Genomics-Based Exploration of Virulence Determinants and Host-Specific Adaptations of Pseudomonas syringae Strains Isolated from Grasses

Abstract
:1. Introduction
2. Results and Discussion
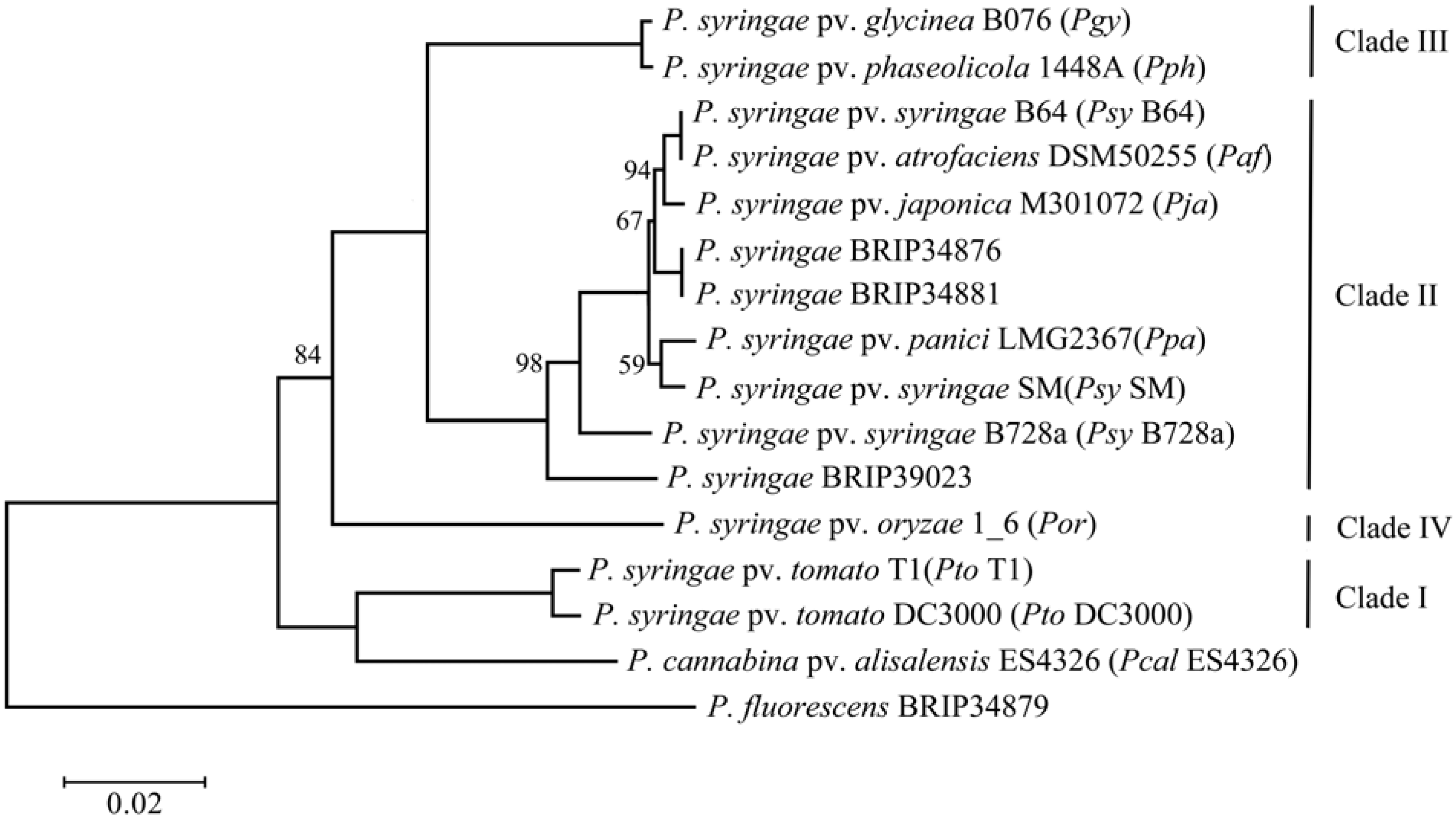
2.1. Phylogenetic Assessment of the Strains
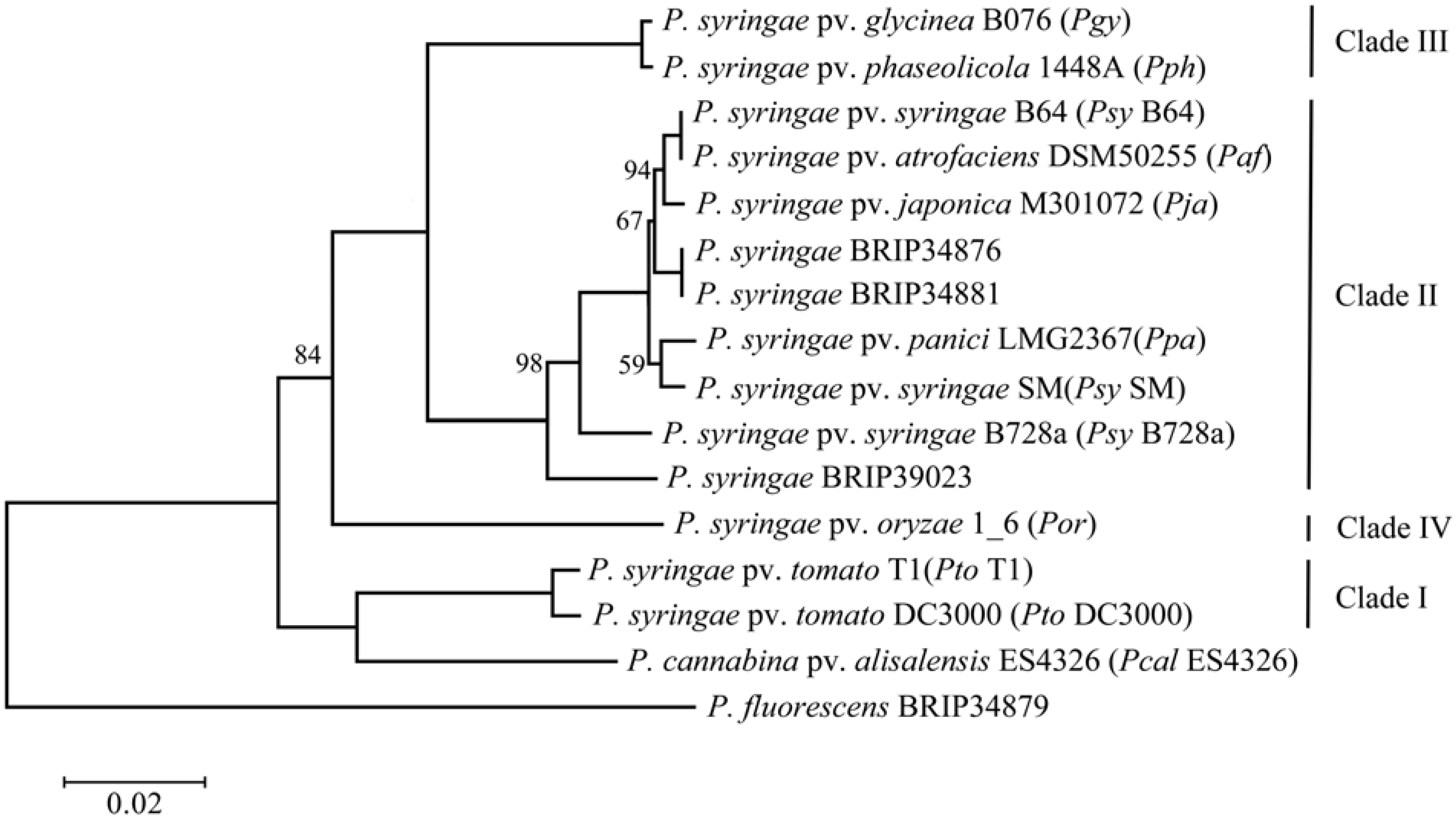
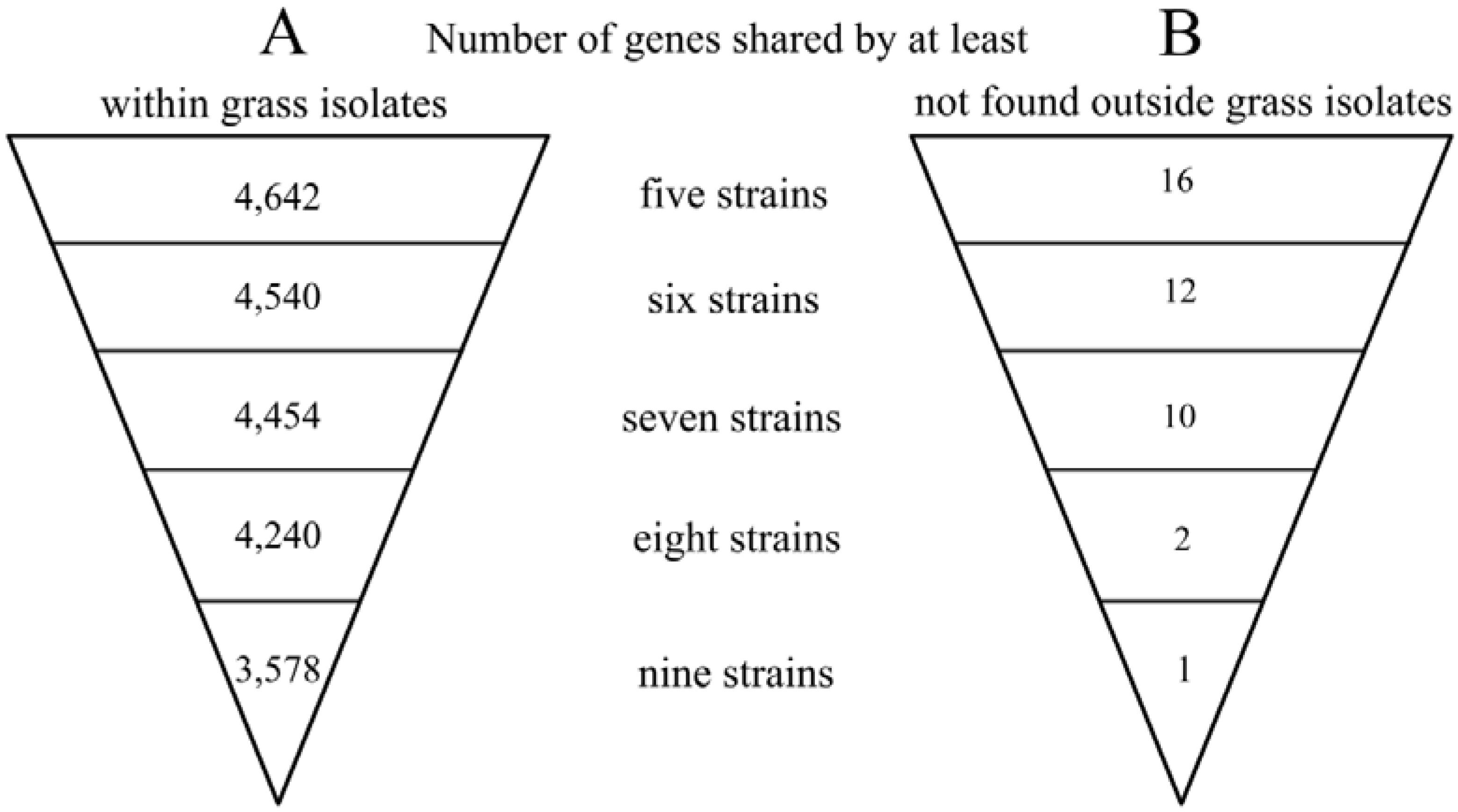
2.2. Genome Comparison and Identification of Poaceae-Specific Genes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ortholog clusters | A (within grass isolates) | B (not found outside grass isolates) |
|---|---|---|
| Unique to Por | 2,333 | 1,566 |
| Unique to BRIP34876 | 11 | 8 |
| Unique to BRIP34881 | 13 | 7 |
| Unique to BRIP39023 | 349 | 121 |
| Unique to Pja | 3,657 | 2,563 |
| Unique to Psy SM | 178 | 75 |
| Unique to Psy B64 | 37 | 9 |
| Unique to Ppa | 484 | 322 |
| Unique to Paf | 216 | 188 |
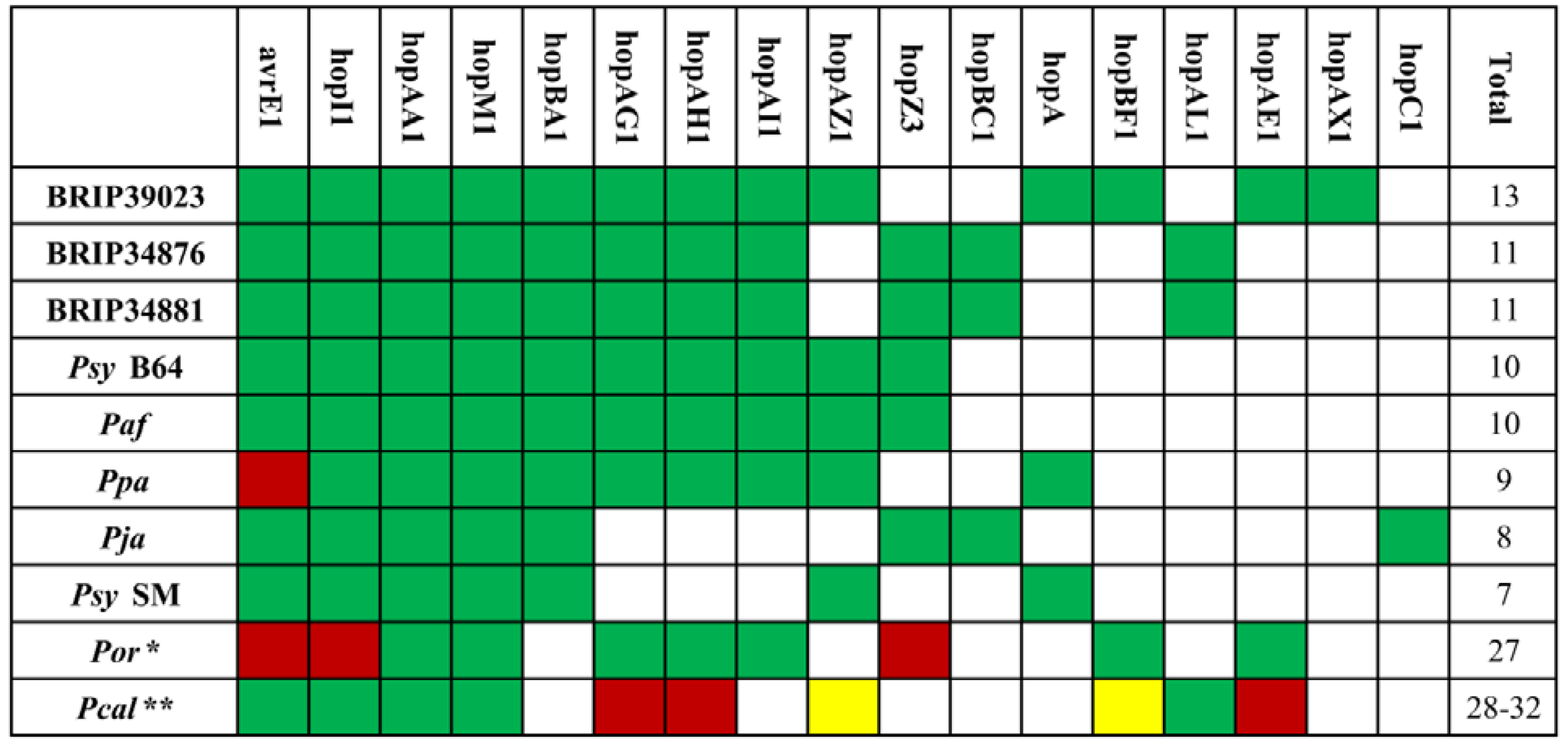
2.3. The Type III Secretion System and Effector Repertoire

2.4. Other Virulence Factors
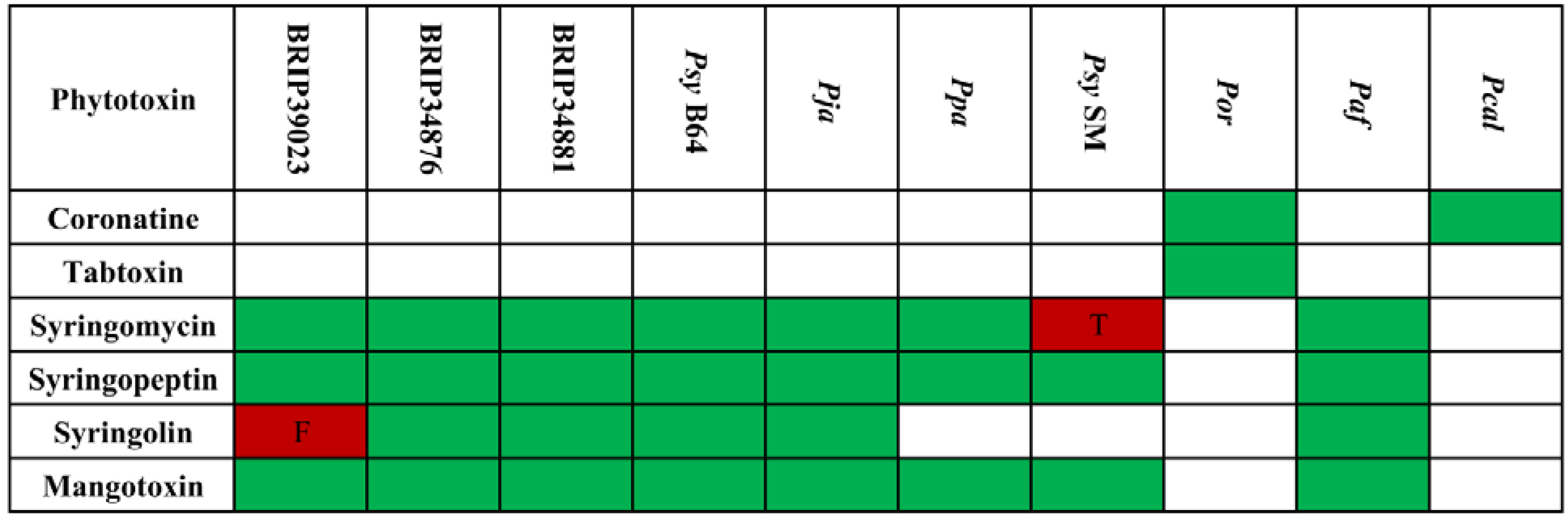
2.4.1. Phytotoxins and Other Small Secreted Molecules
2.4.2. Quorum Sensing
2.4.3. Exopolysaccharides
2.4.4. Type VI Secretion System
2.5. Mobile and Extrachromosomal Elements
2.6. Other Notable Genome Components
2.6.1. Defense Mechanisms against Foreign DNA
2.6.2. Bacteriocins
| Strain | Bacteriocin | Location | Strain | Bacteriocin | Location |
|---|---|---|---|---|---|
| BRIP34876 | Syringacin M | A979_09507 | Psy SM | S-type pyocin | PssSM_0863 |
| BRIP34881 | Syringacin M | A987_14290 | Colicin-DNAse | PssSM_0293 1 | |
| BRIP39023 | S-type pyocin | A988_14544 | Bacteriocin | PssSM_4261 | |
| Colicin-DNAse | A988_07204 1 | Ppa | Syringacin M | ALAC01000012 | |
| Cyclized peptide | A988_02638 2 | S-type pyocin | ALAC01000003 | ||
| Pja | Syringacin M | PSYJA_15707 | Psy B64 | Syringacin M | PssB64_01273 |
| Bacteriocin | PSYJA_00994 | S-type pyocin | PssB64_04860 | ||
| Lasso peptide | PSYJA_17421 | Por | Colicin-DNAse | POR16_04054 1 | |
| Colicin-DNAse | AEAH01000987 1,3 | Bacteriocin | POR16_27431 | ||
| Paf | Syringacin M | AWUI01000310 | Lasso peptide | DS996947 4 | |
| S-type pyocin | AWUI01000345 |
2.6.3. Non-Ribosomal Peptide Synthetases/Polyketide Synthetases
3. Experimental Section
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bradbury, J.F. Pseudomonas syringae pv. syringae. In Guide to Plant Pathogenic Bacteria; CAB International Mycological Institute: Kew, England, 1986; pp. 175–177. [Google Scholar]
- Gironde, S.; Manceau, C. Housekeeping gene sequencing and multilocus variable-number tandem-repeat analysis to identify subpopulations within Pseudomonas syringae pv. maculicola and Pseudomonas syringae pv. tomato that correlate with host specificity. Appl. Environ. Microbiol. 2012, 78, 3266–3279. [Google Scholar] [CrossRef]
