WGS Analysis and Interpretation in Clinical and Public Health Microbiology Laboratories: What Are the Requirements and How Do Existing Tools Compare?

Abstract
:1. Introduction
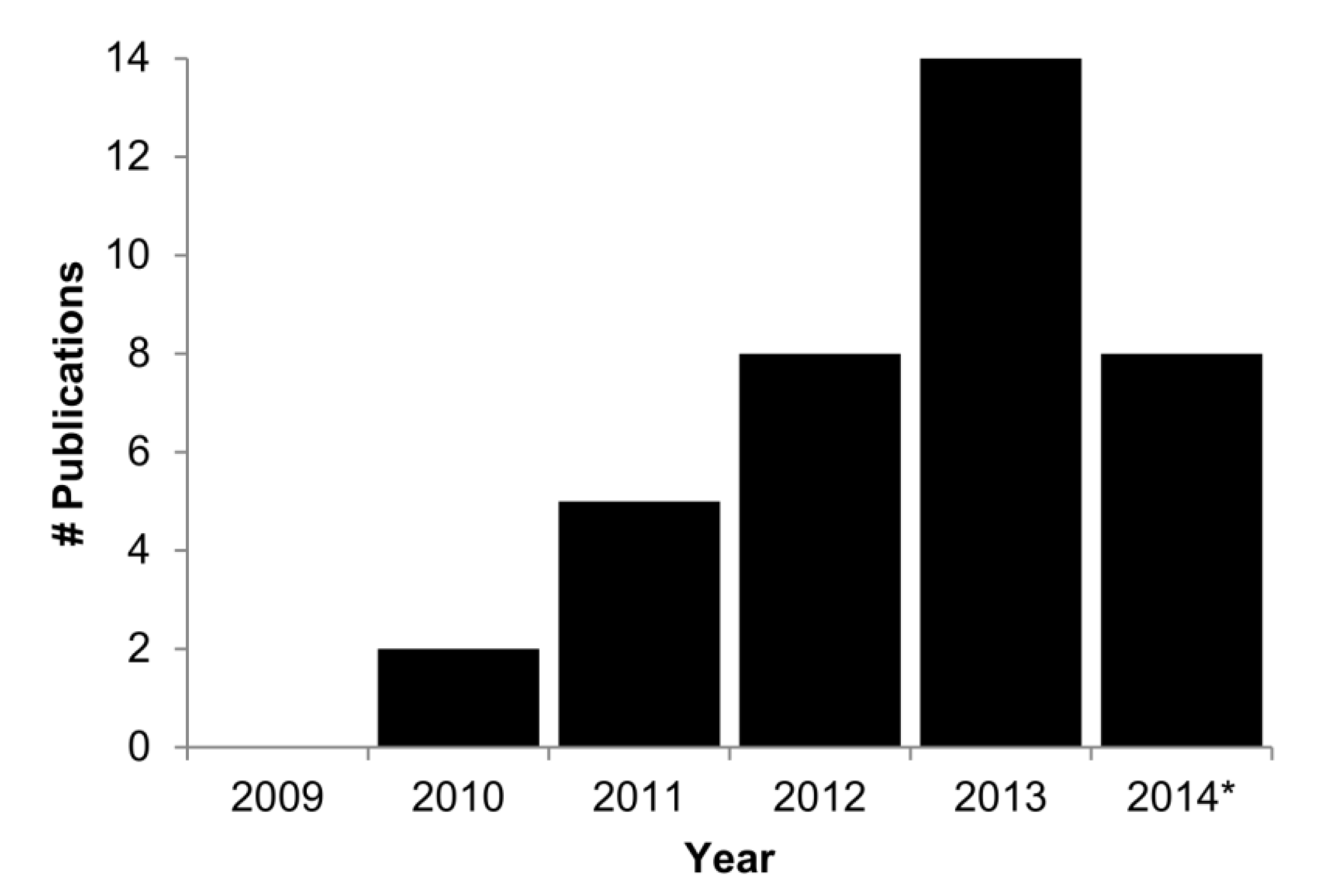
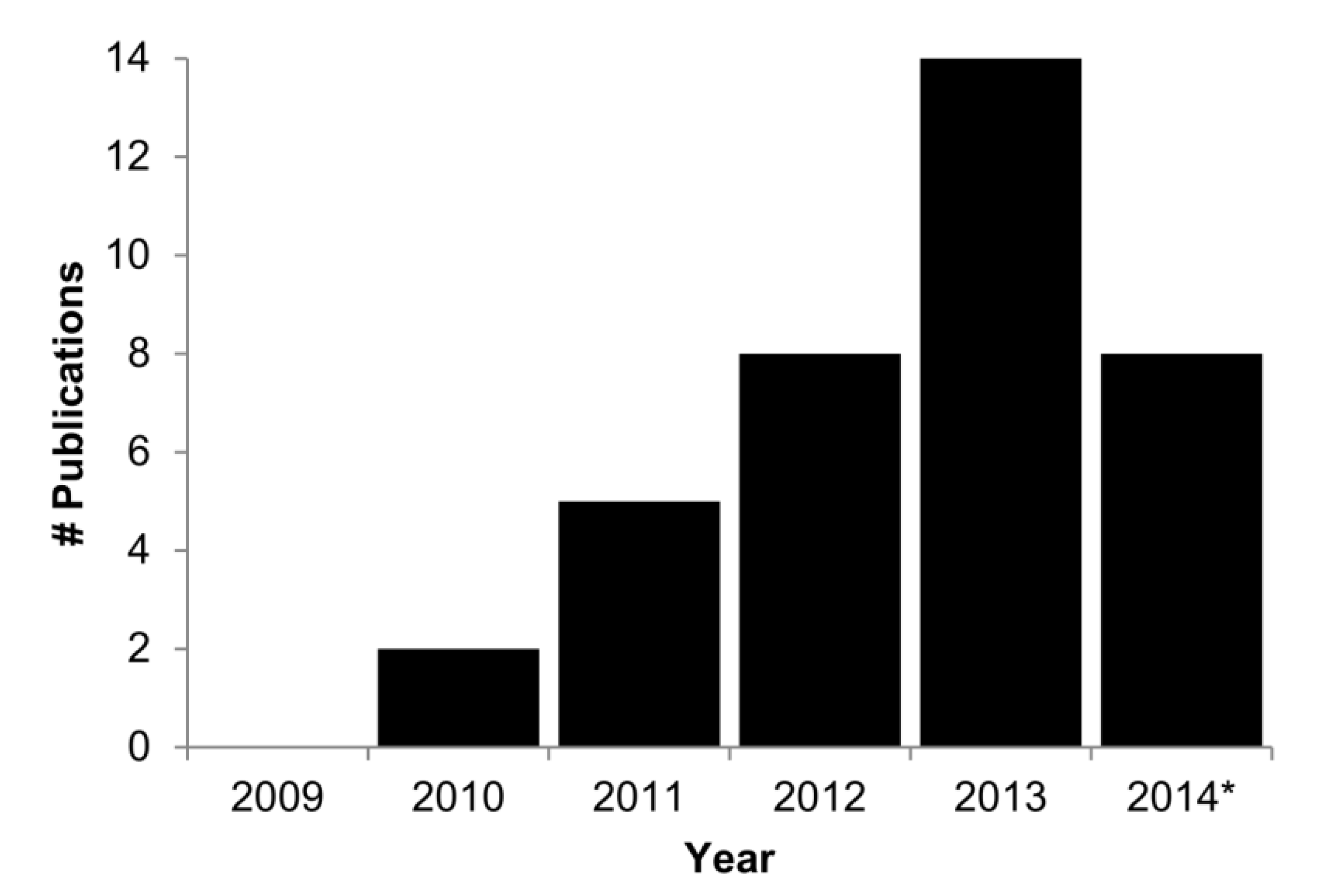
2. What Types of Analyses Are Required?

{kind=link}
| Solution | Date of Publication/First Release | Upload/Analyse Raw Sequence Data | Reference-Based Mapping | de novo Assembly | Variant Calling | Typing analyses (e.g., MLST) | Comparative Typing Analyses | Multiple Sequence Alignment | Phylogenetic Tree/Network Construction |
|---|---|---|---|---|---|---|---|---|---|
| Generic NGS Analysis Solutions: | |||||||||
| BioNumerics a | 1992 | Yes | Yes | Yes | Yes | No c | Yes | Yes | Yes |
| CLC Genomics Workbench a | 2008 | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| Galaxy b | 2007 | Yes | Yes | Yes | Yes | No | No | Yes | Yes |
| Specific Bacterial NGS Analysis Solutions: | |||||||||
| BIGSdb | 2010 | No | No | No | Yes | Yes | Yes | Yes | Yes |
| Center for Genomic Epidemiology Web Portal | 2011 | Yes | No | Yes | Yes | Yes | Yes | No d | Yes |
| Ridom SeqSphere+ a | 2013 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| snp-search | 2013 | No | No | No | No | No | Yes | No e | Yes |
3. Computational Requirements in the Multi-Core, Multi-Processor Era
4. Other Requirements and Considerations
5. Current Techniques for Bacterial WGS Analyses
| Species | Focus a | Type(s) of Methods | Tools b | Ref. |
|---|---|---|---|---|
| Clostridium difficile/Escherichia coli | Transmission | Mapping, variant calling, phylogeny | Independent and/or custom | [15] |
| MLST | de novo assembly, sequence search | Independent and/or custom | ||
| Antibiotic resistance genes | de novo assembly, sequence search | Independent and/or custom | ||
| E. coli | Diversity/Resolution | Mapping, variant calling, phylogeny | Independent and/or custom | [33] |
| Relationship to historical isolates | de novo assembly, multiple sequence alignment | Independent and/or custom | ||
| Gene content comparison | de novo assembly, annotation | Independent and/or custom | ||
| E. coli | Differentiation | Mapping, variant calling | CLC Genomics Workbench | [34] |
| MLST | de novo assembly, sequence search | CLC Genomics Workbench | ||
| Antibiotic resistance genes | de novo assembly, sequence search | CLC Genomics Workbench/custom | ||
| Relationship to historical isolates | Mapping, variant calling, network/de novo assembly | CLC Genomics Workbench/custom | ||
