
Role of α and β Transmembrane Domains in Integrin Clustering


1
Department of Mechanical Engineering, Sharif University of Technology, Tehran, Iran
2
Department of Electrical Engineering, York University, Toronto, M3J1P3, Canada
*
Authors to whom correspondence should be addressed.
Actuators 2015, 4(4), 267-280; https://doi.org/10.3390/act4040267
Submission received: 1 October 2015
/
Accepted: 11 November 2015
/
Published: 20 November 2015
(This article belongs to the Special Issue Biophysical Micro- and Nano-Actuators)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Integrins are transmembrane proteins playing a crucial role in the mechanical signal transduction from the outside to the inside of a cell, and vice versa. Nevertheless, this signal transduction could not be implemented by a single protein. Rather, in order for integrins to be able to participate in signal transduction, they need to be activated and produce clusters first. As integrins consist of α- and β-subunits that are separate in the active state, studying both subunits separately is of a great importance, for, in the active state, the distance between α- and β-subunits is long enough that they do not influence one another significantly. Thus, this study aims to investigate the tendency of transmembrane domains of integrins to form homodimers. We used both Steered and MARTINI Coarse-grained molecular dynamics method to perform our simulations, mainly because of a better resolution and computational feasibility that each of these methods could provide to us. Using the Steered molecular dynamics method for α- and β-subunits, we found that the localized lipid packing prevented them from clustering. Nonetheless, the lipid packing phenomenon was found to be an artifact after investigating this process using a coarse grained (CG) model. Exploiting the coarse-grained molecular dynamics simulations, we found that α- and β-subunits tend to form a stable homo-dimer.
1. Introduction
Integrins are transmembrane receptors, helping cells to embed to their surrounding environments, for instance, other cells, and the extracurricular matrix (ECM). In signal transduction, integrins transfer information about the chemical composition and the conditions of ECM to the inside of the cell. This not only causes integrins to participate in the mechanical force transduction throughout the membrane, but also it plays a crucially important role in regulating the cell cycle, migration, survival, differentiation, movement, and proliferation.
In order to regulate cells’ functions, signals are both transferred from one cell to the other cells and received from the extracellular matrix (ECM). Focal adhesion formation is one of the cells’ critical functions that occurs when cells require to transfer mechanical signals to the other cells or attach to the extracellular matrix. Primarily, the initiators of this process are integrins which commence the signaling process by attaching to ligands or RGD sequence of ECM proteins in order to transfer signals.
Integrin-ligand rigidity of binding can influence focal adhesion formation. Paszek et al. suggested the cooperative nature of the integrin-ligand interaction. They also found that that this cooperation is enough to drive integrin clustering [1]. Integrins often influence one another and interact together, thereby they participate in regulating a cell’s function. Integrins consist of α- and β-subunits that enable them to perform multiple functions [2]. αIIbβ3 integrin is one of the vital receptors that initiates focal adhesion formation [3]. It is believed that in integrin activation, β subunits cytoplasmic domains are of a vital importance, while α cytoplasmic domain has mainly a regulatory function [4]. Nonetheless, the interaction of α-β transmembrane domains varies in different integrins [5]. In the inactive state, α- and β-subunits are bound together, but, in the active state, they are distant from each other and are able to form a cluster [6,7]. Thus, both α- and β-subunits play a crucially important role in studying the mechanism of focal adhesion formation. In addition, studying of α- and β-subunits together and separately are important, because clustering of these subunits in different manners can cause variations in the strength of the focal adhesions and consequently can affect cell signal transduction.
It has been observed that the α-subunit of many integrins has several binding sites for divalent cations, such as Ca2+, which is believed to facilitate focal adhesion formation [8,9]. In this study, the clustering of α- and β-subunits is simulated by exploiting molecular dynamics technique to investigate their potential function in focal adhesion formation.
There are some recent research studies in which the homo-dimerization of α- and β-subunits have been investigated. For instance, Li et al. suggested that α- and β-subunits have a tendency to form dimers and trimmers, respectively. On the other hand, Wei Wang et al. proposed that α- and β-subunits do not have any tendencies to form homo-oligomers [10,11]. Moreover, Mehrbod et al. used the normal mode analysis so as to ignore the effects of the relatively big ectodomain of the integrin, modeling of which increases the computational cost of the integrin simulation significantly. They suggested that the lipid packing impedes integrin clustering and the hydrophobic residues in α subunits are more available than in β subunits [12]. We observed that the so-called lipid packing phenomenon is not natural and mainly caused because of the SMD approach used in their simulations. In addition, Kalli, et al. investigated α- and β-subunits hetero-dimerization by CG MARTINI method and compared their simulation results with NMR structure of these subunits. They showed that the computational and experimental results they obtained are in a good agreement [13]. Chng, et al. have investigated the theories and biological applications of the coarse-graining methods [14]. They further studied αLβ2 integrins heterodimer using CG MARTINI method and then compared it with αIIbβ3 integrins heterodimer [15]. Hence, CG MARTINI method is turned out to be a robust simulation method and is used extensively for the investigation of heterodimer conformation of α- and β-subunits. Following Shamloo et al. [7] previous work on the homo-oligomerization of transmembrane α domain, we investigate α- and β-subunit cluster formation employing CG MARTINI molecular dynamics method.
2. Material and Method
2.1. Method
In this section, we briefly review some basic concepts of the MARTINI coarse-graining method. The current investigation uses MARTINI coarse-grained mapping to construct the model [16]. The MARTINI force field includes parameters for a wide spectrum of biomolecules, such as various lipids and all amino acids. Furthermore, it is a protocol to model proteins and peptides. The performance and accuracy of the MARTINI model was compared with experimental results, and it has been found that the model usually performs efficiently for a variety of systems [17]. In this technique, first, the all-atom structure is constructed completely and then it is mapped onto the CG structure by a standard protocol [16]. In this mapping, four relatively heavy atoms (non-hydrogen) are bundled as a single “bead”. Four water molecules are mapped onto a bead. Amino acids are embodied by two to five beads, one for backbone and the others for side chains. The mass of each bead is equal to the mass of the atoms exist in each group of atoms mapped. The mass of each CG bead is supposed to be equal to 72 amu so as to provide computational efficiency for lipids, ions, and water [18]. However, this kind of mapping has smoother “potentials” compared to the all-atom model simulations, for molecular friction is missing. In the following, the interaction among the beads in bonded and none-bonded forms are discussed. Bonded interactions are defined as a set of potential energy functions defined as follows:
In Equations (1–4), the acting bonded sites are represented as indexes i,j,k,l with equilibrium distance db, angle ϕa, and dihedral angles θd and θid. The force constants K are mostly weak, implying flexibility of CG mapping. Vb represents chemical bond potential, and Va is used for the chain stiffness. Vd is a proper dihedral, representing the secondary structure of the peptide backbone and the improper dihedral angle potential. Vid is used to eliminate out-of-plane distortions of planar groups.
None-bonded interactions are divided into two groups. Each pairs of particles i, j at distance rij interact via Lenard-Jones (LJ) potential which can be written as the following:
Well-depth εij represents the strength of interaction depending on the interacting particles types. The effective size of the particles is governed by the LJ parameter σ = 0.47 nm for almost all normal particles. For the special class of particles such as ring-like molecules, slightly reduced parameters are set to model ring–ring interactions; σ = 0.43 nm, and εij is scaled to 75% of the standard value [17]. The full interaction matrix can be found in Reference [19].
Charged groups interact via a Coulombic energy function as below:
Where relative dielectric constant εrel is considered 15. In order to gain smoother potential for none-bonded potential, interactions are cut off at rcut and begin to shift at rshift.