- Martín-Sanz, A.; de la Vega, M.P.; Murillo, J.; Caminero, C. Strains of Pseudomonas syringae pv. syringae from pea are phylogenetically and pathogenically diverse. Phytopathology 2013, 103, 673–681. [Google Scholar] [CrossRef]
- Ramos, C.; Matas, I.M.; Bardaji, L.; Aragón, I.M.; Murillo, J. Pseudomonas savastanoi pv. savastanoi: some like it knot. Mol. Plant Pathol. 2012, 13, 998–1009. [Google Scholar] [CrossRef]
- Arnold, D.L.; Lovell, H.C.; Jackson, R.W.; Mansfield, J.W. Pseudomonas syringae pv. phaseolicola: from “has bean” to supermodel. Mol. Plant Pathol. 2011, 12, 617–627. [Google Scholar] [CrossRef]
- McCann, H.C.; Rikkerink, E.H.A.; Bertels, F.; Fiers, M.; Lu, A.; Rees-George, J.; Andersen, M.T.; Gleave, A.P.; Haubold, B.; Wohlers, M.W.; et al. Genomic Analysis of the Kiwifruit Pathogen Pseudomonas syringae pv. actinidiae Provides Insight into the Origins of an Emergent Plant Disease. PLoS Pathog. 2013, 9, e1003503. [Google Scholar] [CrossRef]
- Green, S.; Studholme, D.J.; Laue, B.E.; Dorati, F.; Lovell, H.; Arnold, D.; Cottrell, J.E.; Bridgett, S.; Blaxter, M.; Huitema, E.; et al. Comparative genome analysis provides insights into the evolution and adaptation of Pseudomonas syringae pv. aesculi on Aesculus hippocastanum. PLoS One 2010, 5, e10224. [Google Scholar] [CrossRef]
- Qi, M.; Wang, D.; Bradley, C.A.; Zhao, Y. Genome sequence analyses of Pseudomonas savastanoi pv. glycinea and subtractive hybridization-based comparative genomics with nine Pseudomonads. PLoS One 2011, 6, e16451. [Google Scholar]
- Young, J.M. Taxonomy of Pseudomonas syringae. J. Plant Pathol. 2010, 92, S1.5–S1.14. [Google Scholar]
- Sarkar, S.F.; Guttman, D.S. Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen. Appl. Environ. Microbiol. 2004, 70, 1999–2012. [Google Scholar] [CrossRef]
- Hwang, M.S.H.; Morgan, R.L.; Sarkar, S.F.; Wang, P.W.; Guttman, D.S. Phylogenetic characterization of virulence and resistance phenotypes of Pseudomonas syringae. Appl. Environ. Microbiol. 2005, 71, 5182–5191. [Google Scholar] [CrossRef]
- Gardan, L.; Shafik, H.; Belouin, S.; Broch, R.; Grimont, F.; Grimont, P.A.D. DNA relatedness among the pathovars of Pseudomonas syringae and description of Pseudomonas tremae sp. nov. and Pseudomonas cannabina sp. nov. (ex Sutic and Dowson 1959). Int. J. Syst. Bacteriol. 1999, 49, 469–478. [Google Scholar] [CrossRef]
- O’Brien, H.E.; Thakur, S.; Gong, Y.; Fung, P.; Zhang, J.; Yuan, L.; Wang, P.W.; Yong, C.; Scortichini, M.; Guttman, D.S. Extensive remodeling of the Pseudomonas syringae pv. avellanae type III secretome associated with two independent host shifts onto hazelnut. BMC Microbiol. 2012, 12, 141. [Google Scholar] [CrossRef]
- Lindeberg, M.; Cunnac, S.; Collmer, A. The evolution of Pseudomonas syringae host specificity and type III effector repertoires. Mol. Plant Pathol. 2009, 10, 767–775. [Google Scholar] [CrossRef]