| Klebsiella pneumoniae | Transmission | Mapping, variant calling, minimum spanning tree | Independent and/or custom | [17] |
| Legionella pneumophilia | Differentiation | Mapping, variant calling, phylogeny | Independent and/or custom | [35] |
| Mycobacterium tuberculosis | Diversity/Resolution | Mapping, variant calling, phylogeny | Independent and/or custom | [36] |
| Neisseria meningitidis | Diversity/Resolution | de novo assembly, sequence search | Independent/BIGSdb | [16] |
| MLST, other typing | de novo assembly, sequence search | Independent/BIGSdb | ||
| Shigella sonnei | Differentiation | Mapping, variant calling, phylogeny | Independent and/or custom | [37] |
| Staphylococcus aureus | Differentiation | Mapping, variant calling, multiple sequence alignment (references), phylogeny | Independent and/or custom | [13] |
| S. aureus | Differentiation | Mapping, variant calling, phylogeny | Independent and/or custom | [14] |
| MLST | Mapping, read depth analysis, variant calling | Independent and/or custom | ||
| Toxin genes | Mapping, read depth analysis, variant calling | Independent and/or custom | ||
| Antibiotic resistance genes | Mapping, read depth analysis, variant calling | Independent and/or custom |
6. Generic NGS Analysis Solutions
6.1. BioNumerics
6.2. CLC Genomics Workbench
6.3. Galaxy
6.4. Tools from Sequencing Technology Vendors
7. Specific Bacterial WGS Analysis Solutions
7.1. BIGSdb
7.2. Center for Genomic Epidemiology Web Portal
7.3. Ridom SeqSphere+
7.4. Snp-Search
8. Workflow Management Systems
| Application | Native App/Web-based | Security/Authentication Available? | Workflow Design | Analysis Progress Monitoring | Ability to Stop/Pause/Resume Workflows? | Rerun Workflows with Different Parameters? |
|---|---|---|---|---|---|---|
| Anduril | Native | Unclear | AndurilScript | Yes | Stop only | Yes |
| Biomolecular Hub | Native | Yes | Drag-and-drop | Yes | Stop only | Unclear |
| Chipster | Both | Yes | List | Yes | No | Yes |
| NG6 | Web-based | Yes | Drag-and-drop | Yes | No | No |
| VIBE | Client/server | Yes | Drag-and-drop | Yes | Stop only | No |
| Wildfire | Native | No | Drag-and-drop | Yes | No | Yes |
| Kepler | Native | No | Drag-and-drop | Yes | Yes | Yes |
| KNIME | Both | Yes | Drag-and-drop | Yes | Yes | Yes |
| Pegasus | Native | No | Dax files | Yes | Stop only | Yes |
| Pipeline Pilot | Native | Yes | Drag-and-drop | Yes | Yes | Yes |
| Platform Process Manager (PPM) | Native | Yes | Drag-and-drop | Yes | Yes | Yes |
| Taverna | Native | Yes | Drag-and-drop | Yes | Yes | Yes |