2.2. Models and Simulation Details
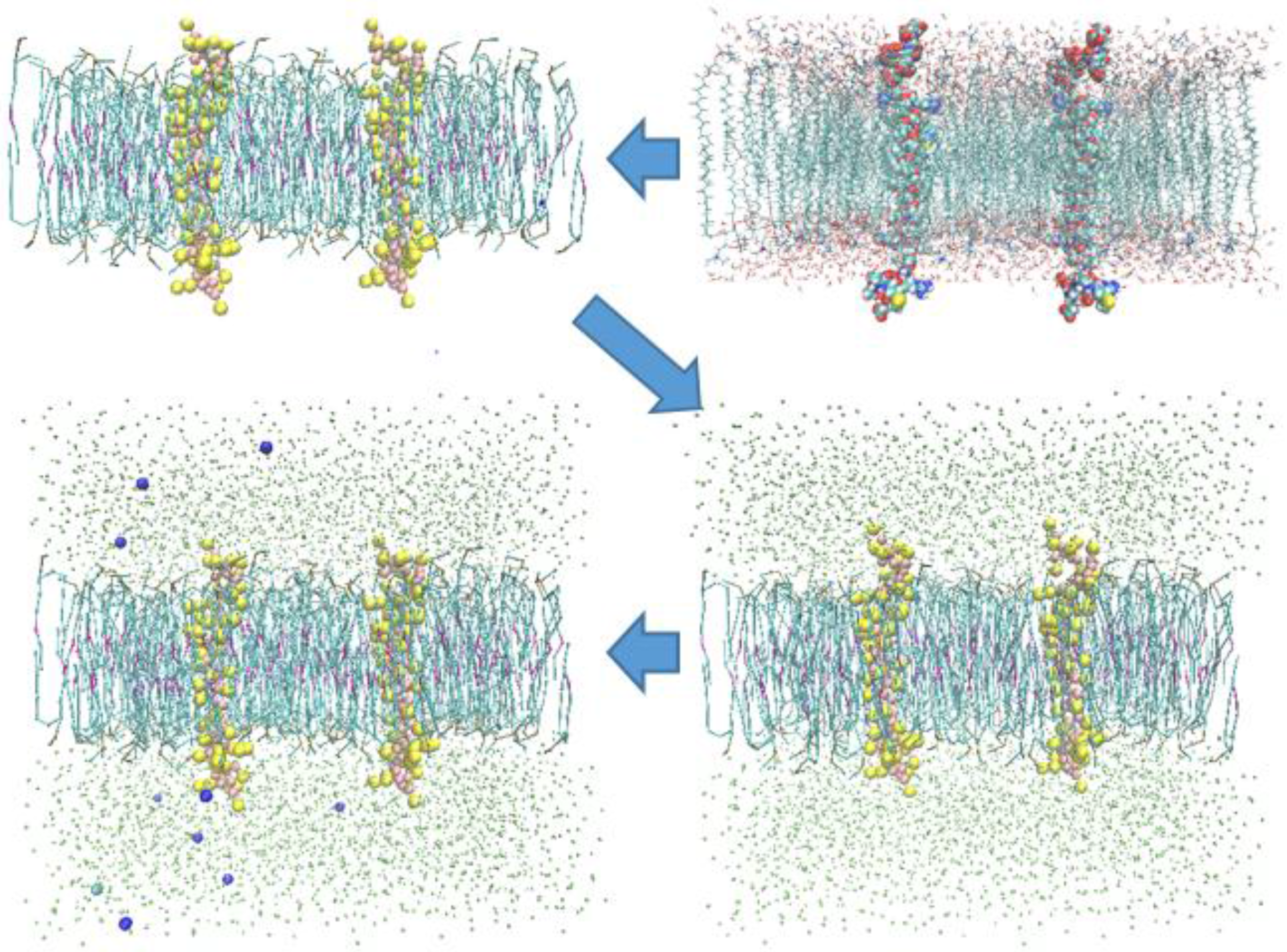
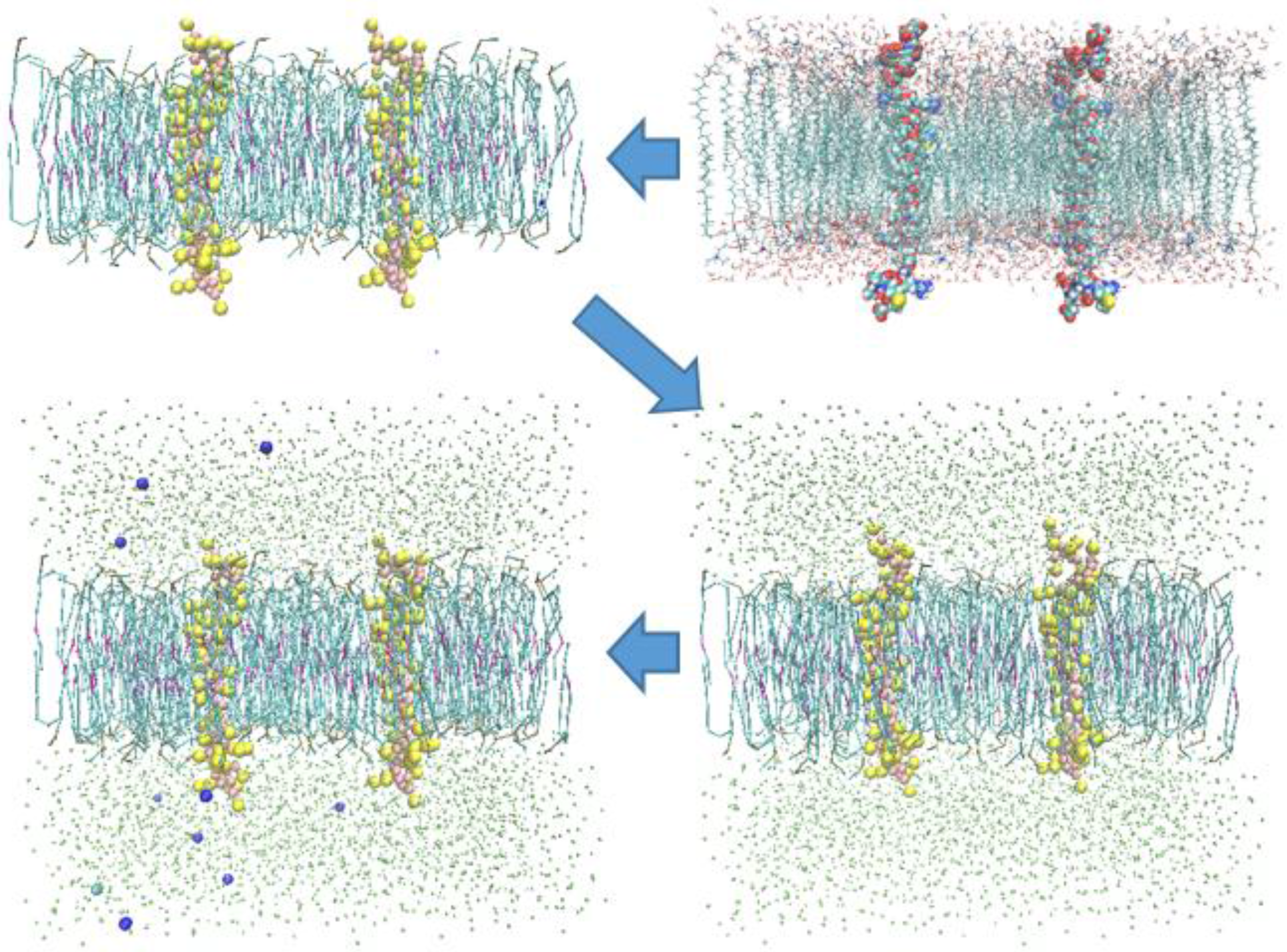
Models for the transmembrane-cytoplasmic α and β domain of integrin αIIbβ3 were downloaded from the published αIIbβ3 structure (Protein Data Bank ID 2KNC). Two identical α and three identical β subdomains were implanted 50 Å apart from one another along the center line joining their geometrical centers in the 100 × 50 Å patch of POPC lipid bilayer and overlapping lipid chains were removed using software VMD [20]. The all-atom models were then converted to their coarse-grained counterparts. The system is solvated and the water box was extended ±15 Å from the proteins. The system was then ionized with NaCl having concentration of 8 mM [21]. Figure 1 shows the procedure of preparing the α-subunit model. The cutoff radius of 12 Å was chosen to calculate none-bonded interactions with a shifting starts at 9 Å to apply a smooth cutoff. Initial system was minimized (relaxed) for 5000 steps, with 30 fs as the time step of the performed simulations. The constant temperature of 323 K was maintained using Langevin dynamics with a damping coefficient of 1 ps−1. The constant pressure of 1 atm was applied with a Nose–Hoover Langevin piston with the period of 2000 fs and a decay time of 1000 fs. It is important to point out that the simulations were implemented in NPT ensemble, and the CG models were obtained from the all-atom models via using VMD software v1.9.1 [20]. Moreover, all of the simulations were performed using Nightly Build of NAMD molecular dynamics program [7,22]. The fundamental idea of using the Steered Molecular Dynamics method is to apply external forces to the steered atoms while other atoms are fixed. We can either attach a virtual spring to each of steered atoms and pull them with a constant speed, or we can apply constant forces to the SMD atoms [22]. Here, we used the two-point steering method, as the four-point steering method would deform the steered subunit to such an extent that the lipid bilayer was not able to accommodate it anymore.
Figure 1.
First, the all-atom model is prepared, then it is converted to the CG model. Subsequently, CG water is added, and the system is solvated. Finally, the system is ionized with 8 mM NaCl solution.
Figure 1.
First, the all-atom model is prepared, then it is converted to the CG model. Subsequently, CG water is added, and the system is solvated. Finally, the system is ionized with 8 mM NaCl solution.
3. Results and Discussion
3.1. α-Subunit Homo-Oligomerization Simulations
Since α- and β-subunits are distant enough in the active state, it is reasonable to assume that they do not interact significantly. Thus, we may employ molecular dynamics simulations to study the homomeric interactions between two α subunits when they are in the active state. In each simulation, at first, the two α subunits were positioned at a distance of approximately 50 Å away from one another. This separation distance between the alpha subunits is larger than the cutoff radius for electrostatic and van der Waals interactions. Hence, no interaction exists between monomers at the beginning of the simulation, i.e., the initial position does not favor the dimer formation.
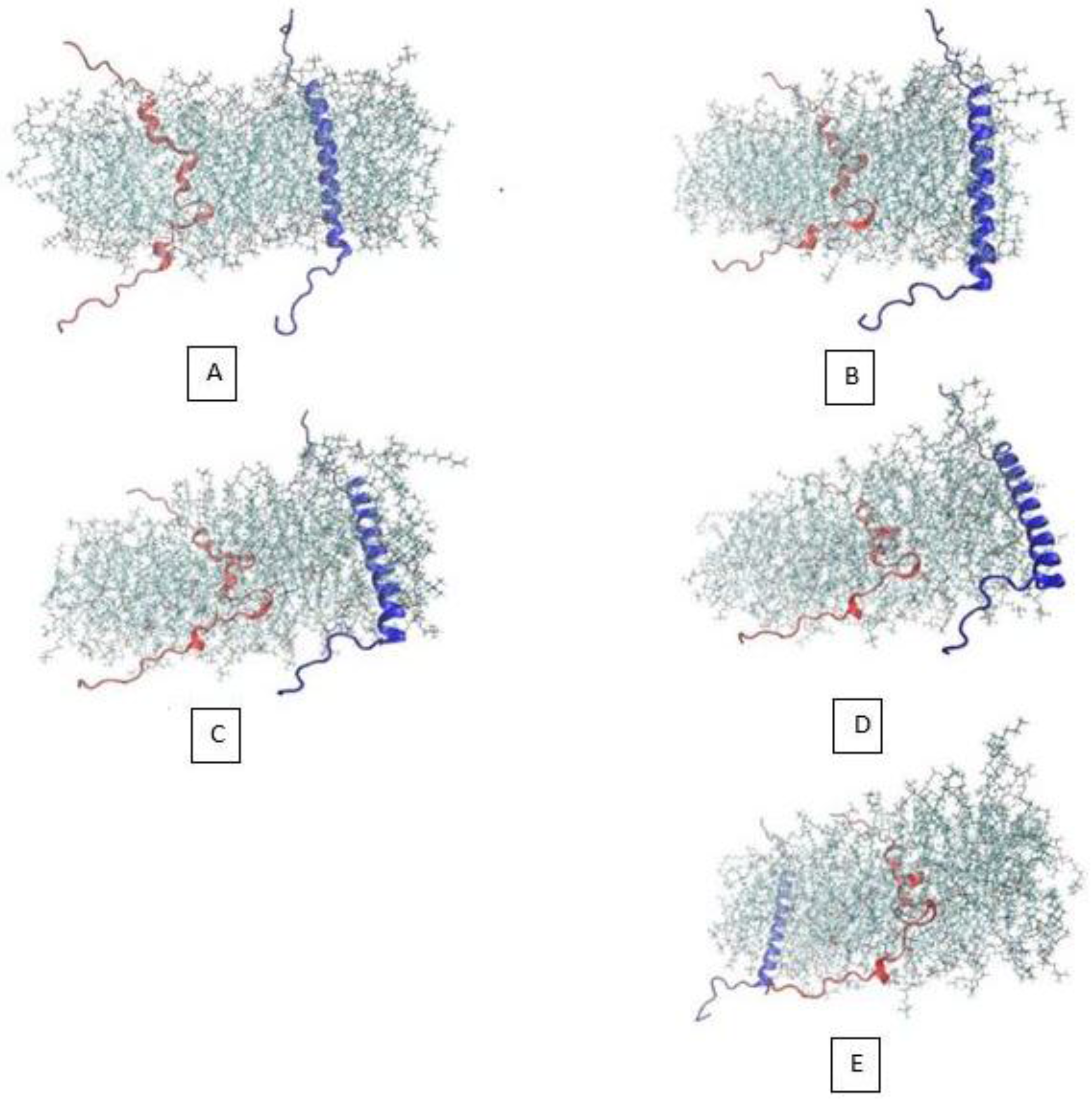
In order to investigate the dimerization of the two α-subunit, we decided to use the all-atom steered molecular dynamics method, which could be a computationally feasible approach if steered atoms were steered with a sufficiently high speed. We placed the two α-subunit monomers in 5 nm from one another, when one is free and the other is steered toward it with the velocity of 0.025 Å/ps. The 5 nm distance is long enough to ensure that the two monomer would not interact at the beginning of the simulation. After 2.5 ns simulation, as it can be observed in Figure 2, because of the considerable steering velocity that the red monomer is being pulled with toward the blue one, the two monomers have approximately maintained in the primary distance. Maintaining in this distance is due to the lipids that have been packed between the two α monomers. The distance of the center of mass of the two monomers is shown in Figure 3b. Figure 3a, shows the van der Waals energy of the two α subunits. In Figure 2E, the location of the two α subunits has changed due to the periodic box condition imposed during the simulation. Intuitively, one can say that the two α-subunits were not given enough time to see one another, interact, and possibly make a stable dimer, which would be detected if the CG molecular dynamics method was employed.