- Rodríguez-Palenzuela, P.; Matas, I.M.; Murillo, J.; López-Solanilla, E.; Bardaji, L.; Pérez-Martínez, I.; Rodríguez-Moskera, M.E.; Penyalver, R.; López, M.M.; Quesada, J.M.; et al. Annotation and overview of the Pseudomonas savastanoi pv. savastanoi NCPPB 3335 draft genome reveals the virulence gene complement of a tumour-inducing pathogen of woody hosts. Environ. Microbiol. 2010, 12, 1604–1620. [Google Scholar]
- Haapalainen, M.; Mosorin, H.; Dorati, F.; Wu, R.-F.; Roine, E.; Taira, S.; Nissinen, R.; Mattinen, L.; Jackson, R.; Pirhonen, M.; et al. Hcp2, a secreted protein of the phytopathogen Pseudomonas syringae pv. tomato DC3000, is required for fitness for competition against bacteria and yeasts. J. Bacteriol. 2012, 194, 4810–4822. [Google Scholar] [CrossRef]
- Lindeberg, M.; Cunnac, S.; Collmer, A. Pseudomonas syringae type III effector repertoires: Last words in endless arguments. Trends Microbiol. 2012, 20, 199–208. [Google Scholar] [CrossRef]
- Schellenberg, B.; Ramel, C.; Dudler, R. Pseudomonas syringae Virulence Factor Syringolin A Counteracts Stomatal Immunity by Proteasome Inhibition. Mol. Plant-Microbe Interact. 2010, 23, 1287–1293. [Google Scholar] [CrossRef]
- Bender, C.L.; Alarcón-Chaidez, F.; Gross, D.C. Pseudomonas syringae phytotoxins: Mode of action, regulation, and biosynthesis by peptide and polyketide synthetases. Microbiol. Mol. Biol. Rev. MMBR 1999, 63, 266–292. [Google Scholar]
- Denny, T.P. Involvement of bacterial polysaccharides in plant pathogenesis. Annu. Rev. Phytopathol. 1995, 33, 173–197. [Google Scholar] [CrossRef]
- Misas-Villamil, J.C.; Kolodziejek, I.; Crabill, E.; Kaschani, F.; Niessen, S.; Shindo, T.; Kaiser, M.; Alfano, J.R.; van der Hoorn, R.A.L. Pseudomonas syringae pv. syringae uses proteasome inhibitor syringolin A to colonize from wound infection sites. PLoS Pathog. 2013, 9, e1003281. [Google Scholar]
- Yu, J.; Penaloza-Vazquez, A.; Chakrabarty, A.M.; Bender, C.L. Involvement of the exopolysaccharide alginate in the virulence and epiphytic fitness of Pseudomonas syringae pv. syringae. Mol. Microbiol. 1999, 33, 712–720. [Google Scholar] [CrossRef]
- Rodríguez-Moreno, L.; Barceló-Muñoz, A.; Ramos, C. In vitro analysis of the interaction of Pseudomonas savastanoi pvs. savastanoi and nerii with micropropagated olive plants. Phytopathology 2008, 98, 815–822. [Google Scholar] [CrossRef]
- Arrebola, E.; Cazorla, F.M.; Perez-García, A.; de Vicente, A. Chemical and metabolic aspects of antimetabolite toxins produced by Pseudomonas syringae pathovars. Toxins (Basel). 2011, 3, 1089–1110. [Google Scholar] [CrossRef]
- Galán, J.E.; Wolf-Watz, H. Protein delivery into eukaryotic cells by type III secretion machines. Nature 2006, 444, 567–573. [Google Scholar] [CrossRef]
