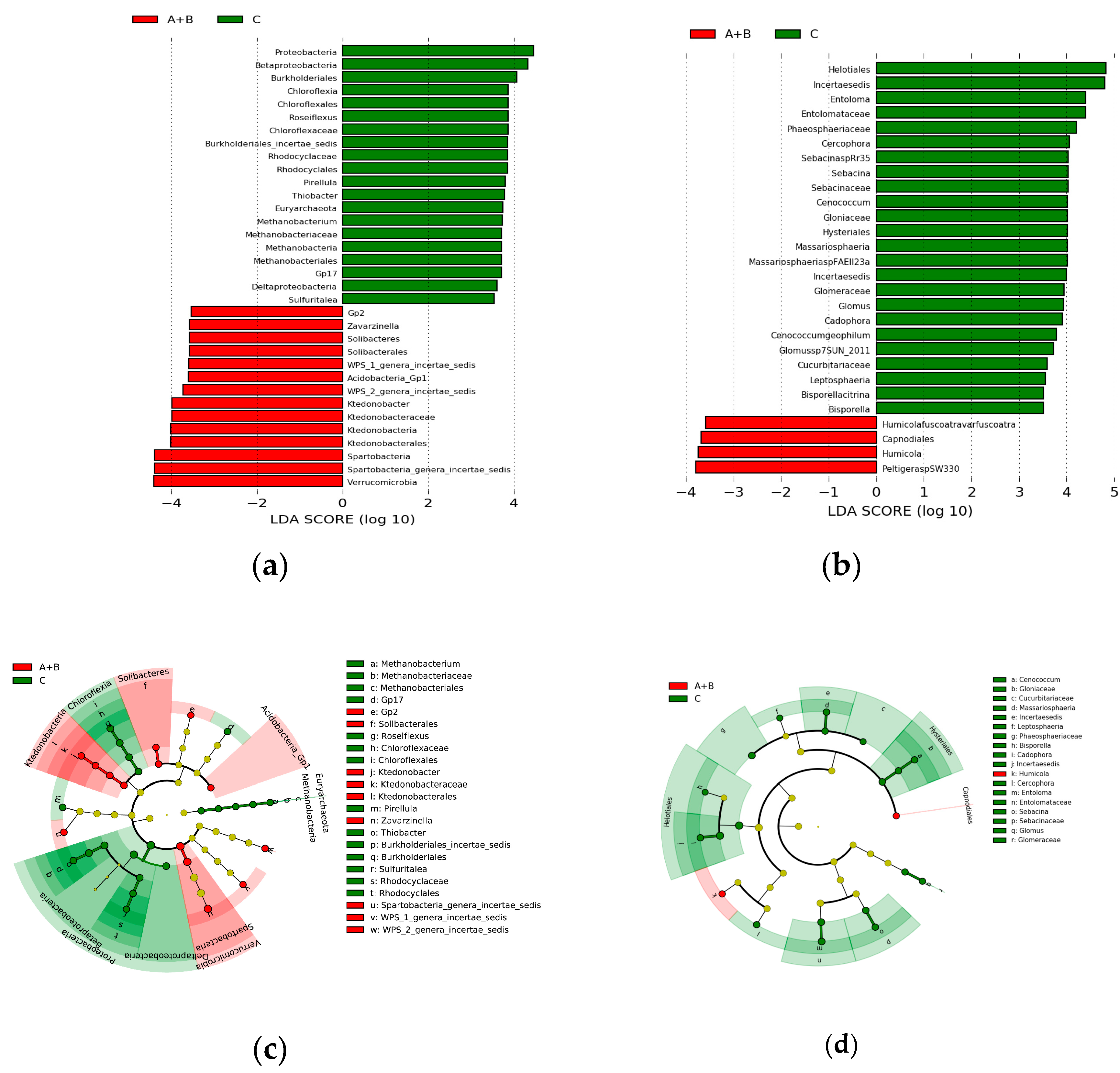
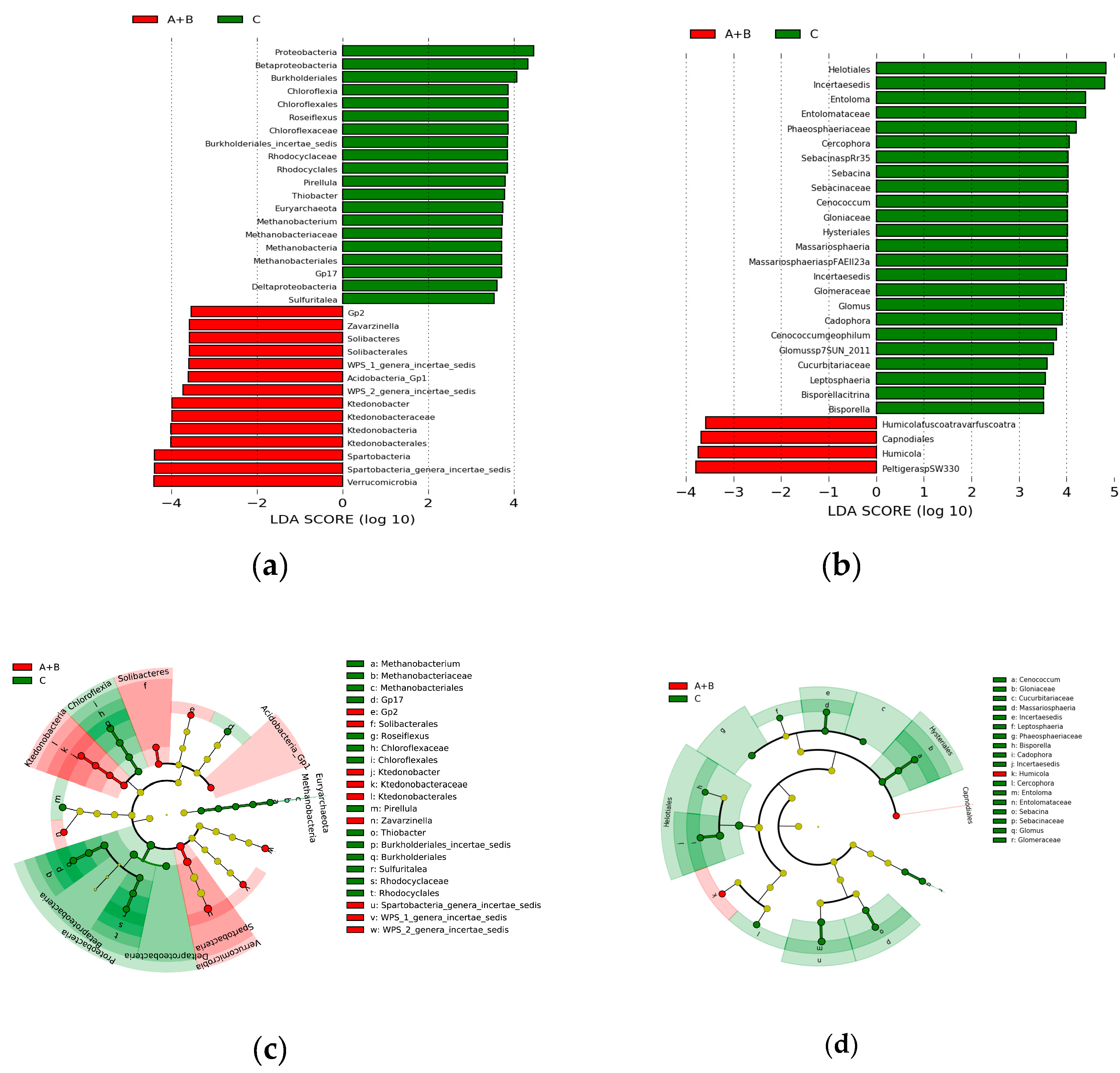
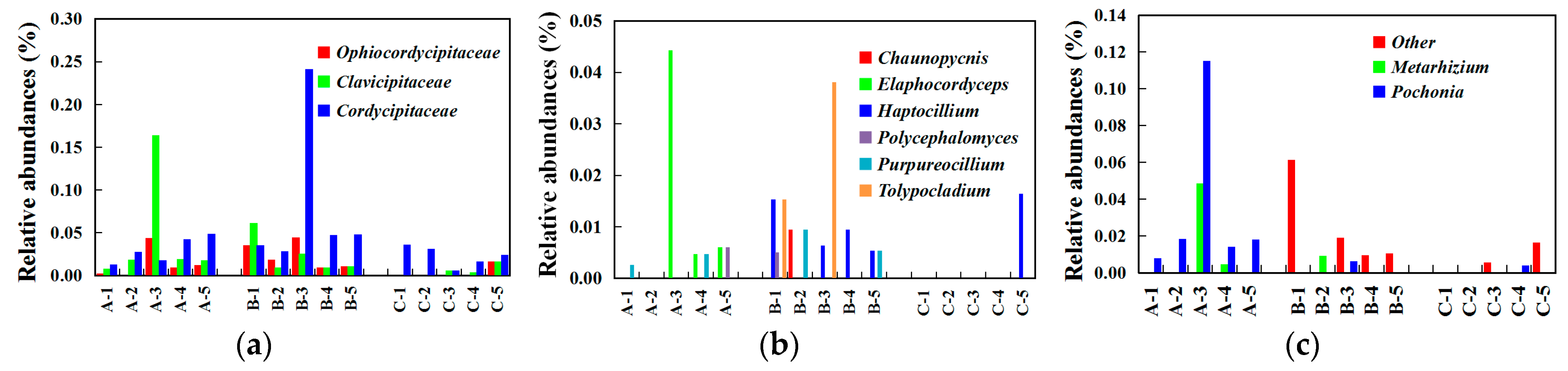
3.4. Differential Operational Taxonomic Units (OTUs) Related to the Occurrence of Chinese Cordyceps
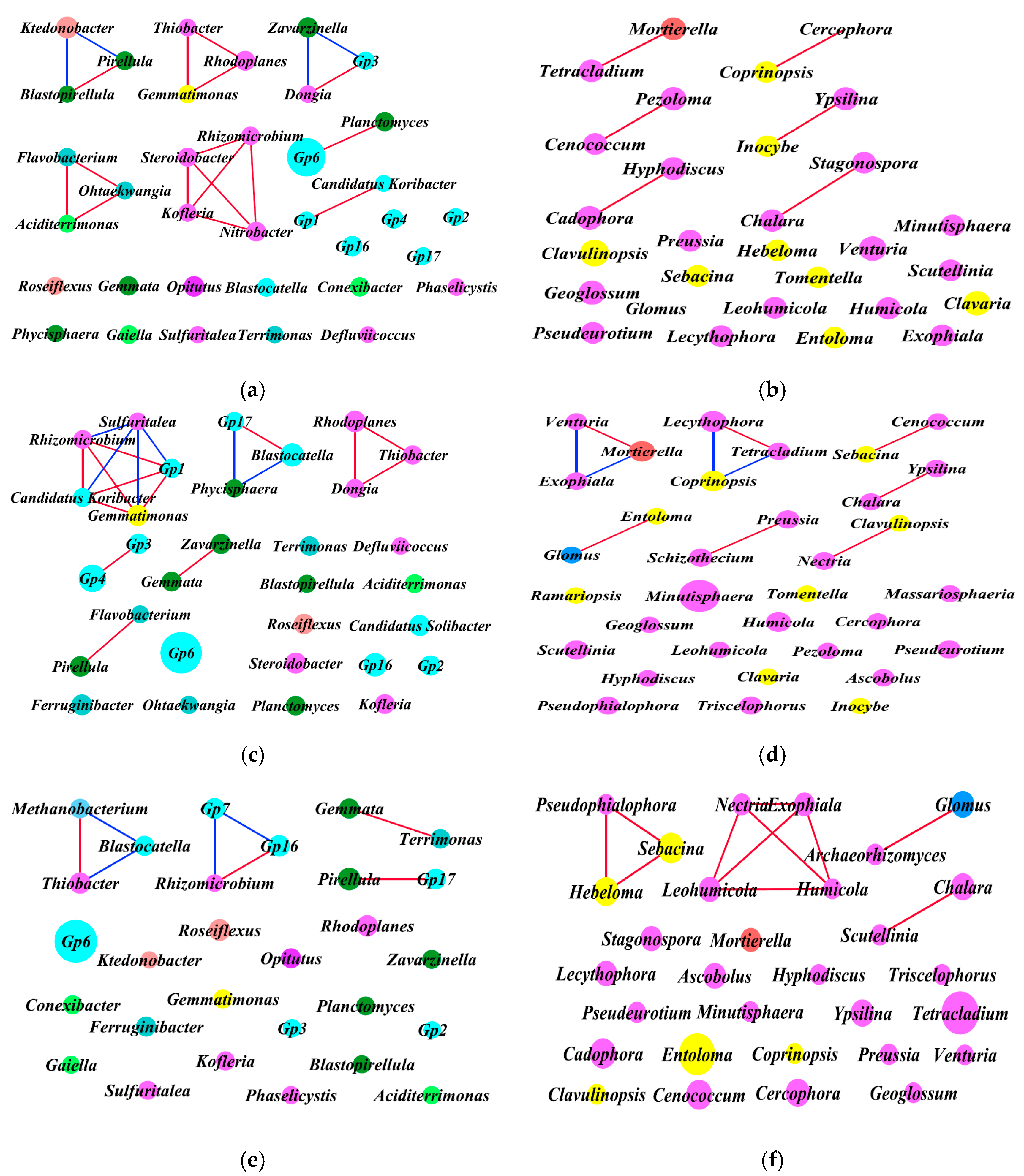
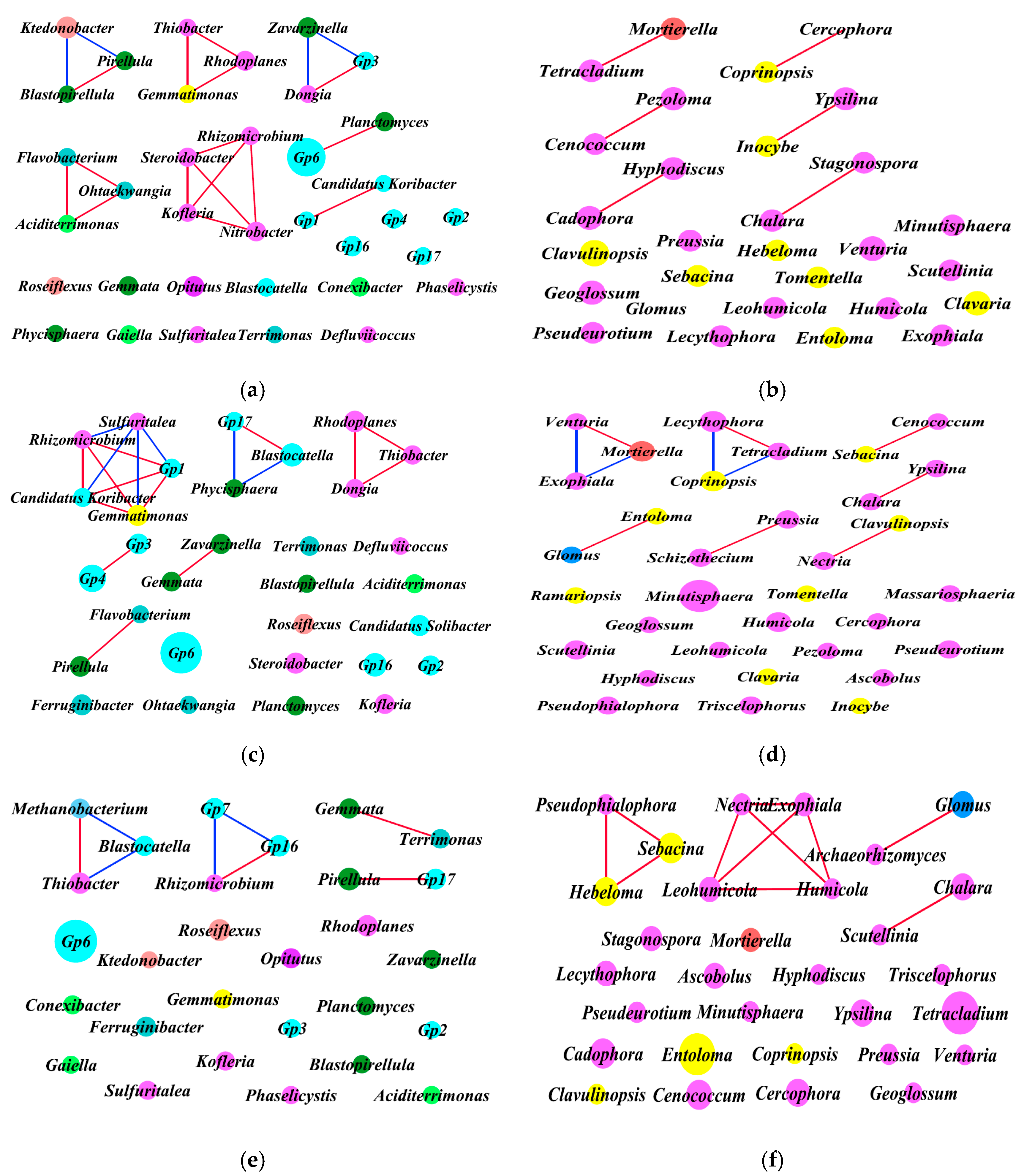
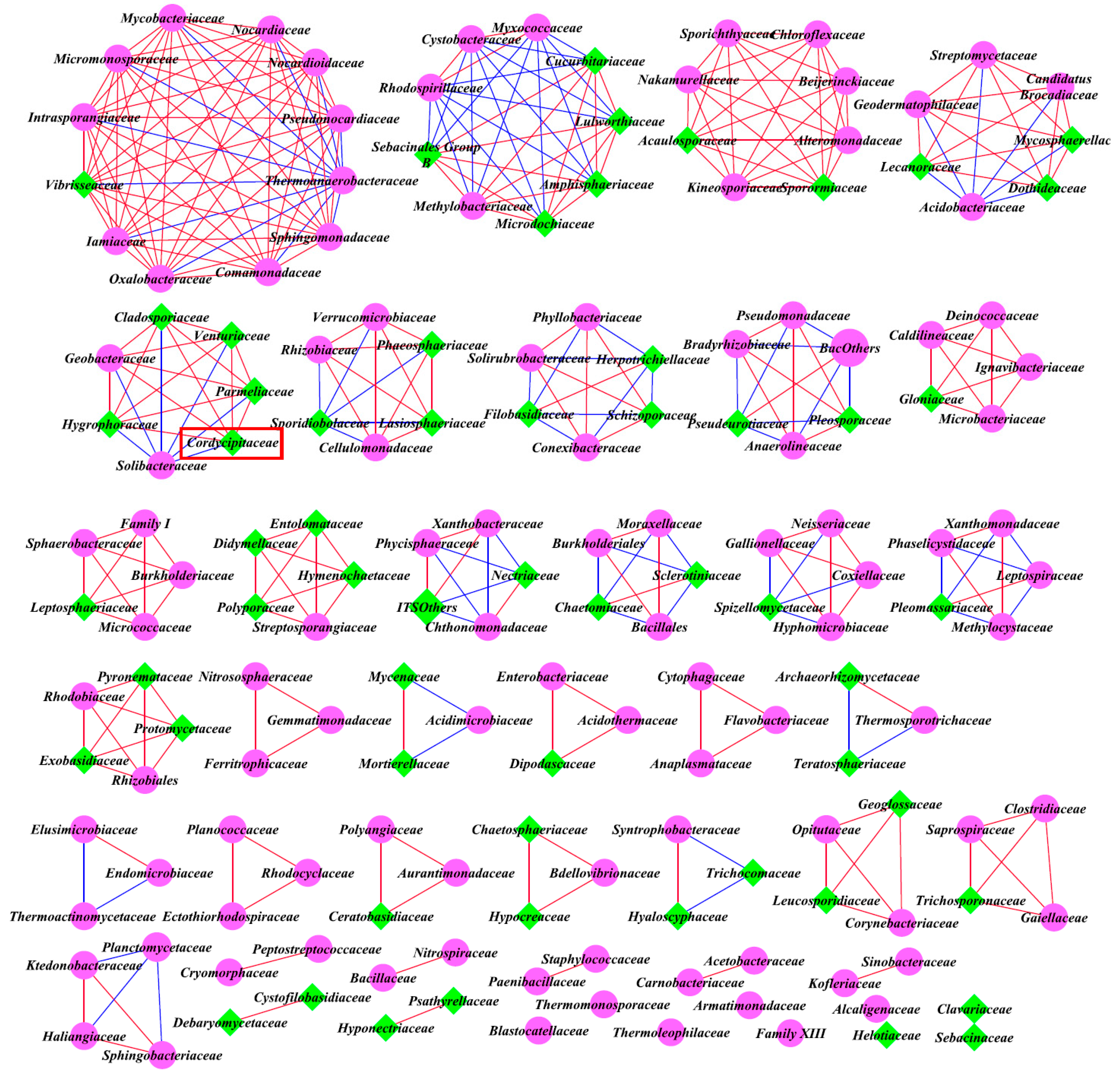
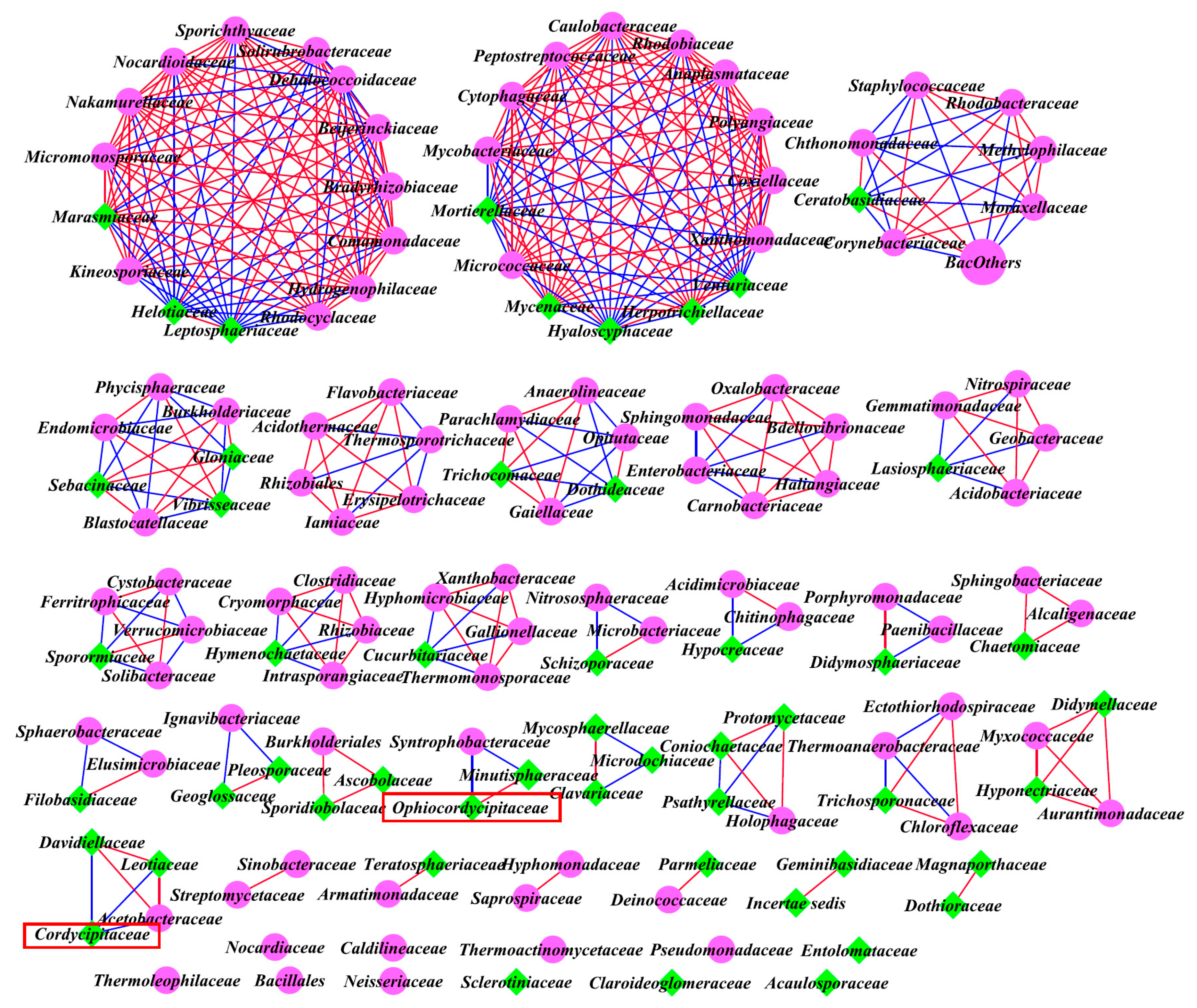
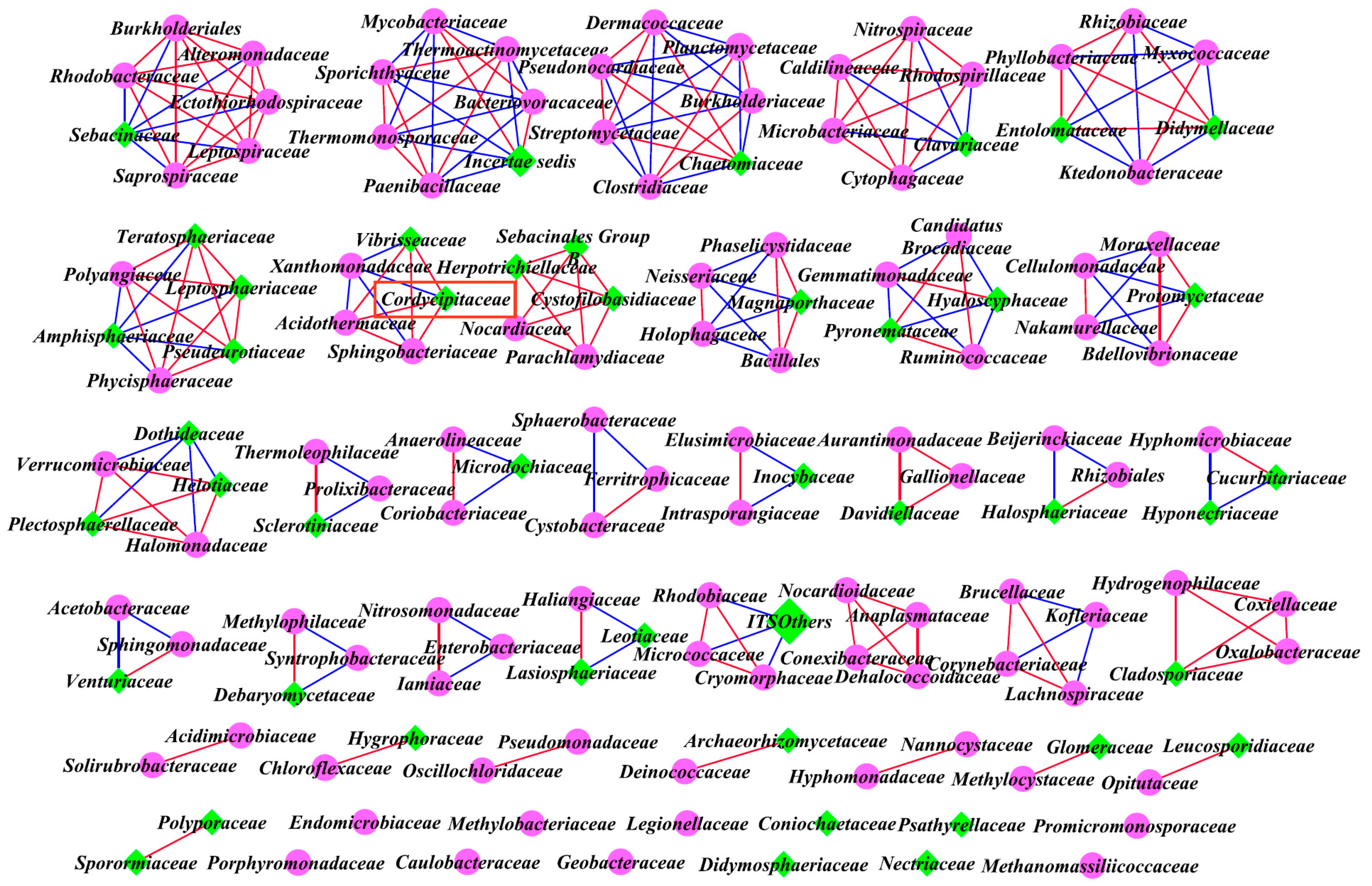
3.5. Intra-kingdom Co-Occurrence Analysis
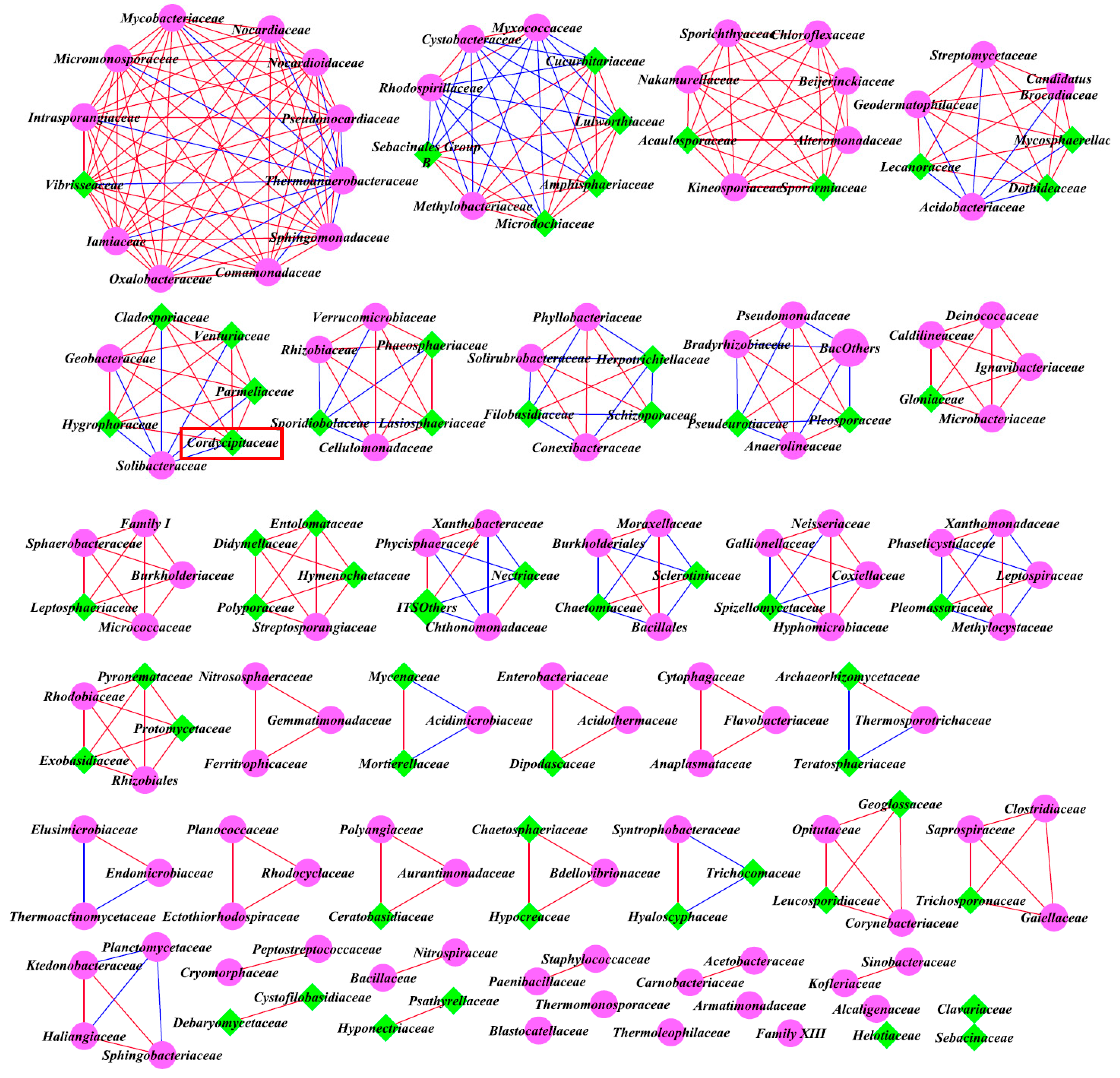
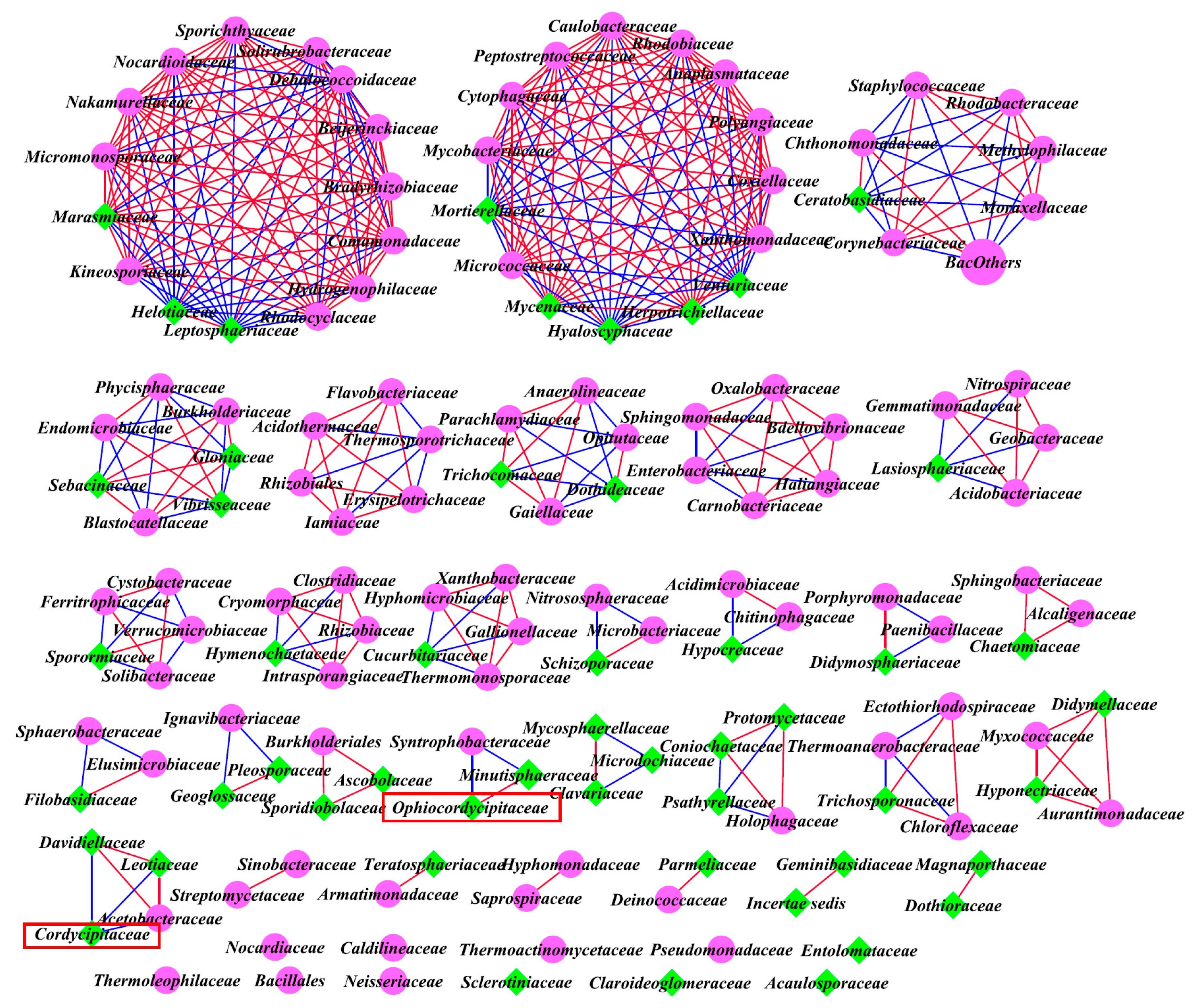
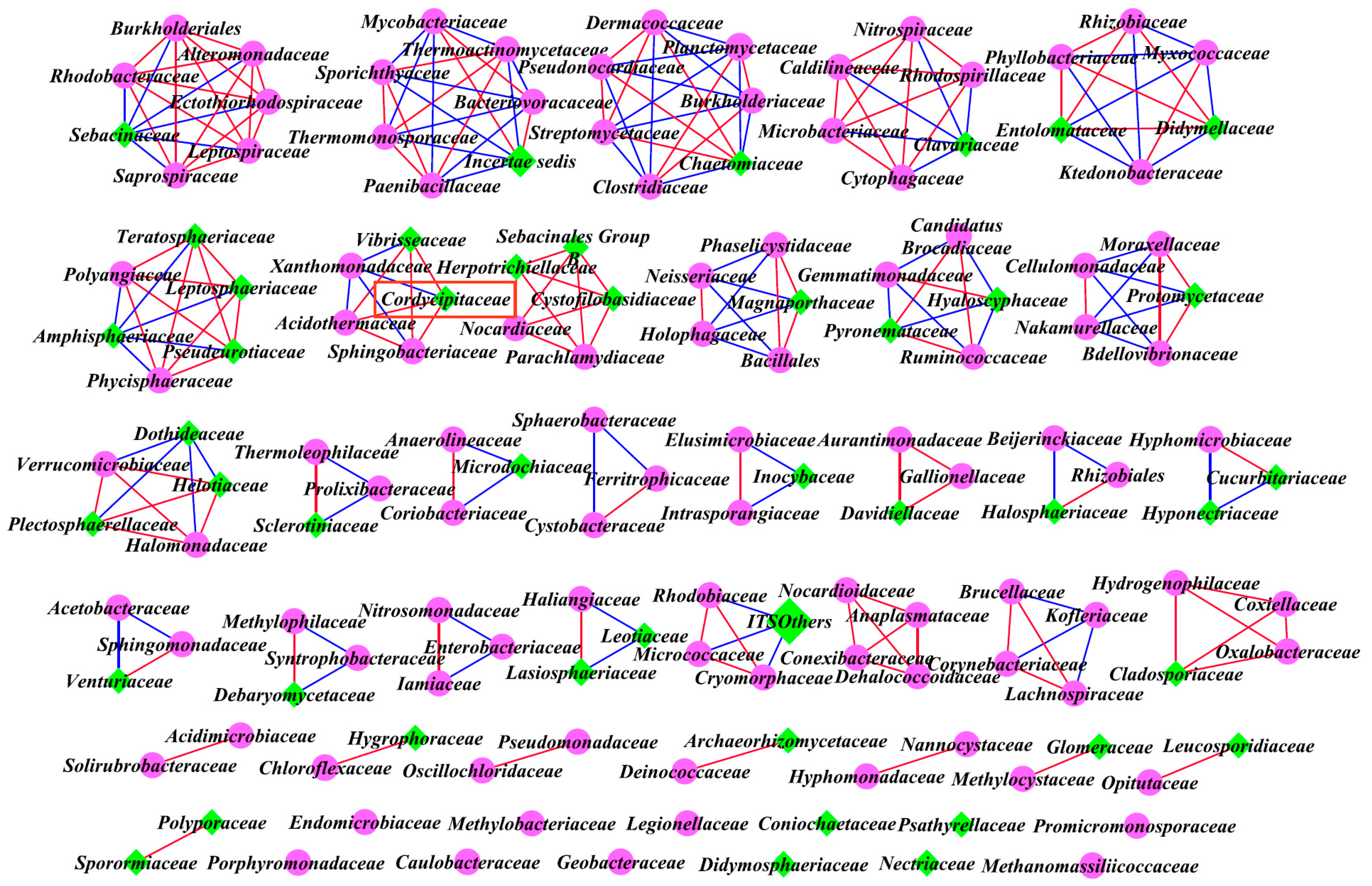
3.6. Inter-kingdom Co-Occurrence Analysis and Determination of Hub Taxa
4. Discussion
4.1. Soil Physicochemical Properties
4.2. Soil Microbial Structure
4.3. Co-Occurrence Interactions of Soil Microbial Community
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Y.J.; Li, E.W.; Wang, C.S.; Li, Y.L.; Liu, X.Z. Ophiocordyceps sinensis, the flagship fungus of China: Terminology, life strategy and ecology. Mycology 2012, 3, 2–10. [Google Scholar]
- Xia, E.H.; Yang, D.R.; Jiang, J.J.; Zhang, Q.J.; Liu, Y.; Liu, Y.L.; Zhang, Y.; Zhang, H.B.; Shi, C.; Tong, Y.; et al. The caterpillar fungus, Ophiocordyceps sinensis, genome provides insights into highland adaptation of fungal pathogenicity. Sci. Rep. 2017, 7, 1806. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.W.; Li, L.J.; Tian, E.W. Advances in research of the artificial cultivation of Ophiocordyceps sinensis in China. Crit. Rev. Biotechnol. 2013, 34, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Baral, B.; Shrestha, B. da Silva JAT. A review of Chinese Cordyceps with special reference to Nepal, focusing on conservation. Environ. Exp. Biol. 2015, 13, 61–73. [Google Scholar]
- Guo, L.X.; Zhang, G.W.; Wang, J.T.; Zhong, Y.P.; Huang, Z.G. Determination of arsenic species in Ophiocordyceps sinensis from major habitats in China by HPLC-ICP-MS and the edible hazard assessment. Molecules 2018, 23, e1012. [Google Scholar] [CrossRef] [PubMed]