Figure 2.
Different simulation stages of all-atom α-subunit SMD method.
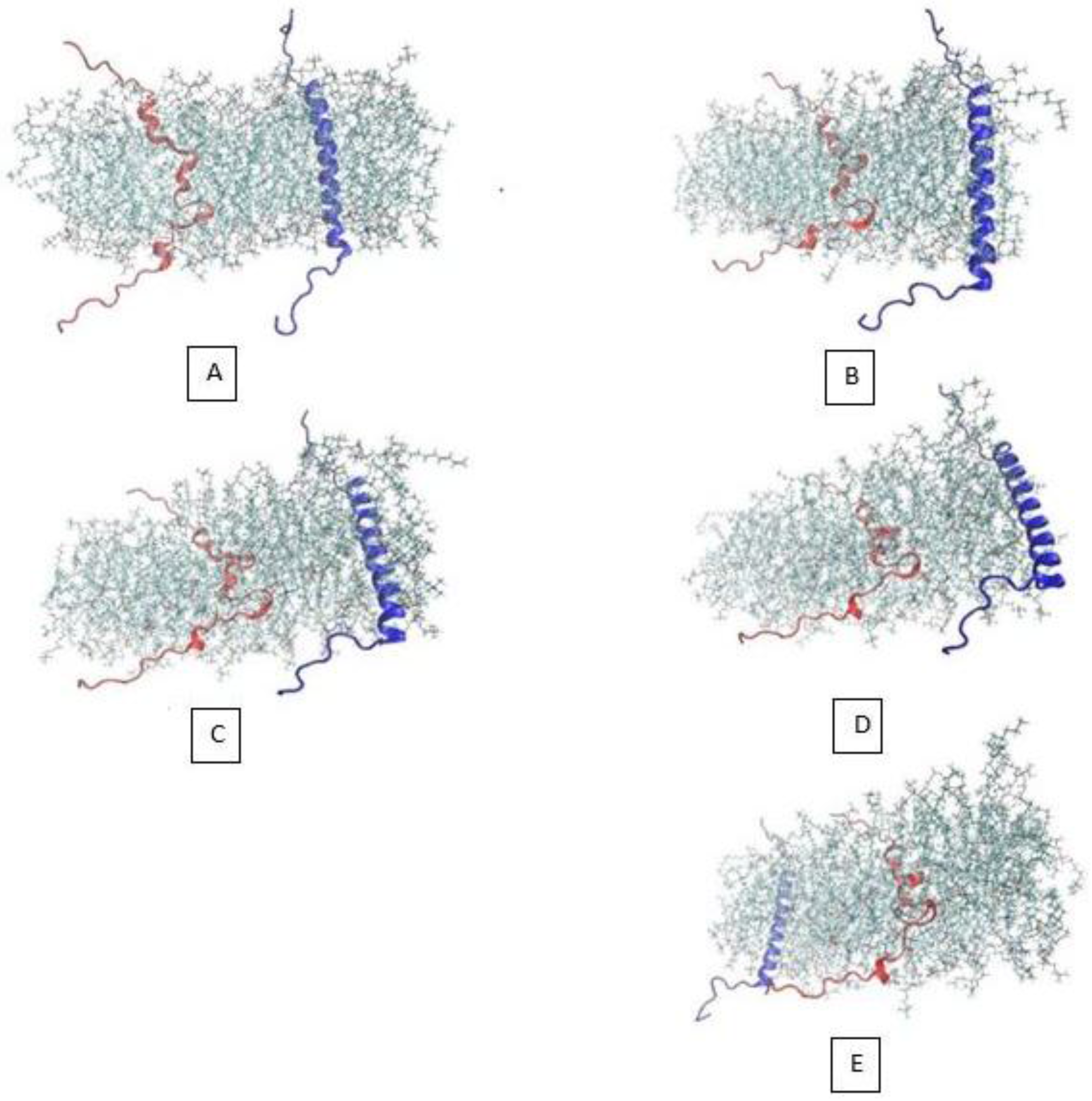
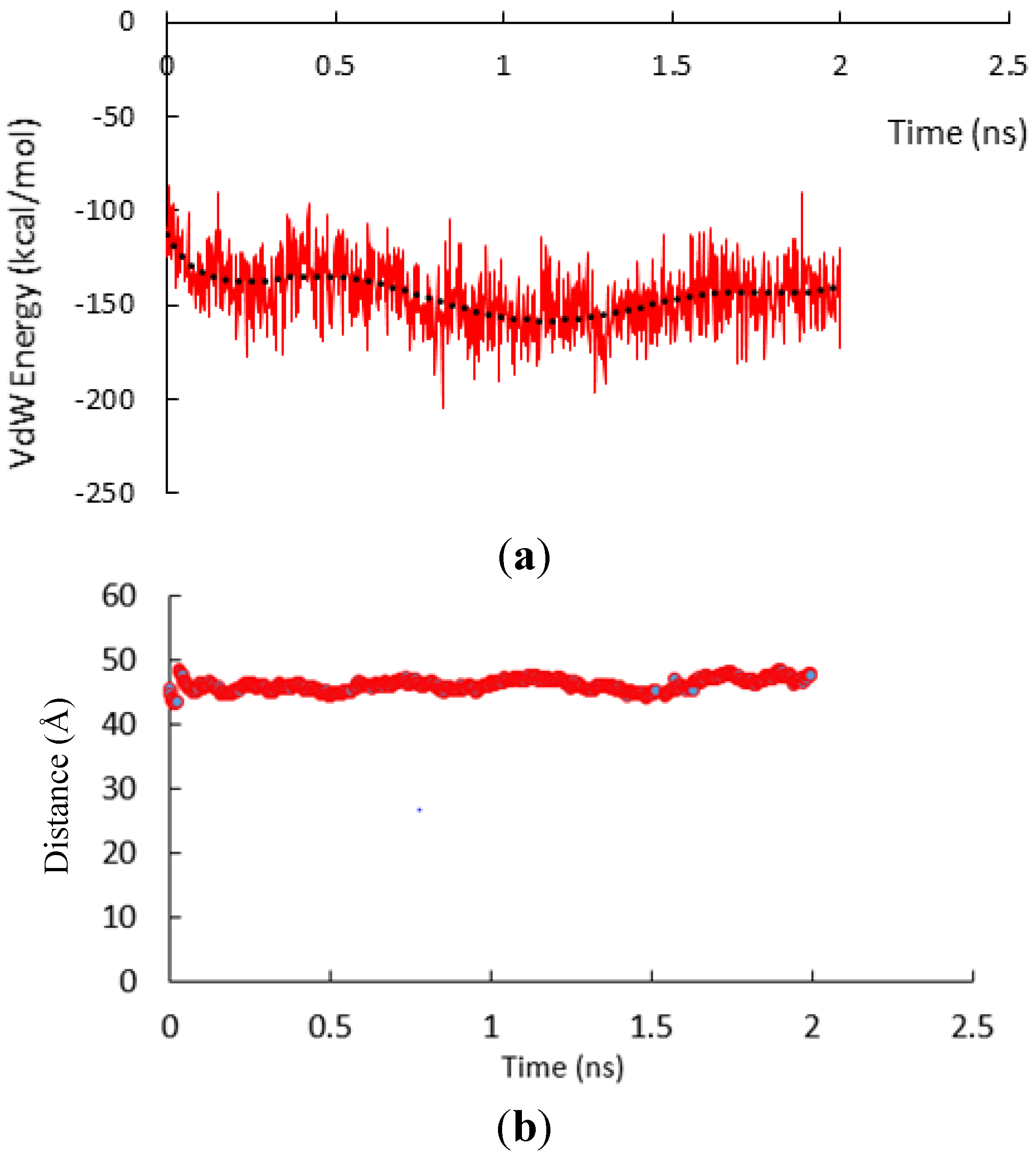
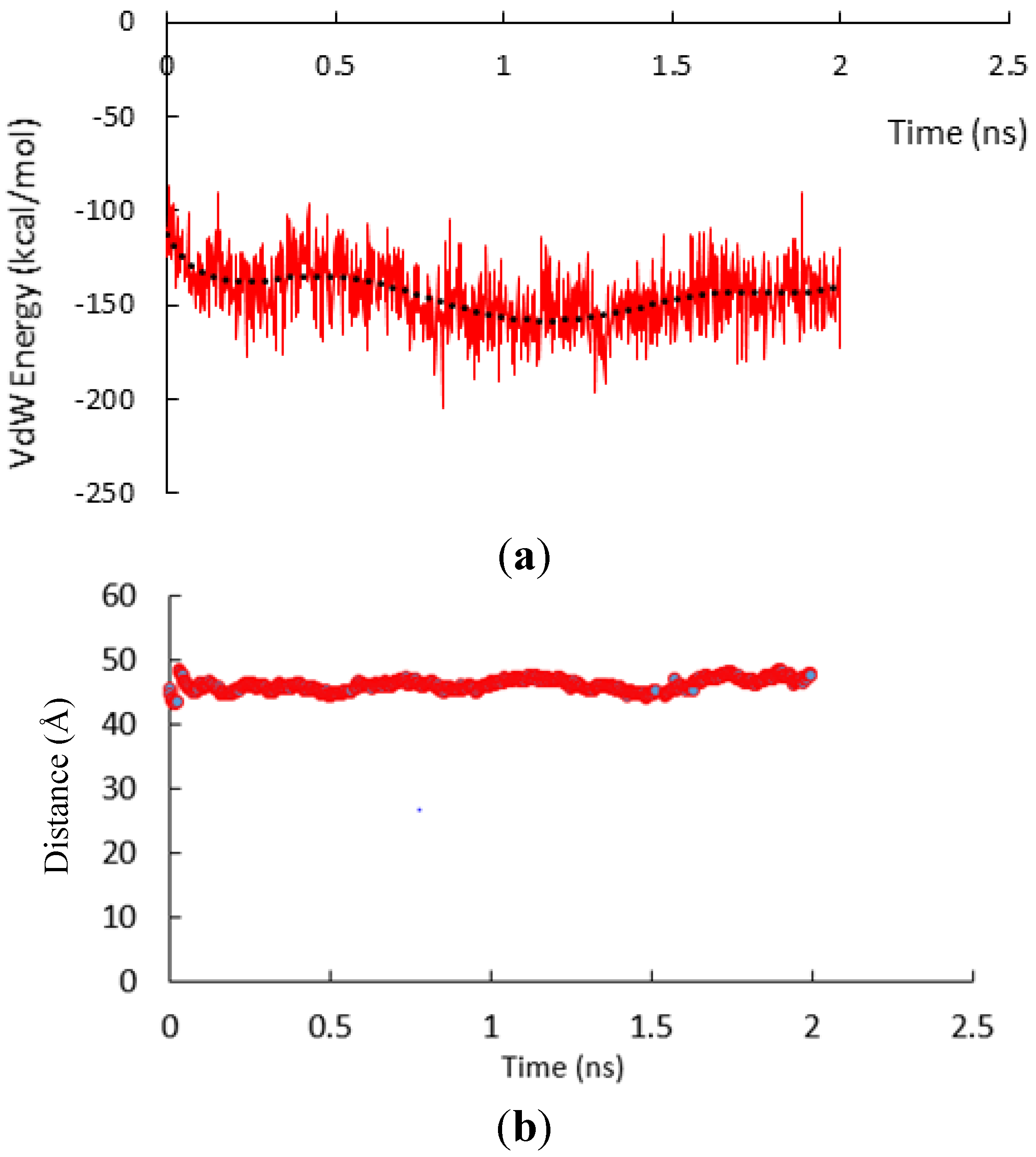
Figure 3.