- Büttner, D. Protein export according to schedule: architecture, assembly, and regulation of type III secretion systems from plant- and animal-pathogenic bacteria. Microbiol. Mol. Biol. Rev. 2012, 76, 262–310. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Cunnac, S.; Lindeberg, M.; Collmer, A. Pseudomonas syringae type III secretion system effectors: Repertoires in search of functions. Curr. Opin. Microbiol. 2009, 12, 53–60. [Google Scholar] [CrossRef]
- Munkvold, K.R.; Martin, M.E.; Bronstein, P.A.; Collmer, A. A survey of the Pseudomonas syringae pv. tomato DC3000 type III secretion system effector repertoire reveals several effectors that are deleterious when expressed in Saccharomyces cerevisiae. Mol. Plant-Microbe Interact. 2008, 21, 490–502. [Google Scholar] [CrossRef]
- Salomon, D.; Bosis, E.; Dar, D.; Nachman, I.; Sessa, G. Expression of Pseudomonas syringae type III effectors in yeast under stress conditions reveals that HopX1 attenuates activation of the high osmolarity glycerol MAP kinase pathway. Microbiology 2012, 158, 2859–2869. [Google Scholar] [CrossRef]
- Baltrus, D.A.; Nishimura, M.T.; Romanchuk, A.; Chang, J.H.; Mukhtar, M.S.; Cherkis, K.; Roach, J.; Grant, S.R.; Jones, C.D.; et al. Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog. 2011, 7, e1002132. [Google Scholar] [CrossRef]
- Göhre, V.; Spallek, T.; Häweker, H.; Mersmann, S.; Mentzel, T.; Boller, T.; de Torres, M.; Mansfield, J.W.; Robatzek, S. Plant pattern-recognition receptor FLS2 is directed for degradation by the bacterial ubiquitin ligase AvrPtoB. Curr. Biol. 2008, 18, 1824–1832. [Google Scholar] [CrossRef]
- Nicaise, V.; Joe, A.; Jeong, B.-R.; Korneli, C.; Boutrot, F.; Westedt, I.; Staiger, D.; Alfano, J.R.; Zipfel, C. Pseudomonas HopU1 modulates plant immune receptor levels by blocking the interaction of their mRNAs with GRP7. EMBO J. 2013, 32, 701–712. [Google Scholar] [CrossRef]
- Rodríguez-Herva, J.J.; González-Melendi, P.; Cuartas-Lanza, R.; Antúnez-Lamas, M.; Río-Alvarez, I.; Li, Z.; López-Torrejón, G.; Díaz, I.; del Pozo, J.C.; Chakravarthy, S.; et al. A bacterial cysteine protease effector protein interferes with photosynthesis to suppress plant innate immune responses. Cell. Microbiol. 2012, 14, 669–681. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Halane, M.K.; Kim, S.H.; Gassmann, W. Pathogen effectors target Arabidopsis EDS1 and alter its interactions with immune regulators. Science 2011, 334, 1405–1408. [Google Scholar] [CrossRef]
- Lee, A.H.-Y.; Hurley, B.; Felsensteiner, C.; Yea, C.; Ckurshumova, W.; Bartetzko, V.; Wang, P.W.; Quach, V.; Lewis, J.D.; Liu, Y.C.; et al. A bacterial acetyltransferase destroys plant microtubule networks and blocks secretion. PLoS Pathog. 2012, 8, e1002523. [Google Scholar] [CrossRef]
- Jiang, S.; Yao, J.; Ma, K.-W.; Zhou, H.; Song, J.; He, S.Y.; Ma, W. Bacterial Effector Activates Jasmonate Signaling by Directly Targeting JAZ Transcriptional Repressors. PLoS Pathog. 2013, 9, e1003715. [Google Scholar]