| Product | Generic vs Genetic Analysis-Specific | NGS Analysis components? | Integration of third-party analyses? | Languages for Integration of New Tools | Reference and/or Vendor |
|---|---|---|---|---|---|
| Anduril | Specific | Limited | Yes | XML | [60] |
| Biomolecular Hub | Specific | No | Unclear | Java | IDBS, Guildford, UK |
| Chipster | Specific | Limited | Yes | R/configuration changes | [61] |
| NG6 | Specific | Yes | Yes | Python (through XML)/Smarty template | [62] |
| VIBE | Specific | No | Yes | XML/Java (through API) | INCOGEN Inc., Williamsburg, VA, USA |
| Wildfire | Specific | No | Yes | Extended ACD syntax (through API) | [58] |
| Kepler | Generic | No | Yes | Java | [63] |
| KNIME | Generic | No | Yes | Eclipse Plugins | KNIME.com AG, Zurich, Switzerland |
| Pegasus | Generic | No | Yes | DAX workflow definitions | [64] |
| Pipeline Pilot | Generic | Yes | Yes | Java/Perl/VBScript/SOAP | Accelrys, Inc., San Diego, CA, USA |
| Platform Process Manager (PPM) | Generic | No | Yes | Custom scripts | IBM Corp., Armonk, NY, USA |
| Taverna | Generic | Yes | Yes | SOAP/template library | [65] |
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Croucher, N.J.; Harris, S.R.; Fraser, C.; Quail, M.A.; Burton, J.; van der Linden, M.; McGee, L.; von Gottberg, A.; Song, J.H.; Ko, K.S.; et al. Rapid pneumococcal evolution in response to clinical interventions. Science 2011, 331, 430–434. [Google Scholar] [CrossRef]
- Castillo-Ramírez, S.; Corander, J.; Marttinen, P.; Aldeljawi, M.; Hanage, W.P.; Westh, H.; Boye, K.; Gulay, Z.; Bentley, S.D.; Parkhill, J.; et al. Phylogeographic variation in recombination rates within a global clone of methicillin-resistant Staphylococcus aureus. Genome Biol. 2012. [Google Scholar] [CrossRef]
- Bryant, J.M.; Schürch, A.C.; van Deutekom, H.; Harris, S.; de Beer, J.L.; de Jager, V.; Kremer, K.; van Hijum, S.A.F.T.; Siezen, R.J.; Borgdoff, M.; et al. Inferring patient to patient transmission of Mycobacterium tuberculosis from whole genome sequencing data. BMC Infect. Dis. 2013, 13, 110. [Google Scholar] [CrossRef] [Green Version]
- Holt, K.E.; Baker, S.; Weill, F.-X.; Holmes, E.C.; Kitchen, A.; Yu, J.; Sangai, V.; Brown, D.J.; Coia, J.E.; Kim, D.W.; et al. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nat. Genet. 2012, 44, 1056–1059. [Google Scholar] [CrossRef] [Green Version]
- Chewapreecha, C.; Harris, S.R.; Croucher, N.J.; Turner, C.; Marttinen, P.; Cheng, L.; Pessia, A.; Aanensen, D.M.; Mather, A.E.; Page, A.J.; et al. Dense genomic sampling identifies highways of pneumococcal recombination. Nat. Genet. 2014, 46, 305–309. [Google Scholar] [CrossRef]