(a) Van der Waals energy interaction between the two subunits. (b) Distance between the central of mass of the two α-subunit vs time in all atom SMD simulation.
Figure 3.
(a) Van der Waals energy interaction between the two subunits. (b) Distance between the central of mass of the two α-subunit vs time in all atom SMD simulation.
Giving more credence to our hypothesis regarding the artificial nature of lipid packing phenomenon observed due to the usage of SMD, we employed the CG Steered molecular dynamics method with the same steering speed. As it was expected the lipid packing was observed again (Figure 4a). Thus, we have concluded that this phenomenon is actually an artifact. Figure 4b, depicts the pulling force versus frame numbers and the oscillatory nature of the steering force. It can be suggested that the system dynamics are so sensitive to the possible external forces, which are applied to expedite the relaxation process to such an extent that the behavior of the system deviates from what is suggested by the CG molecular dynamics approach.
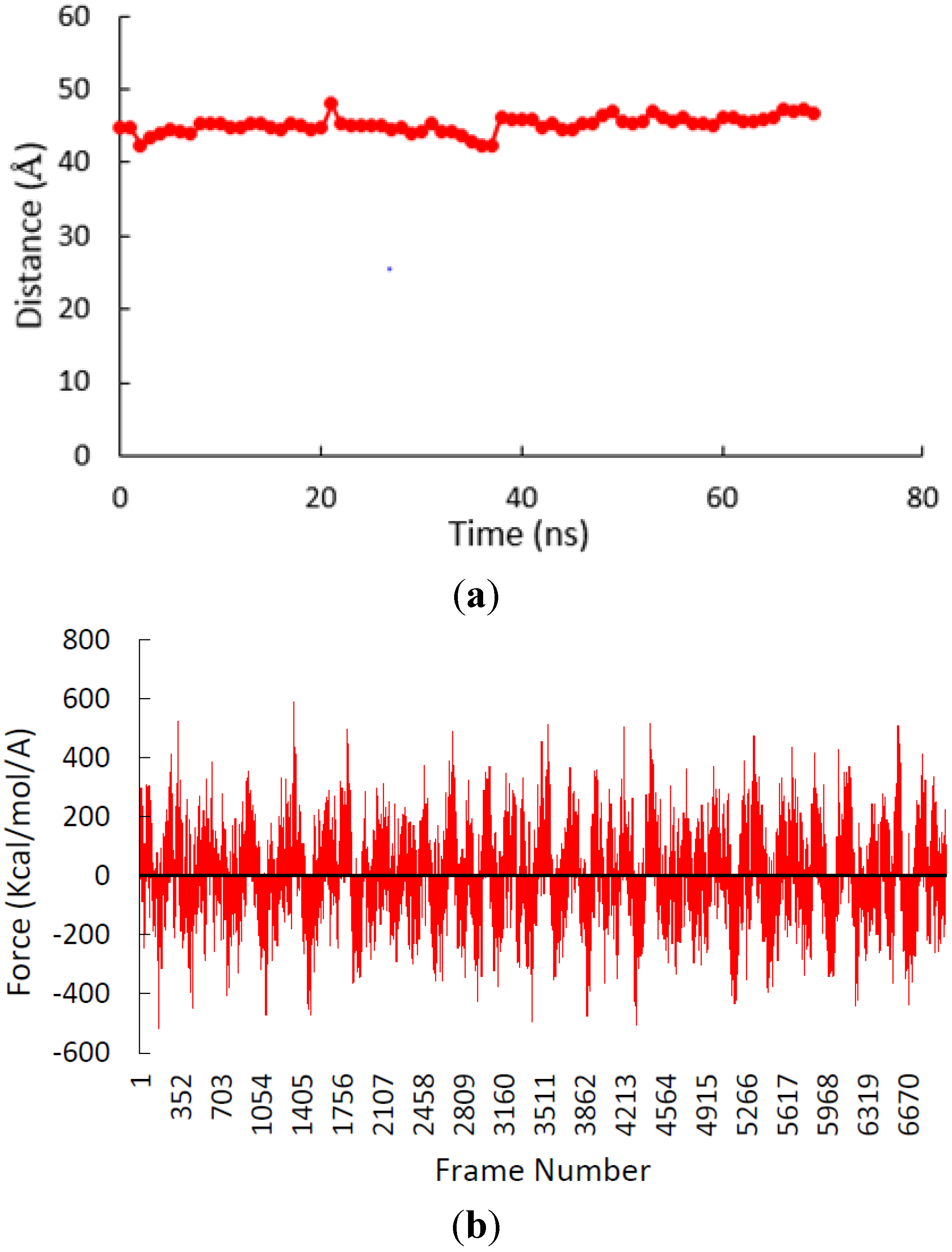
Figure 4.
(a) Distance between the central of mass of the two α-subunit vs time in the CG SMD simulation. (b) Steered force of the CG SMD simulation.
Figure 4.
(a) Distance between the central of mass of the two α-subunit vs time in the CG SMD simulation. (b) Steered force of the CG SMD simulation.
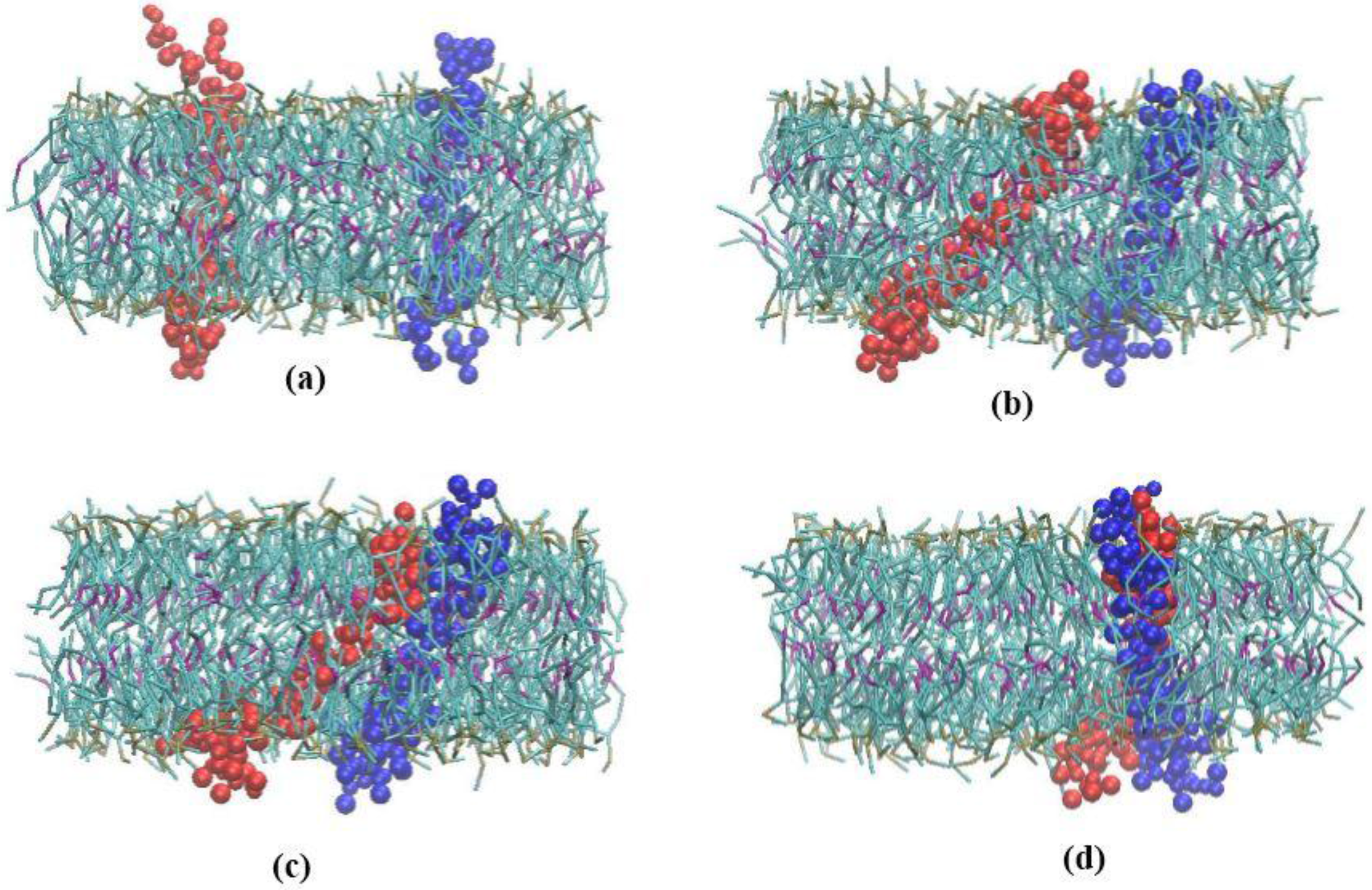
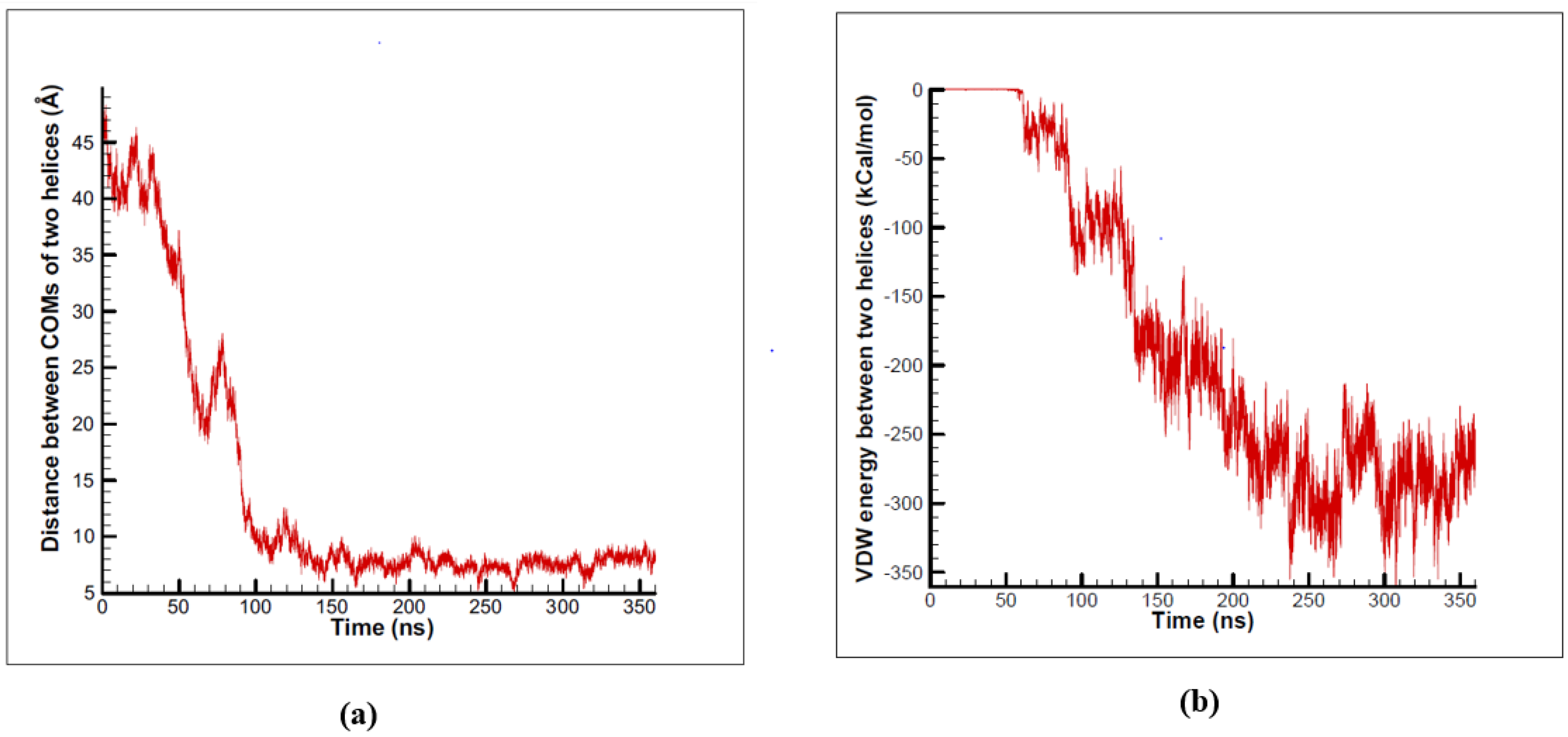