- Cunnac, S.; Chakravarthy, S.; Kvitko, B.H.; Russell, A.B.; Martin, G.B.; Collmer, A. Genetic disassembly and combinatorial reassembly identify a minimal functional repertoire of type III effectors in Pseudomonas syringae. Proc. Natl. Acad. Sci. USA 2011, 108, 2975–2980. [Google Scholar] [CrossRef]
- Almeida, N.F.; Yan, S.; Lindeberg, M.; Studholme, D.J.; Schneider, D.J.; Condon, B.; Liu, H.; Viana, C.J.; Warren, A.; Evans, C.; et al. A draft genome sequence of Pseudomonas syringae pv. tomato T1 reveals a type III effector repertoire significantly divergent from that of Pseudomonas syringae pv. tomato DC3000. Mol. Plant Microbe. Interact. 2009, 22, 52–62. [Google Scholar] [CrossRef]
- Buell, C.R.; Joardar, V.; Lindeberg, M.; Selengut, J.; Paulsen, I.T.; Gwinn, M.L.; Dodson, R.J.; Deboy, R.T.; Durkin, A.S.; Kolonay, J.F.; et al. The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc. Natl. Acad. Sci. USA 2003, 100, 10181–10186. [Google Scholar] [CrossRef]
- Wei, C.-F.; Kvitko, B.H.; Shimizu, R.; Crabill, E.; Alfano, J.R.; Lin, N.-C.; Martin, G.B.; Huang, H.-C.; Collmer, A. A Pseudomonas syringae pv. tomato DC3000 mutant lacking the type III effector HopQ1-1 is able to cause disease in the model plant Nicotiana benthamiana. Plant J. 2007, 51, 32–46. [Google Scholar] [CrossRef]
- Feil, H.; Feil, W.S.; Chain, P.; Larimer, F.; DiBartolo, G.; Copeland, A.; Lykidis, A.; Trong, S.; Nolan, M.; Goltsman, E.; et al. Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc. Natl. Acad. Sci. USA 2005, 102, 11064–11069. [Google Scholar] [CrossRef]
- Joardar, V.; Lindeberg, M.; Jackson, R.W.; Selengut, J.; Dodson, R.; Brinkac, L.M.; Daugherty, S.C.; Deboy, R.; Durkin, A.S.; Giglio, M.G.; et al. Whole-genome sequence analysis of Pseudomonas syringae pv. phaseolicola 1448A reveals divergence among pathovars in genes involved in virulence and transposition. J. Bacteriol. 2005, 187, 6488–6498. [Google Scholar] [CrossRef]
- Dudnik, A.; Dudler, R. High-Quality Draft Genome Sequence of Pseudomonas syringae pv. Syringae Strain SM, Isolated from Wheat. Genome Announc. 2013, 1, e00610–e00613. [Google Scholar]
- Dudnik, A.; Dudler, R. Non contiguous-finished genome sequence of Pseudomonas syringae pathovar syringae strain B64 isolated from wheat. Stand. Genomic Sci. 2013, 8, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, D.M.; Stiller, J.; Covarelli, L.; Lindeberg, M.; Shivas, R.G.; Manners, J.M. Genome Sequences of Pseudomonas spp. Isolated from Cereal Crops. Genome Announc. 2013, 1, e00209–e00213. [Google Scholar]
- Reinhardt, J.A.; Baltrus, D.A.; Nishimura, M.T.; Jeck, W.R.; Jones, C.D.; Dangl, J.L. De novo assembly using low-coverage short read sequence data from the rice pathogen Pseudomonas syringae pv. oryzae. Genome Res. 2009, 19, 294–305. [Google Scholar]
- Liu, H.; Qiu, H.; Zhao, W.; Cui, Z.; Ibrahim, M.; Jin, G.; Li, B.; Zhu, B.; Xie, G.L. Genome sequence of the plant pathogen Pseudomonas syringae pv. panici LMG 2367. J. Bacteriol. 2012, 194, 5693–5694. [Google Scholar] [CrossRef]