- Quan, Q.M.; Chen, L.L.; Wang, X.; Li, S.; Yang, X.L.; Zhu, Y.G.; Wang, M.; Cheng, Z. Genetic diversity and distribution patterns of host insects of caterpillar fungus Ophiocordyceps sinensis in the Qinghai-Tibet Plateau. PLoS ONE 2014, 9, e92293. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, U.B.; Bawa, K.S. Impact of climate change on potential distribution of Chinese caterpillar fungus (Ophiocordyceps sinensis) in Nepal Himalaya. PLoS ONE 2014, 9, e106405. [Google Scholar] [CrossRef] [PubMed]
- He, J. Harvest and trade of caterpillar mushroom (Ophiocordyceps sinensis) and the implications for sustainable use in the Tibet region of southwest China. J. Ethnopharmacol. 2018, 221, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Q.; Li, W.J.; Li, Q.P.; Qian, Z.G.; Liu, X.Z.; Dong, C.Y. A breakthrough in the artificial cultivation of Chinese Cordyceps on a large-scale and its impact on science, the economy, and industry. Crit. Rev. Biotechnol. 2019, 39, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.L.; Zhou, G.L.; Zhang, H.; Meng, Q.; Zhang, J.H.; Wang, H.T.; Miao, L.; Li, X. Obstacles and approaches in artificial cultivation of Chinese Cordyceps. Mycology 2018, 9, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, S.; Wang, M.; Bai, F.; Liu, X. High diversity of the fungal community structure in naturally-occurring Ophiocordyceps sinensis. PLoS ONE 2010, 5, e15570. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Ma, L.; Zhao, K.; Yang, J.; Chang, Y. Survey analysis of soil physicochemical factors that influence the distribution of Cordyceps in the Xiahe Region of Gansu Province. Open Life Sci. 2017, 12, 76–81. [Google Scholar] [CrossRef]
- Yu, B.; Liang, L.; Li, X.; Liu, P.; Liu, X.; Zhu, L. Biological characteristics analysis of Ophiocordyceps sinensis. Plant Divers. Resour. 2012, 34, 478–482. [Google Scholar] [CrossRef]
- De, B.W.; Verheggen, P.; Klein Gunnewiek, P.J.; Kowalchuk, G.A.; van Veen, J.A. Microbial community composition affects soil fungistasis. Appl. Environ. Microbiol. 2003, 69, 835–844. [Google Scholar]