- Casali, N.; Nikolayevskyy, V.; Balabanova, Y.; Harris, S.R.; Ignatyeva, O.; Kontsevaya, I.; Corander, J.; Bryant, J.; Parkhill, J.; Nejentsev, S.; et al. Evolution and transmission of drug-resistant tuberculosis in a russian population. Nat. Genet. 2014, 46, 279–286. [Google Scholar] [CrossRef]
- Köser, C.U.; Ellington, M.J.; Cartwright, E.J.P.; Gillespie, S.H.; Brown, N.M.; Farrington, M.; Holden, M.T.G.; Dougan, G.; Bentley, S.D.; Parkhill, J.; et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 2012, 8, e1002824. [Google Scholar] [CrossRef] [Green Version]
- Didelot, X.; Bowden, R.; Wilson, D.J.; Peto, T.E.A.; Crook, D.W. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 2012, 13, 601–612. [Google Scholar]
- Török, M.E.; Peacock, S.J. Rapid whole-genome sequencing of bacterial pathogens in the clinical microbiology laboratory—pipe dream or reality? J. Antimicrob. Chemother. 2012, 67, 2307–2308. [Google Scholar] [CrossRef]
- Aarestrup, F.M.; Brown, E.W.; Detter, C.; Gerner-Smidt, P.; Gilmour, M.W.; Harmsen, D.; Hendriksen, R.S.; Hewson, R.; Heymann, D.L.; Johansson, K.; et al. Integrating genome-based informatics to modernize global disease monitoring, information sharing, and response. Emerg. Infect. Dis. 2012. [Google Scholar] [CrossRef]
- Caboche, S.; Audebert, C.; Hot, D. High-throughput sequencing, a versatile weapon to support genome-based diagnosis in infectious diseases: Applications to clinical bacteriology. Pathogens 2014, 3, 258–279. [Google Scholar] [CrossRef]
- Van Belkum, A.; Durand, G.; Peyret, M.; Chatellier, S.; Zambardi, G.; Schrenzel, J.; Shortridge, D.; Engelhardt, A.; Dunne, W.M. Rapid clinical bacteriology and its future impact. Ann. Lab. Med. 2013, 33, 14–27. [Google Scholar] [CrossRef]
- Harris, S.R.; Cartwright, E.J.P.; Török, M.E.; Holden, M.T.G.; Brown, N.M.; Ogilvy-Stuart, A.L.; Ellington, M.J.; Quail, M.A.; Bentley, S.D.; Parkhill, J.; et al. Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: A descriptive study. Lancet Infect. Dis. 2012, 13, 130–136. [Google Scholar]
- Köser, C.U.; Holden, M.T.G.; Ellington, M.J.; Cartwright, E.J.P.; Brown, N.M.; Ogilvy-Stuart, A.L.; Hsu, L.Y.; Chewapreecha, C.; Croucher, N.J.; Harris, S.R.; et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N. Engl. J. Med. 2012, 366, 2267–2275. [Google Scholar] [CrossRef]
- Eyre, D.W.; Golubchik, T.; Gordon, N.C.; Bowden, R.; Piazza, P.; Batty, E.M.; Ip, C.L.C.; Wilson, D.J.; Didelot, X.; O’Connor, L.; et al. A pilot study of rapid benchtop sequencing of Staphylococcus aureus and Clostridium difficile for outbreak detection and surveillance. BMJ Open 2012, 2, e001124. [Google Scholar]