- Sarris, P.F.; Trantas, E.A.; Baltrus, D.A.; Bull, C.T.; Wechter, W.P.; Yan, S.; Ververidis, F.; Almeida, N.F.; Jones, C.D.; Dangl, J.L.; et al. Comparative genomics of multiple strains of Pseudomonas cannabina pv. alisalensis, a potential model pathogen of both monocots and dicots. PLoS One 2013, 8, e59366. [Google Scholar] [CrossRef]
- Baltrus, D.A.; Dougherty, K.; Beckstrom-Sternberg, S.M.; Beckstrom-Sternberg, J.S.; Foster, J.T. Incongruence between multi-locus sequence analysis (MLSA) and whole-genome-based phylogenies: Pseudomonas syringae pathovar pisi as a cautionary tale. Mol. Plant Pathol. 2014. [Google Scholar] [CrossRef]
- Elasri, M.; Delorme, S.; Lemanceau, P.; Stewart, G.; Laue, B.; Glickmann, E.; Oger, P.M.; Dessaux, Y. Acyl-homoserine lactone production is more common among plant-associated Pseudomonas spp. than among soilborne Pseudomonas spp. Appl. Environ. Microbiol. 2001, 67, 1198–1209. [Google Scholar] [CrossRef]
- Marcelletti, S.; Ferrante, P.; Petriccione, M.; Firrao, G.; Scortichini, M. Pseudomonas syringae pv. actinidiae draft genomes comparison reveal strain-specific features involved in adaptation and virulence to Actinidia species. PLoS One 2011, 6, e27297. [Google Scholar]
- Deng, W.-L.; Preston, G.; Collmer, A.; Chang, C.-J.; Huang, H.-C. Characterization of the hrpC and hrpRS operons of Pseudomonas syringae pathovars syringae, tomato, and glycinea and analysis of the ability of hrpF, hrpG, hrcC, hrpT, and hrpV mutants to elicit the hypersensitive response and disease in plants. J. Bacteriol. 1998, 180, 4523–4531. [Google Scholar]
- Sarkar, S.F.; Gordon, J.S.; Martin, G.B.; Guttman, D.S. Comparative genomics of host-specific virulence in Pseudomonas syringae. Genetics 2006, 174, 1041–1056. [Google Scholar] [CrossRef]
- Cintas, N.A.; Koike, S.T.; Bull, C.T. A New pathovar, Pseudomonas syringae pv. alisalensis pv. nov., proposed for the causal agent of bacterial blight of broccoli and broccoli raab. Plant Dis. 2002, 86, 992–998. [Google Scholar] [CrossRef]
- Ishiyama, Y.; Yamagishi, N.; Ogiso, H.; Fujinaga, M.; Takikawa, Y. Bacterial brown spot on Avena storigosa Schereb. caused by Pseudomonas syringae pv. alisalensis. J. Gen. Plant Pathol. 2013, 79, 155–157. [Google Scholar] [CrossRef]
- Gazi, A.D.; Sarris, P.F.; Fadouloglou, V.E.; Charova, S.N.; Mathioudakis, N.; Panopoulos, N.J.; Kokkinidis, M. Phylogenetic analysis of a gene cluster encoding an additional, rhizobial-like type III secretion system that is narrowly distributed among Pseudomonas syringae strains. BMC Microbiol. 2012, 12, 188. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, H.; Sun, M.; Zhang, Q.; Guo, D. T3DB: An integrated database for bacterial type III secretion system. BMC Bioinform. 2012, 13, 66. [Google Scholar] [CrossRef]