- Wu, M.; Zhang, H.; Li, X.; Zhang, Y.; Su, Z.; Zhang, C. Soil fungistasis and its relations to soil microbial composition and diversity: A case study of a series of soils with different fungistasis. J. Environ. Sci. 2008, 20, 871–877. [Google Scholar] [CrossRef]
- Bonanomi, G.; Gaglione, S.A.; Incerti, G.; Zoina, A. Biochemical quality of organic amendments affects soil fungistasis. Appl. Soil Ecol. 2013, 72, 135–142. [Google Scholar] [CrossRef]
- Dutta, S.; Chatterjee, M.; Teknos, T.N.; Carlson, R.W. Predictable bacterial composition and hydrocarbon degradation in Arctic soils following diesel and nutrient disturbance. ISME J. 2013, 7, 1200–1210. [Google Scholar]
- Urbanová, M.; Šnajdr, J.; Baldrian, P. Composition of fungal and bacterial communities in forest litter and soil is largely determined by dominant trees. Soil Biol. Biochem. 2015, 84, 53–64. [Google Scholar] [CrossRef]
- Freilich, M.A.; Wieters, E.; Broitman, B.R.; Marquet, P.A.; Navarrete, S.A. Species co-occurrence networks: Can they reveal trophic and non-trophic interactions in ecological communities? Ecology 2018, 99, 690–699. [Google Scholar] [CrossRef]
- Freilich, S.; Kreimer, A.; Meilijson, I.; Gophma, U.; Sharan, R.; Ruppin, E. The large-scale organizaiton of the bacterial network of ecological co-occurrence interactions. Nucleic Acids Res. 2010, 38, 3857–3868. [Google Scholar] [CrossRef]
- Bonfante, P.; Anca, I.A. Plants, mycorrhizal fungi, and bacteria: A network of interactions. Annu. Rev. Microbiol. 2009, 63, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2018, 6, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, F. Diversity and co-occurrence patterns of soil bacterial and fungal communities in seven intercropping systems. Front. Microbiol. 2018, 9, 1521. [Google Scholar] [CrossRef]
- Yang, R.H.; Wang, X.L.; Su, J.H.; Li, Y.; Jiang, S.P.; Gu, F.; Yao, Y.J. Bacterial diversity in native habitats of the medicinal fungus Ophiocordyceps sinensis on Tibetan Plateau as determined using Illumina sequencing data. FEMS Microbiol 2015, 362, fnu044. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Chen, X.; Guo, M.Y.; Bai, X.H.; Liu, Y.; Shen, G.R.; Li, Y.L.; Lin, J.; Zhou, X.W. High-throughput sequencing-based analysis of endogenetic fungal communities inhabiting the Chinese Cordyceps reveals unexpectedly high fungal diversity. Sci. Rep. 2016, 6, 33437. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.D. Soil Analysis in Agricultural Chemistry, 3rd ed.; China Agricultural Press: Beijing, China, 2005. (In Chinese) [Google Scholar]
- Xu, L.; Wang, C.Y.; Zhu, J.X.; Gao, Y.; Li, M.L.; Lv, Y.L.; Yu, G.R.; He, N.P. Latitudinal patterns and influencing factors of soil humic carbon fractions from tropical to temperate forests. J. Geogr. Sci. 2017, 28, 15–30. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Knight, R. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Lemos, L.; Fulthorpe, R.; Triplett, E.; Roesch, L. Rethinking microbial diversity analysis in the high throughput sequencing era. J. Microb. Methods. 2011, 86, 42–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmaso, N. Seasonal variation in the composition and rate of change of the phytoplankton community in a deep subalpine lake (lake garda, northern Italy): An application of nonmetric multidimensional scaling and cluster analysis. Hydrobiologia 1996, 337, 49–68. [Google Scholar] [CrossRef]
- Yan, Q.; Yu, Y.; Feng, W.; Yu, Z.; Chen, H. Plankton community composition in the Three Gorges Reservoir Region revealed by PCR-DGGE and its relationships with environmental factors. J. Environ. Sci. 2008, 20, 732–738. [Google Scholar] [CrossRef]
- Zheng, P.; Xia, Y.; Zhang, S.; Wang, C. Genetics of Cordyceps and related fungi. Appl. Microbiol. Biotechnol. 2013, 97, 2797–2804. [Google Scholar] [CrossRef] [PubMed]
- Ambika, M.; Corinna, M.; Stefan, R.; Volker, R.; Andreas, S.; Massimiliano, C.; Sylvia, S. Diversity, specificity, co-occurrence and hub taxa of the bacterial–fungal pollen microbiome. FEMS Microbiol. Ecol. 2018, 94, fiy112. [Google Scholar]
- Wu, Q.G.; Su, Z.X.; Su, R.J.; Hu, J.Y.; Wang, H. The dominant factors of habitat selection of Cordyceps sinensis. Guihaia 2009, 29, 331–336. [Google Scholar]