In order to eliminate artificial effects of SMD method, we employed CG method to study the homomeric interactions between two alpha subunits, but this time, they were permitted to diffuse freely in the lipid bilayer membrane. The total number of 10 equilibrium simulations were performed for 100–500 ns [7]. Within 100–300 ns of all of the ten independent αIIbβ3 simulations, transmembrane α homodimer formation was observed (Figure 5). A diagram of the distance between the center of mass of the two monomers is shown for one of the simulations in Figure 6a. The primary distance of the two monomers was 50 Å, as time passed, this distance decreased. After 150 ns, the two monomers stopped moving towards each other and maintained at the distance of 8 Å, which is equal to the total radius of the two monomers. Hence, α subunits tend to form a stable dimer (Figure 5d) [7].
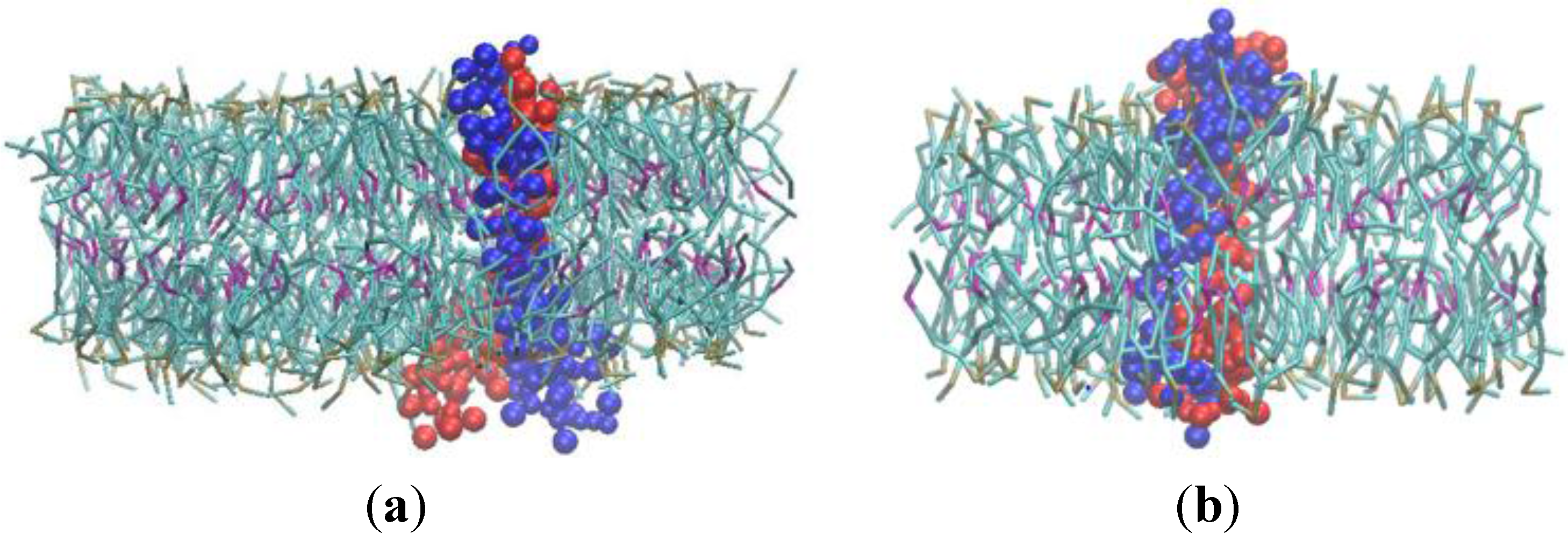
Figure 5.
(a) Initial position of the two α subunits within lipid bilayer when they are positioned 50 Å apart from each other. (b) Simulation snapshot at 50 ns (c) Simulation snapshot at 90 ns and (d) Formation of α homodimer at 150 ns. (Water molecules have been removed).
Figure 5.
(a) Initial position of the two α subunits within lipid bilayer when they are positioned 50 Å apart from each other. (b) Simulation snapshot at 50 ns (c) Simulation snapshot at 90 ns and (d) Formation of α homodimer at 150 ns. (Water molecules have been removed).
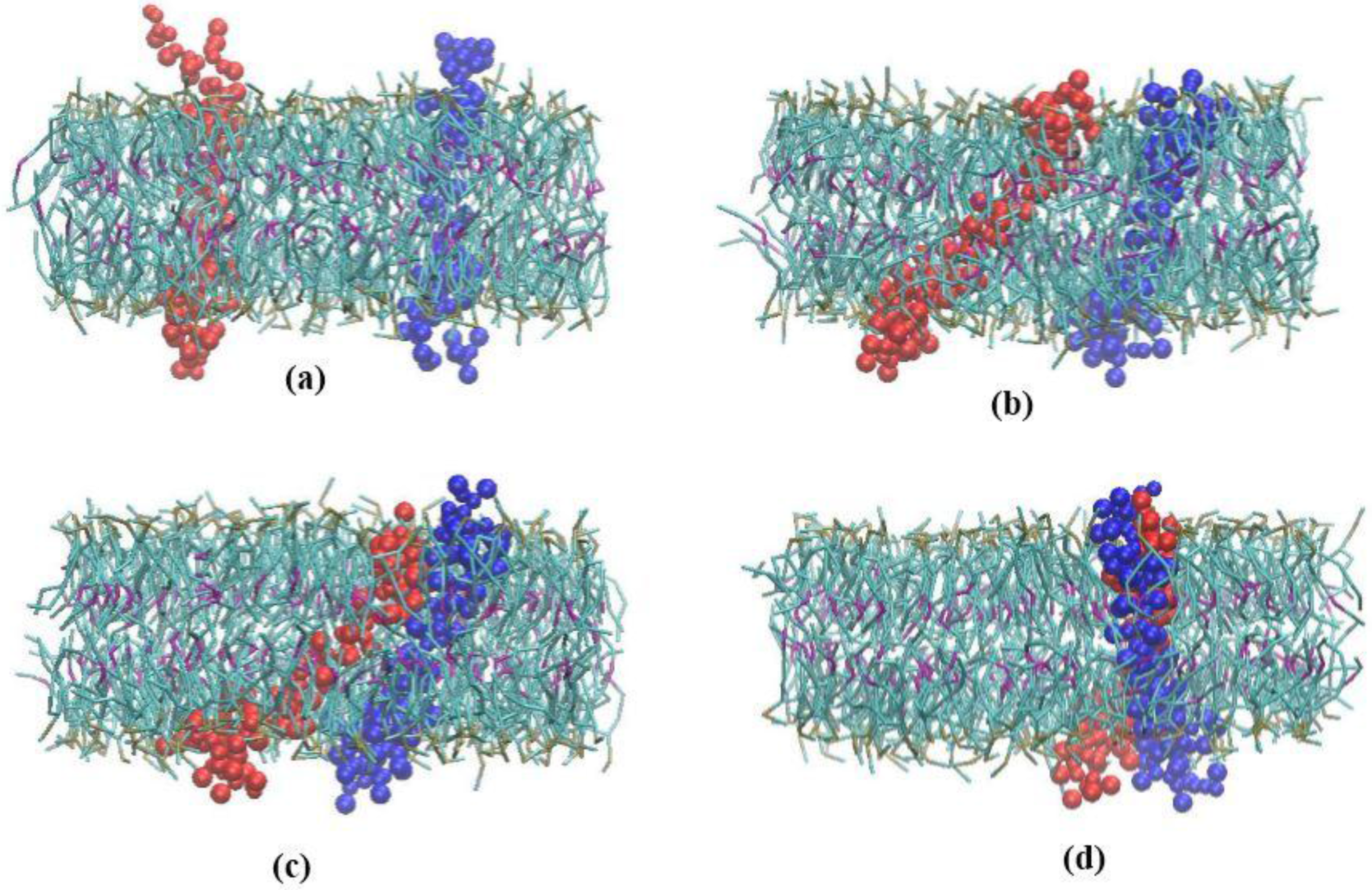
Figure 6.