- Jolley, K.A.; Hill, D.M.C.; Bratcher, H.B.; Harrison, O.B.; Feavers, I.M.; Parkhill, J.; Maiden, M.C.J. Resolution of a meningococcal disease outbreak from whole-genome sequence data with rapid web-based analysis methods. J. Clin. Microbiol. 2012, 50, 3046–3053. [Google Scholar] [CrossRef]
- Snitkin, E.S.; Zelazny, A.M.; Thomas, P.J.; Stock, F.; NISC Comparative Sequencing Program; Henderson, D.K.; Palmore, T.N.; Segre, J.A. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci. Transl. Med. 2012, 4, 148ra116. [Google Scholar]
- Török, M.E.; Reuter, S.; Bryant, J.; Köser, C.U.; Stinchcombe, S.V.; Nazareth, B.; Ellington, M.J.; Bentley, S.D.; Smith, G.P.; Parkhill, J.; et al. Rapid whole-genome sequencing for investigation of a suspected tuberculosis outbreak. J. Clin. Microbiol. 2013, 51, 611–614. [Google Scholar]
- Loman, N.J.; Constantinidou, C.; Christner, M.; Rohde, H.; Chan, J.Z.; Quick, J.; Weir, J.C.; Quince, C.; Smith, G.P.; Betley, J.R.; et al. A culture-independent sequence-based metagenomics approach to the investigation of an outbreak of shiga-toxigenic Escherichia coli O104:H4. JAMA 2013, 309, 1502–1510. [Google Scholar] [CrossRef]
- Fricke, W.F.; Rasko, D.A. Bacterial genome sequencing in the clinic: Bioinformatic challenges and solutions. Nat. Rev. Genet. 2014, 15, 49–55. [Google Scholar] [CrossRef]
- Maiden, M.C.J.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.J.; Zurth, K.; Caugant, D.A.; et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 1998, 95, 3140–3145. [Google Scholar] [CrossRef]
- Bennedsen, M.; Stuer-Lauridsen, B.; Danielsen, M.; Johansen, E. Screening for antimicrobial resistance genes and virulence factors via genome sequencing. Appl. Environ. Microbiol. 2011, 77, 2785–2787. [Google Scholar] [CrossRef]
- Inouye, M.; Conway, T.C.; Zobel, J.; Holt, K.E. Short read sequence typing (SRST): Multi-locus sequence types from short reads. BMC Genomics 2012. [Google Scholar] [CrossRef]
- Stoesser, N.; Batty, E.M.; Eyre, D.W.; Morgan, M.; Wyllie, D.H.; Del Ojo Elias, C.; Johnson, J.R.; Walker, A.S.; Peto, T.E.; Crook, D.W. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J. Antimicrob. Chemother. 2013, 68, 2234–2244. [Google Scholar]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Edwards, D.J.; Bio21 Institute, University of Melbourne, 30 Flemington Road, Melbourne, Parkville, VIC 3052, Australia. Personal communication, 2014.