- Newbound, M.; Bennett, L.T.; Tibbits, J.; Kasel, S. Soil chemical properties, rather than landscape context, influence woodland fungal communities along an urban-rural gradient. Austral. Ecol. 2012, 37, 236–247. [Google Scholar] [CrossRef]
- Zhang, J.; Zeng, G.; Chen, Y.; Yu, M.; Yu, Z.; Li, H.; Yu, Y.; Huang, H. Effects of physico-chemical parameters on the bacterial and fungal communities during agricultural waste composting. Bioresour. Technol. 2011, 102, 2950–2956. [Google Scholar] [CrossRef]
- Zhu, J.S.; Gao, L.; Li, X.H.; Yao, Y.S.; Zhao, J.Q.; Zhou, Y.J.; Lu, J.H. Maturational alteration of oppositely orientated rDNA and differential proliferation of GC and AT-biased genotypes of Ophiocordyceps sinensis and Paecilomyces hepiali in natural Cordyceps sinensis. Am. J. Biomed. Sci. 2010, 2, 217–238. [Google Scholar] [CrossRef]
- Loy, A.; Schulz, C.; Lücker, S.; Schöpfer-Wendels, A.; Stoecker, K.; Baranyi, C.; Lehner, A.; Wagner, M. 16S rRNA gene-based oligonucleotide microarray for environmental monitoring of the betaproteobacterial order “Rhodocyclales”. Appl. Environ. Microbiol. 2005, 71, 1373–1386. [Google Scholar] [CrossRef]
- Bauld, J.; Brock, T.D. Ecological studies of Chloroflexis, a gliding photosynthetic bacterium. Archiv. Mikrobiol. 1973, 92, 267–284. [Google Scholar] [CrossRef]
- Granada, C.; Hasan, C.; Marder, M.; Konrad, L.; Vargas, L.; Passaglia, L.; Giongo, A.; Oliveira, R.; Pereira, L.; Trindade, F.; et al. Biogas from slaughterhouse wastewater anaerobic digestion is driven by the archaeal family Methanobacteriaceae and bacterial families Porphyromonadaceae, and Tissierellaceae. Renew. Energy 2018, 118, 840–846. [Google Scholar]
- Rizvi, S.Z.; Raman, A. Epiphyas postvittana (Lepidoptera: Tortricidae)-Botrytis cinerea (Helotiales: Sclerotiniaceae)-Vitis vinifera (Vitales: Vitaceae) interaction: The role of B. cinerea on the development of E. postvittana in synthetic nutritional media. J. Econ. Entomol. 2015, 108, 1646–1654. [Google Scholar] [CrossRef]