(a) Distance between center of masses of two alpha subunits decreases during simulation time until it forms a dimer and the distance reaches a stable amount. (b) The Van Der Waals interaction energy of the two alpha subunits decreases during the simulation time.
Figure 6.
(a) Distance between center of masses of two alpha subunits decreases during simulation time until it forms a dimer and the distance reaches a stable amount. (b) The Van Der Waals interaction energy of the two alpha subunits decreases during the simulation time.
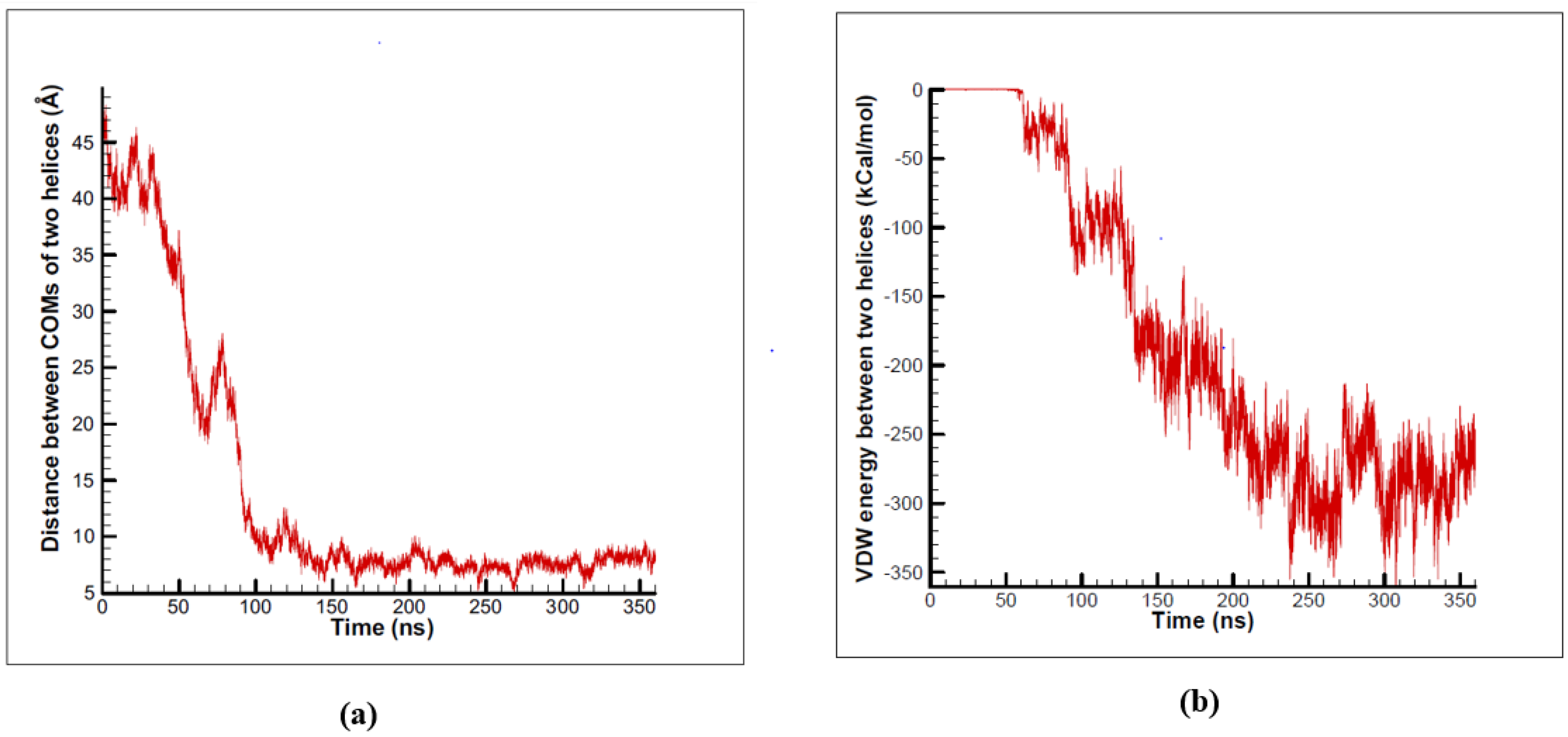
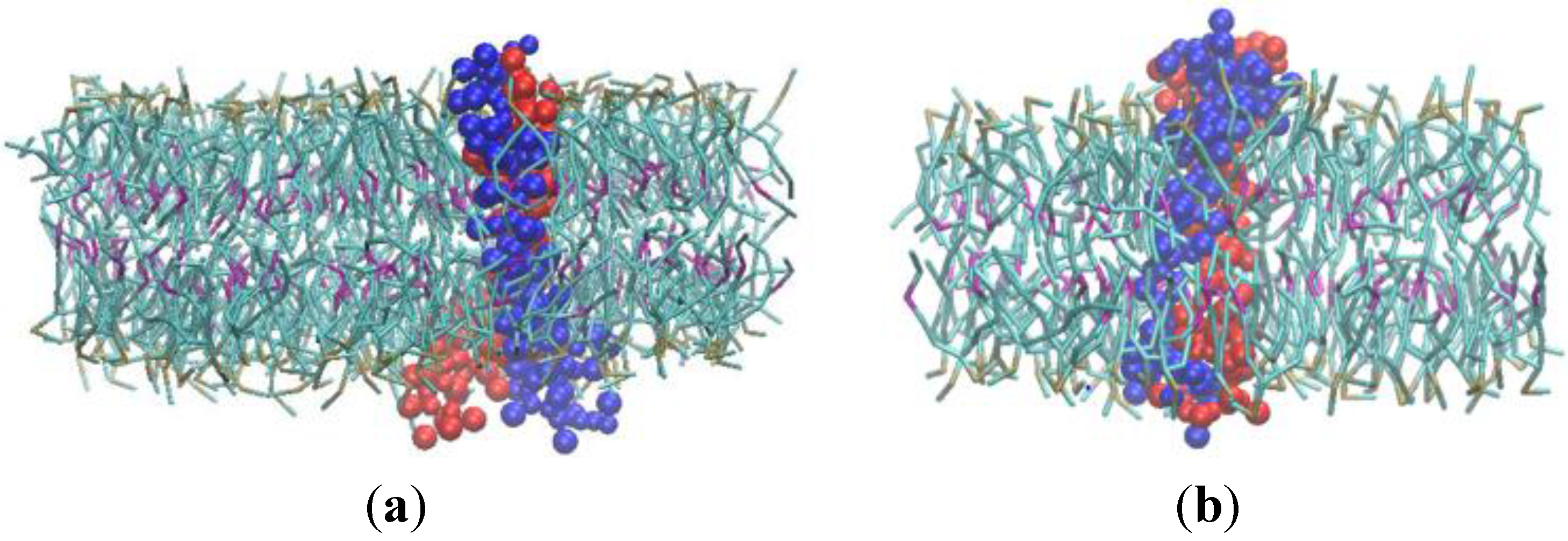
Diagram of the van der Waals interaction energy of the two monomers is shown in Figure 6b. Obviously, at the primary stages of the simulation, the two monomers do not have any van der Waals interactions, and hence, the van der Waals energy of the two is zero. Nevertheless, as time passes, the two monomers get closer and the van der Waals interaction energy decreases gradually. After some fluctuations, finally, van der Waals interaction energy reaches a significantly negative amount, which is an indication that a stable dimer is formed during the simulation [7]. Furthermore, the results of the simulations revealed that α subunits tend to form a dimer with a left-hand structure. Figure 7 shows the structure of a left-hand and right-hand dimer.
Figure 7.
(a) A dimer with a left-hand structure. (b) A dimer with a right-hand structure.