- Cai, R.; Lewis, J.; Yan, S.; Liu, H.; Clarke, C.R.; Campanile, F.; Almeida, N.F.; Studholme, D.J.; Lindeberg, M.; Schneider, D.; et al. The plant pathogen Pseudomonas syringae pv. tomato is genetically monomorphic and under strong selection to evade tomato immunity. PLoS Pathog. 2011, 7, e1002130. [Google Scholar] [CrossRef]
- Murillo, J.; Bardaji, L.; Navarro de la Fuente, L.; Führer, M.E.; Aguilera, S.; Álvarez-Morales, A. Variation in conservation of the cluster for biosynthesis of the phytotoxin phaseolotoxin in Pseudomonas syringae suggests at least two events of horizontal acquisition. Res. Microbiol. 2011, 162, 253–261. [Google Scholar] [CrossRef]
- Arrebola, E.; Cazorla, F.M.; Codina, J.C.; Gutiérrez-Barranquero, J.A.; Pérez-García, A.; de Vicente, A. Contribution of mangotoxin to the virulence and epiphytic fitness of Pseudomonas syringae pv. syringae. Int. Microbiol. 2009, 12, 87–95. [Google Scholar]
- Groll, M.; Schellenberg, B.; Bachmann, A.S.; Archer, C.R.; Huber, R.; Powell, T.K.; Lindow, S.; Kaiser, M.; Dudler, R. A plant pathogen virulence factor inhibits the eukaryotic proteasome by a novel mechanism. Nature 2008, 452, 755–758. [Google Scholar] [CrossRef]
- Carrión, V.J.; Gutiérrez-Barranquero, J.A.; Arrebola, E.; Bardaji, L.; Codina, J.C.; de Vicente, A.; Cazorla, F.M.; Murillo, J. The mangotoxin biosynthetic operon (mbo) is specifically distributed within Pseudomonas syringae genomospecies 1 and was acquired only once during evolution. Appl. Environ. Microbiol. 2013, 79, 756–767. [Google Scholar] [CrossRef]
- Costacurta, A.; Vanderleyden, J. Synthesis of Phytohormones by Plant-Associated Bacteria. Crit. Rev. Microbiol. 2008, 21, 1–18. [Google Scholar] [CrossRef]
- Akiyoshi, D.E.; Regier, D.A.; Gordon, M.P. Cytokinin production by Agrobacterium and Pseudomonas spp. J. Bacteriol. 1987, 169, 4242–4248. [Google Scholar]
- Weingart, H.; Völksch, B.; Ullrich, M.S. Comparison of Ethylene Production by Pseudomonas syringae and Ralstonia solanacearum. Phytopathology 1999, 89, 360–365. [Google Scholar] [CrossRef]
- Glickmann, E.; Gardan, L.; Jacquet, S.; Hussain, S.; Elasri, M.; Petit, A.; Dessaux, Y. Auxin production is a common feature of most pathovars of Pseudomonas syringae. Mol. Plant. Microbe. Interact. 1998, 11, 156–162. [Google Scholar] [CrossRef]
- Roberto, F.F.; Klee, H.; White, F.; Nordeen, R.; Kosuge, T. Expression and fine structure of the gene encoding N epsilon-(indole-3-acetyl)-L-lysine synthetase from Pseudomonas savastanoi. Proc. Natl. Acad. Sci. USA 1990, 87, 5797–5801. [Google Scholar] [CrossRef]
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef]
- Nealson, K.H. Autoinduction of bacterial luciferase. Arch. Microbiol. 1977, 112, 73–79. [Google Scholar] [CrossRef]
- Von Bodman, S.B.; Bauer, W.D.; Coplin, D.L. Quorum sensing in plant-pathogenic bacteria. Annu. Rev. Phytopathol. 2003, 41, 455–482. [Google Scholar] [CrossRef]