- Zhong, X.; Peng, Q.Y.; Li, S.S.; Chen, H.; Sun, H.X.; Zhang, G.R.; Liu, X. Detection of Ophiocordyceps sinensis in the roots of plants in alpine meadows by nested-touchdown polymerase chain reaction. Fungal Biol. 2014, 118, 359–363. [Google Scholar] [CrossRef]
- Li, Y.L.; Yao, Y.S.; Zhang, Z.H.; Liu, X.; Xu, H.F.; Ma, S.L.; Wu, Z.W.; Zhu, J.S. Synergy of fungal complexes isolated from the intestines of Hepialus lagii larvae in increasing infection potency. J. Fungal Res. 2016, 14, 96–112. [Google Scholar]
- de Menezes, A.B.; Richardson, A.E.; Thrall, P.H. Linking fungal-bacterial co-occurrences to soil ecosystem function. Curr. Opin. Microbiol. 2017, 37, 135–141. [Google Scholar] [CrossRef]
- Wu, Y.; Li, R.W.; Huang, H.; Fletcher, A.; Yu, L.; Pham, Q.; Yu, L.; He, Q.; Wang, T.T.Y. Inhibition of tumor growth by dietary indole−3-carbinol in a prostate cancer xenograft model may be associated with disrupted gut microbial interactions. Nutrients 2019, 11, e467. [Google Scholar] [CrossRef]
- Faust, K.; Lima-Mendez, G.; Lerat, J.S.; Sathirapongsasuti, J.F.; Knight, R.; Huttenhower, C.; Lenaerts, T.; Raes, J. Cross-biome comparison of microbial association networks. Front. Microbiol. 2015, 6, 1200. [Google Scholar] [CrossRef] [Green Version]
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662. [Google Scholar] [CrossRef]
- Sovran, B.; Planchais, J.; Jegou, S.; Straube, M.; Lamas, B.; Natividad, J.M.; Agus, A.; Dupraz, L.; Glodt, J.; Da Costa, G.; et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome 2018, 6, 152. [Google Scholar] [CrossRef]
- De Vries, F.T.; Griffiths, R.I.; Bailey, M.; Craig, H.; Girlanda, M.; Gweon, H.S.; Hallin, S.; Kaisermann, A.; Keith, A.M.; Kretzschmar, M.; et al. Soil bacterial networks are less stable under drought than fungal networks. Nat. Commun. 2018, 9, 3033. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Site A | Site B | Site C |
|---|---|---|---|
| Latitude | 29°36′09.6″ N | 29°35′49.6″ N | 29°36′11.0″ N |
| Longitude | 94°36′23.1″ E | 94°36′12.0″ E | 94°36′19.0″ E |
| Elevation | 4166 m | 4170 m | 4173 m |
| Density of Thitarodes larvae | 50 larva/m2 | 70 larva/m2 | 75 larva/m2 |