- Edwards, D.J.; Holt, K. Beginner’s guide to comparative bacterial genome analysis using next-generation sequence data. Microb. Inform. Exp. 2013. [Google Scholar] [CrossRef]
- Li, H.; Homer, N. A survey of sequence alignment algorithms for next-generation sequencing. Brief. Bioinfor. 2010, 11, 473–483. [Google Scholar] [CrossRef]
- Nagarajan, N.; Pop, M. Sequence assembly demystified. Nat. Rev. Genet. 2013, 14, 157–167. [Google Scholar] [CrossRef]
- Church, P.C.; Goscinski, A.; Holt, K.; Inouye, M.; Ghoting, A.; Makarychev, K.; Reumann, M. Design of multiple sequence alignment algorithms on parallel, distributed memory supercomputers. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2011, 2011, 924–927. [Google Scholar]
- Menhorn, F.; Reumann, M. Genome assembly framework on massively parallel, distributed memory supercomputers. Biomed. Tech. 2013. [Google Scholar] [CrossRef]
- Pop, M. Genome assembly reborn: Recent computational challenges. Brief. Bioinfor. 2009, 10, 354–366. [Google Scholar] [CrossRef]
- Grad, Y.H.; Lipsitch, M.; Feldgarden, M.; Arachchi, H.M.; Cerqueira, G.C.; Fitzgerald, M.; Godfrey, P.; Haas, B.J.; Murphy, C.I.; Russ, C.; et al. Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe, 2011. Proc. Natl. Acad. Sci. USA 2012, 109, 3065–3070. [Google Scholar] [CrossRef]
- Sherry, N.L.; Porter, J.L.; Seemann, T.; Watkins, A.; Stinear, T.P.; Howden, B.P. Outbreak investigation using high-throughput genome sequencing within a diagnostic microbiology laboratory. J. Clin. Microbiol. 2013, 51, 1396–1401. [Google Scholar] [CrossRef]
- Reuter, S.; Harrison, T.G.; Köser, C.U.; Ellington, M.J.; Smith, G.P.; Parkhill, J.; Peacock, S.J.; Bentley, S.D.; Török, M.E. A pilot study of rapid whole-genome sequencing for the investigation of a Legionella outbreak. BMJ Open 2013. [Google Scholar] [CrossRef]
- Walker, T.M.; Ip, C.L.; Harrell, R.H.; Evans, J.T.; Kapatai, G.; Dedicoat, M.J.; Eyre, D.W.; Wilson, D.J.; Hawkey, P.M.; Crook, D.W.; et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect. Dis. 2013, 13, 137–146. [Google Scholar] [CrossRef]
- McDonnell, J.; Dallman, T.; Atkin, S.; Turbitt, D.A.; Connor, T.R.; Grant, K.A.; Thomson, N.R.; Jenkins, C. Retrospective analysis of whole genome sequencing compared to prospective typing data in further informing the epidemiological investigation of an outbreak of Shigella sonnei in the UK. Epidemiol. Infect. 2013, 141, 2568–2575. [Google Scholar] [CrossRef]
- Espedido, B.A.; Steen, J.A.; Barbagiannakos, T.; Mercer, J.; Paterson, D.L.; Grimmond, S.M.; Cooper, M.A.; Gosbell, I.B.; van Hal, S.J.; Jensen, S.O. Carriage of an acme II variant may have contributed to methicillin-resistant Staphylococcus aureus sequence type 239-like strain replacement in Liverpool hospital, Sydney, Australia. Antimicrob. Agents Chemother. 2012, 56, 3380–3383. [Google Scholar] [CrossRef] [Green Version]
- Sandegren, L.; Groenheit, R.; Koivula, T.; Ghebremichael, S.; Advani, A.; Castro, E.; Pennhag, A.; Hoffner, S.; Mazurek, J.; Pawlowski, A.; et al. Genomic stability over 9 years of an isoniazid resistant Mycobacterium tuberculosis outbreak strain in Sweden. PLoS One 2011, 6, e16647. [Google Scholar] [CrossRef]