3.2. β-Subunit Homo-Oligomerization Simulations
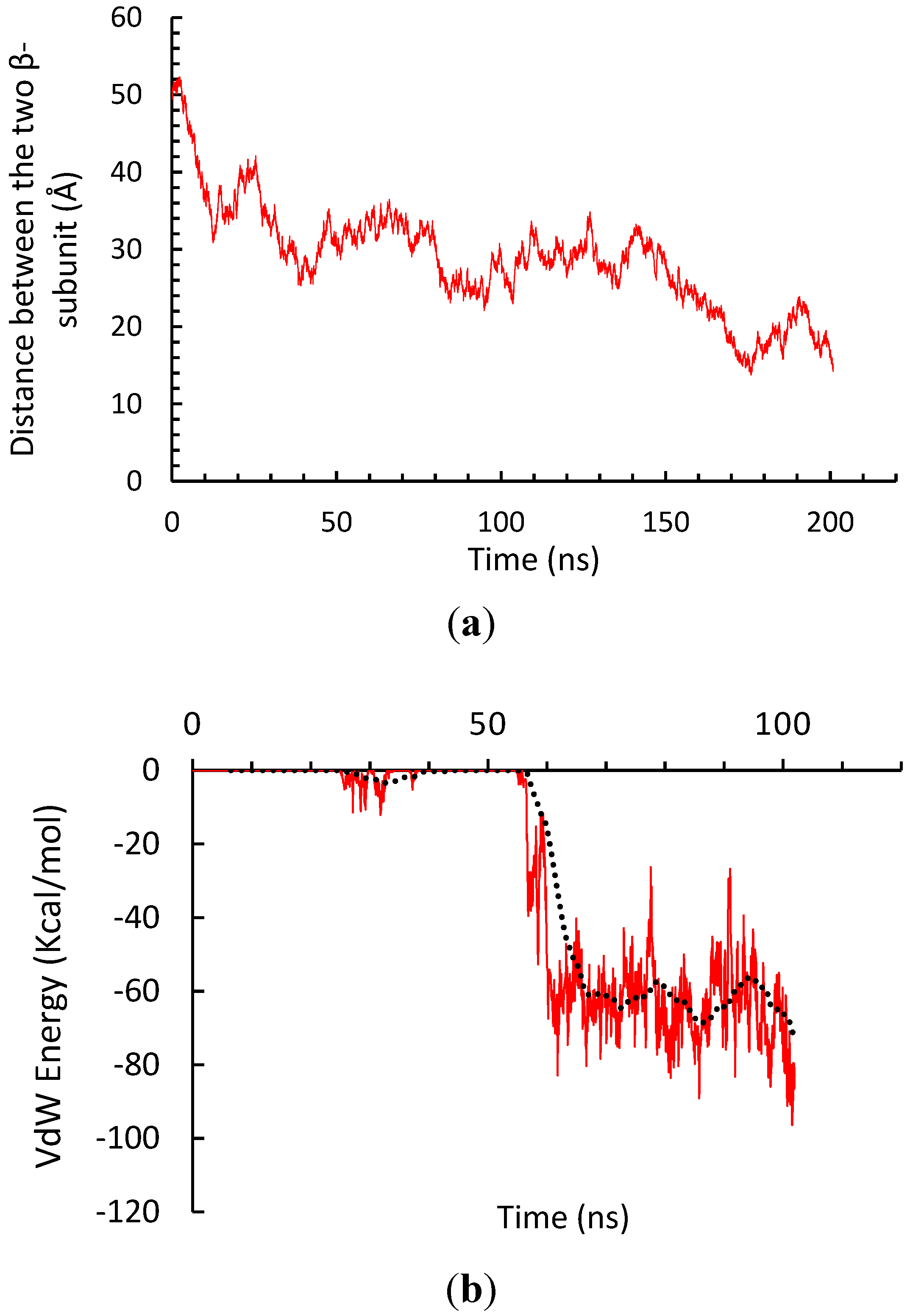
To investigate whether β subunits tend to form a trimer or dimer, we prepared a model which could capture both type of a primary cluster. We placed the three β subunits in the vertices of an equilateral triangle with the side length of 5 nm. Then, similar to the α subunit model, we generated CG lipid and water, and solvated the whole system with NaCl. Figure 8 shows the steps to construct the system. We observed that the two of the three β-subunits had formed a stable dimer. Thus, it is more likely for β-subunit of integrin to make dimers rather than trimers. Figure 9, shows the steps until the stable dimer is formed. Figure 10b, depicts the van der Waals interaction energy of the two β-subunit, which made a dimer. The negative value of this energy shows how stable the dimer is. Figure 10a, shows the distance between the center of mass (COM) of the two β monomers. Similar behavior as the α subunits is exhibited by the two β subunits engaging in the dimer formation.
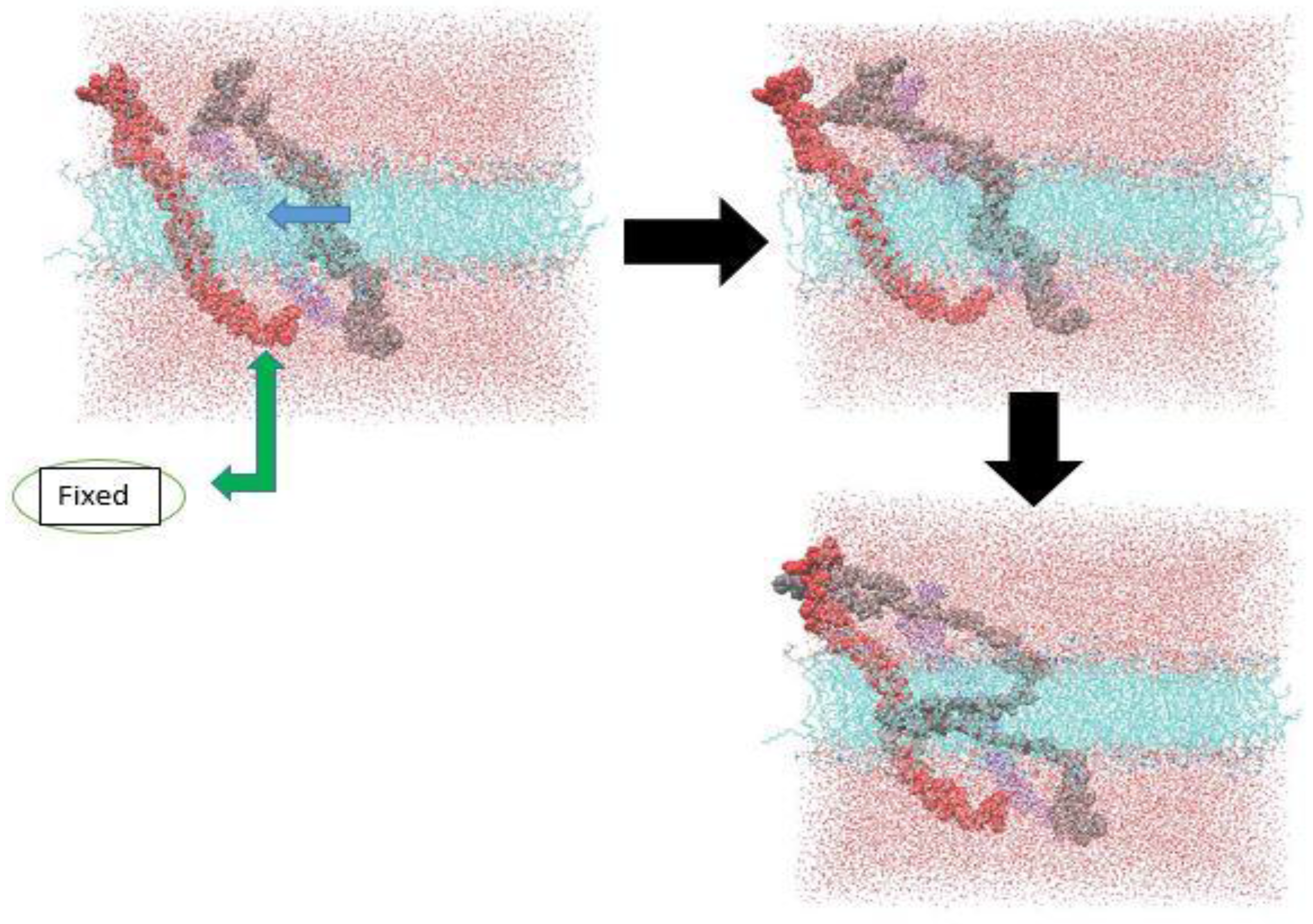
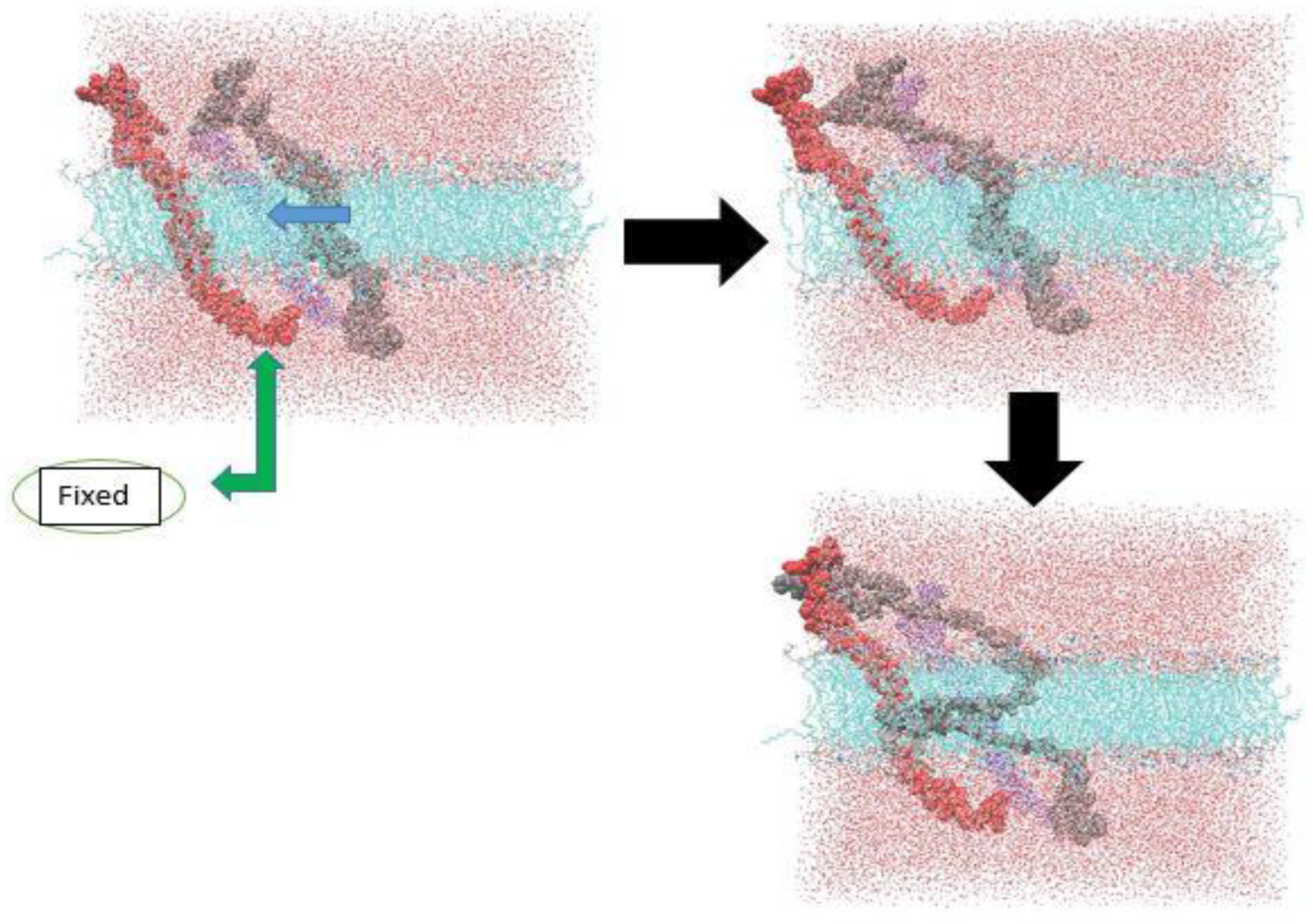
In our last simulation, we decided to use SMD method for investigating the β-subunit dimerization. The all-atom system has about 182,000 atoms, which is very computationally expensive to be run. Therefore, pulling the atoms with a relatively high speed was required to overcome the high cost of the computation. Since the artificial lipid packing phenomenon was observed in the all-atom SMD simulation of the α-subunit, we needed to fix one of the β-subunits and pull another one toward it. If we did not constrain one of the β-subunits, the two β-subunits would move in the same direction and with the same speed, as it was seen for α-subunit monomers. Figure 11, shows the steps and the dimer structure in the initial stages of its formation. The deformation of the gray β-subunit is due to the high speed applied to the steered atoms.