| Density of Chinese Cordyceps | 5 Chinese Cordyceps/m2 | 1 Chinese Cordyceps/m2 | 0 |
| Occurrence rate | 10.00% | 1.40% | 0 |
| Sampling time | Mid-July 2016 | Mid-July 2016 | Mid-July 2016 |
| Sampling depth of the soil | 10–20 cm | 10–20 cm | 10–20 cm |
| Physicochemical Property | Chinese Cordyceps Group | Null Chinese Cordyceps Group | |
|---|---|---|---|
| Site A | Site B | Site C | |
| SWC (%) | 41.0 ± 0.4 | 40.9 ± 0.9 | 38.3 ± 0.7 ##,@@ |
| pH | 5.37 ± 0.18 | 5.41 ± 0.12 | 7.37 ± 0.18 ##,@@ |
| NH4+-N (mg/kg) | 85.15 ± 2.79 | 106.14 ± 6.43 ** | 71.21 ± 5.53 #,@@ |
| NO3−-N (mg/kg) | 6.06 ± 0.48 | 27.11 ± 1.94 ** | 4.41 ± 0.61 #,@@ |
| NH4+-N/NO3−-N | 14.11 ± 0.88 | 3.93 ± 0.33 ** | 16.549 ± 3.73 @@ |
| APH (mg/kg) | 20.96 ± 0.84 | 91.57 ± 2.43 ** | 61.66 ± 2.95 ##,@@ |
| APO (mg/kg) | 202.00 ± 6.16 | 298.00 ± 3.87 ** | 277.60 ± 5.18 ##,@@ |
| EOC (mg/kg) | 19.07 ± 0.93 | 24.17 ± 1.07 ** | 25.03 ± 1.55 ## |
| TN (%) | 5.31 ± 0.39 | 5.64 ± 0.33 | 5.25 ± 0.22 |
| MBC (mg/kg) | 1029.14 ± 21.27 | 2352.61 ± 135.21 ** | 1352.47 ± 35.6 ##,@@ |
| SOC (g/kg) | 18.90 ± 0.99 | 23.48 ± 1.85 * | 25.69 ± 1.58 ## |
| DOC (g/kg) | 0.90 ± 0.10 | 1.58 ± 0.12 ** | 1.10 ± 0.11#,@@ |
| HEC (g/kg) | 23.14 ± 1.10 | 30.92 ± 1.00 ** | 27.24 ± 1.74 #,@ |
| FAC (g/kg) | 13.12 ± 0.78 | 16.7 ± 1.69 * | 13.71 ± 1.69 @ |
| HAC (g/kg) | 10.38 ± 0.62 | 15.45 ± 1.00 ** | 15.30 ± 1.92 # |
| HMC (g/kg) | 86.43 ± 1.08 | 120.34 ± 4.13 ** | 120.36 ± 2.38 ## |
| Classified | Sample Site | Number of Sequences | Number of OTUs | Shannon | Simpson | Chao1 |
|---|---|---|---|---|---|---|
| Bacteria | Site A | 42,129 ± 7501 | 3648 ± 334 | 9.38 ± 0.35 | 0.99 ± 0.00 | 4172 ± 380.85 |
| Site B | 36,130 ± 9977 | 3437 ± 514 | 9.22 ± 0.19 | 0.99 ± 0.00 | 4221.6 ± 211.97 | |
| Site C | 42,276 ± 6043 | 4233 ± 377 *,# | 9.62 ± 0.19# | 1.00 ± 0.00# | 4875.6 ± 279.04 *,# | |
| Fungi | Site A | 21,629.6 ± 9927 | 740 ± 140 | 4.9 ± 1.02 | 0.87 ± 0.06 | 769.4 ± 183.73 |
| Site B | 150,145 ± 4308 | 711 ± 130 | 6.24 ± 0.44 * | 0.95 ± 0.03 * | 806 ± 90.02 | |