- Vogel, U.; Szczepanowski, R.; Claus, H.; Jünemann, S.; Prior, K.; Harmsen, D. Ion Torrent Personal Genome Machine sequencing for genomic typing of Neisseria meningitidis for rapid determination of multiple layers of typing information. J. Clin. Microbiol. 2012, 50, 1889–1894. [Google Scholar] [CrossRef]
- Price, L.B.; Stegger, M.; Hasman, H.; Aziz, M.; Larsen, J.; Andersen, P.S.; Pearson, T.; Waters, A.E.; Foster, J.T.; Schupp, J.; et al. Staphylococcus aureus CC398: Host adaptation and emergence of methicillin resistance in livestock. mBio 2012, 3, e00305–e00311. [Google Scholar]
- Goecks, J.; Nekrutenko, A.; Taylor, J.; Galaxy Team. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010. [Google Scholar] [CrossRef]
- Al-Khedery, B.; Lundgren, A.M.; Stuen, S.; Granquist, E.G.; Munderloh, U.G.; Nelson, C.M.; Alleman, A.R.; Mahan, S.M.; Barbet, A.F. Structure of the type IV secretion system in different strains of Anaplasma phagocytophilum. BMC Genomics 2012. [Google Scholar] [CrossRef]
- Clayton, A.L.; Oakeson, K.F.; Gutin, M.; Pontes, A.; Dunn, D.M.; von Niederhausern, A.C.; Weiss, R.B.; Fisher, M.; Dale, C. A novel human-infection-derived bacterium provides insights into the evolutionary origins of mutualistic insect-bacterial symbioses. PLoS Genet. 2012, 8, e1002990. [Google Scholar] [CrossRef]
- Morin, A.; Urban, J.; Adams, P.D.; Foster, I.; Sali, A.; Baker, D.; Sliz, P. Research priorities. Shining light into black boxes. Science 2012, 336, 159–160. [Google Scholar] [CrossRef]
- Scholtalbers, J.; Rößler, J.; Sorn, P.; de Graaf, J.; Boisguérin, V.; Castle, J.; Sahin, U. Galaxy LIMS for next-generation sequencing. Bioinformatics 2013, 29, 1233–1234. [Google Scholar] [CrossRef]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinfor. 2010, 10, 595. [Google Scholar]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Mellmann, A.; Harmsen, D.; Cummings, C.A.; Zentz, E.B.; Leopold, S.R.; Rico, A.; Prior, K.; Szczepanowski, R.; Ji, Y.; Zhang, W.; et al. Prospective genomic characterization of the german enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PLoS One 2011, 6, e22751. [Google Scholar] [CrossRef]
- Bennett, J.S.; Jolley, K.A.; Earle, S.G.; Corton, C.; Bentley, S.D.; Parkhill, J.; Maiden, M.C. A genomic approach to bacterial taxonomy: An examination and proposed reclassification of species within the genus Neisseria. Microbiology 2012, 158, 1570–1580. [Google Scholar] [CrossRef]
- Wyres, K.L.; Lambertsen, L.M.; Croucher, N.J.; McGee, L.; von Gottberg, A.; Liñares, J.; R, J.M.; Kristinsson, K.G.; Beall, B.W.; Klugman, K.P.; et al. The multidrug-resistant PMEN1 pneumococcus is a paradigm for genetic success. Genome Biol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef]
- Leekitcharoenphon, P.; Nielsen, E.M.; Kaas, R.S.; Lund, O.; Aarestrup, F.M. Evaluation of whole genome sequencing for outbreak detection of Salmonella enterica. PLoS One 2014, 9, e87991. [Google Scholar]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Leekitcharoenphon, P.; Kaas, R.S.; Thomsen, M.C.; Friis, C.; Rasmussen, S.; Aarestrup, F.M. SnpTree—A web-server to identify and construct snp trees from whole genome sequence data. BMC Genomics 2012. [Google Scholar] [CrossRef]
- Jünemann, S.; Sedlazeck, F.J.; Prior, K.; Albersmeier, A.; John, U.; Kalinowski, J.; Mellmann, A.; Goesmann, A.; von Haeseler, A.; Stoye, J.; et al. Updating benchtop sequencing performance comparison. Nat. Biotechnol. 2013, 31, 294–296. [Google Scholar] [CrossRef] [Green Version]