Figure 8.
Preparation of the β-subunit CG simulation.

Figure 9.
Simulation stages of β-subunit dimerization.

Figure 10.
(a) Distance between the center of mass of the two β-subunit in CG simulation. (b) Van der Waals energy of the two β-subunit monomers.
Figure 10.
(a) Distance between the center of mass of the two β-subunit in CG simulation. (b) Van der Waals energy of the two β-subunit monomers.
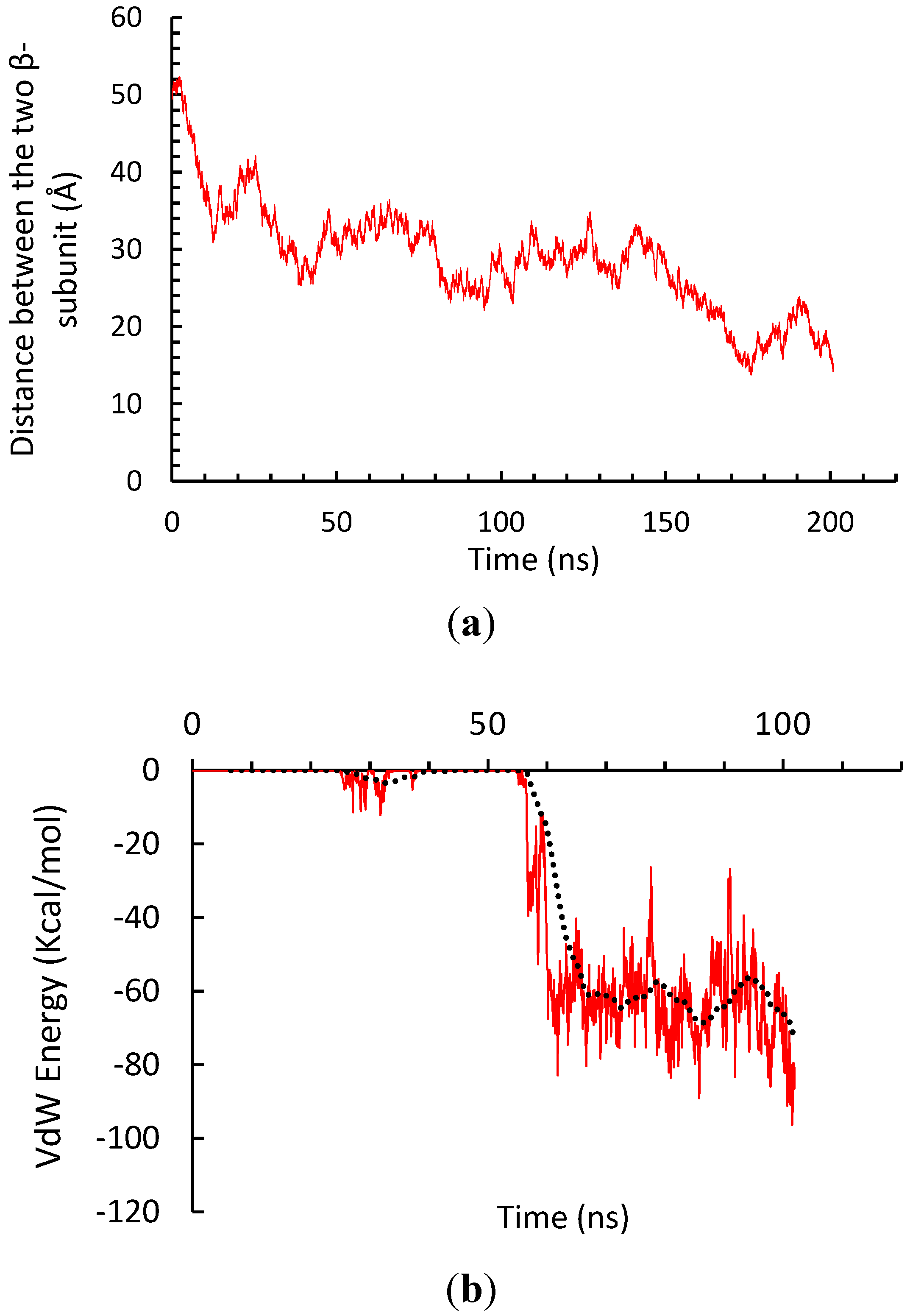
Figure 11.
β-subunit all-atom SMD simulation, when one is fixed.
4. Conclusions
In this paper, homodimer formation of TMC α and β domain of αIIbβ3 integrin were investigated via employing both all-atom SMD and coarse-grained molecular dynamics method. Simulation results demonstrate that α- and β-subunits can form a stable dimer in the membrane bilayer. In fact, the interactions of α- and β-subunits in molecular dynamic simulations can help to have a better grasp of the mechanisms of the formation of focal adhesions, which in turn are important in the development of drug delivery methods. Nonetheless, the possibility of the formation of bindings between α subunit of αIIbβ3 integrin and the heterodimerization of α- and β-subunits have not yet been studied comprehensively. As it was discussed, it is a reasonable assumption to let α- and β-subunits diffuse freely in the lipid bilayer to investigate their homomeric interaction in the active state [7,12]. Nevertheless, it seems of a vital importance to do the simulations while both α- and β-subunits are present in the membrane to better understand the focal adhesion formation. The aforementioned Steered Molecular Dynamics Method applied a relatively high velocity to the steered atoms, which has greatly deviated the system from what actually occurs in the natural system. Therefore, it is crucially important to choose an appropriate steered velocity and spring constant before using this method in the biological systems simulations. According to the simulation results, α- and β-subunits have a tendency to play a role in integrin clustering. Nevertheless, it is not possible to know which of these transmembrane domains contribute more in the integrin clustering process due to the fact that approximately the same amount of time is required for the α- and β-dimers to be formed. In conclusion, the results of this paper may be helpful in investigating the mechanisms of focal adhesion formation.
Acknowledgments
The authors would like to thank the research group members of Mohammad Mofrad at the University of California, Berkeley for their instructions and consultations.
Author Contributions
This paper has been prepared by A. Shampoo, A. Golgoon and A. Ghafar-Zadeh.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Paszek, M.J.; Boettiger, D.; Weaver, V.M.; Hammer, D.A. Integrin Clustering Is Driven by Mechanical Resistance from the Glycocalyx and the Substrate. PLoS Comput. Biol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Engelman, D.M. Involvement of transmembrane domain interactions in signal transduction by α/β integrins. J. Biol. Chem. 2004, 279, 9840–9846. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J.D. Molecular Biology of the Cell; Garland Publishing: New York, NY, USA, 1994; pp. 139–194. [Google Scholar]
- Ginsberg, M.H.; Partridge, A.; Shattil, S.J. Integrin regulation. Curr. Opin. Cell Biol. 2005, 17, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, M.-C. Differences in α–β transmembrane domain interactions among integrins enable diverging integrin signaling. Biochem. Biophys. Res. Commun. 2013, 436, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Srichai, M.B.; Zent, R. Integrin structure and function. In Cell-Extracellular Matrix Interactions in Cancer; Springer: New York, NY, USA, 2010; pp. 19–41. [Google Scholar]
- Shamloo, A.; Nikbin, E.; Mehboudi, N.; Damirchi, B. Homo-oligomerization of transmembrane α-domain of integrin. In Proceedings of the 2014 36th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Chicago, IL, USA, 26–30 August 2014.
- Nelson, D.L.; Lehninger, A.L.; Cox, M.M. Lehninger Principles of Biochemistry; Macmillan: New York, NY, USA, 2008. [Google Scholar]
- Zilly, F.E.; Halemani, N.D.; Walrafen, D.; Spitta, L.; Schreiber, A.; Jahn, R.; Lang, T. Ca2+ induces clustering of membrane proteins in the plasma membrane via electrostatic interactions. EMBO J. 2011, 30, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhu, J.; Springer, T.A.; Luo, B.H. Tests of integrin transmembrane domain homo-oligomerization during integrin ligand binding and signaling. J. Biol. Chem. 2011, 286, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Babu, C.R.; Lear, J.D.; Wand, A.J.; Bennett, J.S.; DeGrado, W.F. Oligomerization of the integrin αIIbβ3: Roles of the transmembrane and cytoplasmic domains. Proc. Natl. Acad. Sci. USA 2001, 98, 12462–12467. [Google Scholar] [CrossRef] [PubMed]
- Mehrbod, M.; Mofrad, M.R. Localized lipid packing of transmembrane domains impedes integrin clustering. PLoS Comput. Biol. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Kalli, A.C.; Hall, B.A.; Campbell, I.D.; Sansom, M.S. A helix heterodimer in a lipid bilayer: Prediction of the structure of an integrin transmembrane domain via multiscale simulations. Structure 2011, 19, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Chng, C.-P.; Yang, L.-W. Coarse-grained models reveal functional dynamics–II. molecular dynamics simulation at the coarse-grained level–theories and biological applications. Bioinform. Biol. Insights 2008, 2, 171–185. [Google Scholar] [PubMed]
- Chng, C.P.; Tan, S.M. Leukocyte integrin αLβ2 transmembrane association dynamics revealed by coarse-grained molecular dynamics simulations. Proteins Struct. Funct. Bioinform. 2011, 79, 2203–2213. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, H.J.; Yefi mov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force-field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Voth, G.A. Coarse-Graining of Condensed Phase and Biomolecular Systems; CRC Press/Taylor and Francis Group: Boca Raton, FL, USA, 2009. [Google Scholar]
- Marrink, S.J.; de Vries, A.H.; Mark, A.E. Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. 2004, 108, 750–760. [Google Scholar] [CrossRef]
- Brouillette, C.; Anantharamaiah, G. Structural models of human apolipoprotein A-I. Biochim. Biophys. Acta 1995, 1256, 103–129. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lodish, H. Molecular Cell Biology; Macmillan: New York, NY, USA, 2008. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shamloo, A.; Golgoon, A.; Zadeh, E.G. Role of α and β Transmembrane Domains in Integrin Clustering. Actuators 2015, 4, 267-280. https://doi.org/10.3390/act4040267
AMA Style
Shamloo A, Golgoon A, Zadeh EG. Role of α and β Transmembrane Domains in Integrin Clustering. Actuators. 2015; 4(4):267-280. https://doi.org/10.3390/act4040267
Chicago/Turabian StyleShamloo, Amir, Ashkan Golgoon, and Ebrahim Ghafar Zadeh. 2015. "Role of α and β Transmembrane Domains in Integrin Clustering" Actuators 4, no. 4: 267-280. https://doi.org/10.3390/act4040267