| Site C | 14,621 ± 6460 | 681 ± 84 | 6.16 ± 0.66 * | 0.95 ± 0.02 * | 780 ± 79.03 |
| Classified | ANOSIM | Adonis | MRPP | |||||
|---|---|---|---|---|---|---|---|---|
| R | P | F | R2 | P | Delta (δ) | Effect Size (A) | P | |
| Bacteria | 0.564 | 0.001 | 3.910 | 0.395 | 0.001 | 0.090 | 0.131 | 0.004 |
| Fungi | 0.545 | 0.001 | 2.697 | 0.310 | 0.001 | 0.318 | 0.106 | 0.001 |
| Classified | Site | Average Degree | Positive Correlations | Negative Correlations | Positive/Negative | Nodes | |
|---|---|---|---|---|---|---|---|
| Intra-kingdom analysis based on top 40 genera | Bacteria | A | 1.143 | 16 | 4 | / | 35 |
| B | 1.256 | 13 | 6 | / | 31 | ||
| C | 0.571 | 4 | 4 | / | 28 | ||
| Fungi | A | 0.414 | 6 | 0 | / | 29 | |
| B | 0.667 | 7 | 4 | / | 33 | ||
| C | 0.733 | 11 | 0 | / | 30 | ||
| Inter-kingdom analysis of all family levels | Bacteria and Fungi | A | 4.241 | 257 | 95 | 2.71 | 166 |
| B | 5.383 | 243 | 158 | 1.54 | 149 | ||
| C | 3.831 | 182 | 157 | 1.16 | 177 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, J.-L.; Lai, B.; Jiang, W.; Wang, J.-T.; Hong, Y.-H.; Chen, F.-B.; Tan, S.-Q.; Guo, L.-X. Diversity and Co-Occurrence Patterns of Soil Bacterial and Fungal Communities of Chinese Cordyceps Habitats at Shergyla Mountain, Tibet: Implications for the Occurrence. Microorganisms 2019, 7, 284. https://doi.org/10.3390/microorganisms7090284
Shao J-L, Lai B, Jiang W, Wang J-T, Hong Y-H, Chen F-B, Tan S-Q, Guo L-X. Diversity and Co-Occurrence Patterns of Soil Bacterial and Fungal Communities of Chinese Cordyceps Habitats at Shergyla Mountain, Tibet: Implications for the Occurrence. Microorganisms. 2019; 7(9):284. https://doi.org/10.3390/microorganisms7090284
Chicago/Turabian StyleShao, Jun-Li, Bei Lai, Wei Jiang, Jia-Ting Wang, Yue-Hui Hong, Fu-Bin Chen, Shao-Qing Tan, and Lian-Xian Guo. 2019. "Diversity and Co-Occurrence Patterns of Soil Bacterial and Fungal Communities of Chinese Cordyceps Habitats at Shergyla Mountain, Tibet: Implications for the Occurrence" Microorganisms 7, no. 9: 284. https://doi.org/10.3390/microorganisms7090284