- Al-Shahib, A.; Underwood, A. Snp-search: Simple processing, manipulation and searching of snps from high-throughput sequencing. BMC Bioinfor. 2013. [Google Scholar] [CrossRef]
- Tang, F.; Chua, C.L.; Ho, L.Y.; Lim, Y.P.; Issac, P.; Krishnan, A. Wildfire: Distributed, grid-enabled workflow construction and execution. BMC Bioinfor. 2005. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. Emboss: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Ovaska, K.; Laakso, M.; Haapa-Paananen, S.; Louhimo, R.; Chen, P.; Aittomäki, V.; Valo, E.; Núñez-Fontarnau, J.; Rantanen, V.; Karinen, S.; et al. Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med. 2010. [Google Scholar] [CrossRef]
- Kallio, M.A.; Tuimala, J.T.; Hupponen, T.; Klemelä, P.; Gentile, M.; Scheinin, I.; Koski, M.; Käki, J.; Korpelainen, E.I. Chipster: User-friendly analysis software for microarray and other high-throughput data. BMC Genomics 2011. [Google Scholar] [CrossRef]
- Mariette, J.; Escudié, F.; Allias, N.; Salin, G.; Noirot, C.; Thomas, S.; Klopp, C. NG6: Integrated next generation sequencing storage and processing environment. BMC Genomics 2012. [Google Scholar] [CrossRef]
- McPhillips, T.; Bowers, S.; Zinn, D.; Ludäscher, B. Scientific workflow design for mere mortals. Future Gener. Comp. Syst. 2009, 25, 541–551. [Google Scholar] [CrossRef]
- Vahi, K.; Harvey, I.; Samak, T.; Gunter, D.; Evans, K.; Rogers, D.; Taylor, I.; Goode, M.; Silva, F.; Al-Shakarachi, E.; et al. A case study into using common real-time workflow monitoring infrastructure for scientific workflows. J. Grid Comput. 2013, 11, 381–406. [Google Scholar] [CrossRef]
- Wolstencroft, K.; Haines, R.; Fellows, D.; Williams, A.; Withers, D.; Owen, S.; Soiland-Reyes, S.; Dunlop, I.; Nenadic, A.; Fisher, P.; et al. The Taverna workflow suite: Designing and executing workflows of web services on the desktop, web or in the cloud. Nucleic Acids Res. 2013, 41, W557–W561. [Google Scholar] [CrossRef]
- Goble, C.A.; Bhagat, J.; Aleksejevs, S.; Cruickshank, D.; Michaelides, D.; Newman, D.; Borkum, M.; Bechhofer, S.; Roos, M.; Li, P.; et al. MyExperiment: A repository and social network for the sharing of bioinformatics workflows. Nucleic Acids Res. 2010, 38, W677–W682. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wyres, K.L.; Conway, T.C.; Garg, S.; Queiroz, C.; Reumann, M.; Holt, K.; Rusu, L.I. WGS Analysis and Interpretation in Clinical and Public Health Microbiology Laboratories: What Are the Requirements and How Do Existing Tools Compare? Pathogens 2014, 3, 437-458. https://doi.org/10.3390/pathogens3020437
Wyres KL, Conway TC, Garg S, Queiroz C, Reumann M, Holt K, Rusu LI. WGS Analysis and Interpretation in Clinical and Public Health Microbiology Laboratories: What Are the Requirements and How Do Existing Tools Compare? Pathogens. 2014; 3(2):437-458. https://doi.org/10.3390/pathogens3020437
Chicago/Turabian StyleWyres, Kelly L., Thomas C. Conway, Saurabh Garg, Carlos Queiroz, Matthias Reumann, Kathryn Holt, and Laura I. Rusu. 2014. "WGS Analysis and Interpretation in Clinical and Public Health Microbiology Laboratories: What Are the Requirements and How Do Existing Tools Compare?" Pathogens 3, no. 2: 437-458. https://doi.org/10.3390/pathogens3020437