- Dumenyo, C.K.; Mukherjee, A.; Chun, W.; Chatterjee, A.K. Genetic and physiological evidence for the production of N-acyl homoserine lactones by Pseudomonas syringae pv. syringae and other fluorescent plant pathogenic Pseudomonas species. Eur. J. Plant Pathol. 1998, 104, 569–582. [Google Scholar] [CrossRef]
- BCCM/LMG bacteria catalogue-Strain details LMG2367. Available online: http://bccm.belspo.be/db/lmg_strain_details.php?NUM=2367 (accessed on 7 November 2013).
- Venturi, V.; Fuqua, C. Chemical signaling between plants and plant-pathogenic bacteria. Annu. Rev. Phytopathol. 2013, 51, 17–37. [Google Scholar] [CrossRef]
- Schikora, A.; Schenk, S.T.; Stein, E.; Molitor, A.; Zuccaro, A.; Kogel, K.-H. N-acyl-homoserine lactone confers resistance toward biotrophic and hemibiotrophic pathogens via altered activation of AtMPK6. Plant Physiol. 2011, 157, 1407–1418. [Google Scholar] [CrossRef]
- Brencic, A.; Winans, S.C. Detection of and response to signals involved in host-microbe interactions by plant-associated bacteria. Microbiol. Mol. Biol. Rev. 2005, 69, 155–194. [Google Scholar] [CrossRef]
- Subramoni, S.; Venturi, V. LuxR-family “solos”: Bachelor sensors/regulators of signalling molecules. Microbiology 2009, 155, 1377–1385. [Google Scholar] [CrossRef]
- Subramoni, S.; Gonzalez, J.F.; Johnson, A.; Péchy-Tarr, M.; Rochat, L.; Paulsen, I.; Loper, J.E.; Keel, C.; Venturi, V. Bacterial subfamily of LuxR regulators that respond to plant compounds. Appl. Environ. Microbiol. 2011, 77, 4579–4588. [Google Scholar] [CrossRef]
- Ferluga, S.; Venturi, V. OryR is a LuxR-family protein involved in interkingdom signaling between pathogenic Xanthomonas oryzae pv. oryzae and rice. J. Bacteriol. 2009, 191, 890–897. [Google Scholar] [CrossRef]
- González, J.F.; Myers, M.P.; Venturi, V. The inter-kingdom solo OryR regulator of Xanthomonas oryzae is important for motility. Mol. Plant Pathol. 2013, 14, 211–221. [Google Scholar] [CrossRef]
- Lu, S.-E.; Scholz-Schroeder, B.K.; Gross, D.C. Characterization of the salA, syrF, and syrG regulatory genes located at the right border of the syringomycin gene cluster of pseudomonas syringae pv. syringae. Mol. Plant-Microbe Interact. 2002, 15, 43–53. [Google Scholar] [CrossRef]
- Mo, Y.Y.; Gross, D.C. Plant signal molecules activate the syrB gene, which is required for syringomycin production by Pseudomonas syringae pv. syringae. J. Bacteriol. 1991, 173, 5784–5792. [Google Scholar]
- Deisinger, P.J.; Hill, T.S.; English, J.C. Human exposure to naturally occurring hydroquinone. J. Toxicol. Environ. Health 1996, 47, 31–46. [Google Scholar] [CrossRef]
- Zakataeva, N.P.; Kutukova, E.A.; Gronskiy, S.V.; Troshin, P.V.; Livshits, V.A.; Aleshin, V.V. Export of metabolites by the proteins of the DMT and RhtB families and its possible role in intercellular communication. Microbiology 2006, 75, 438–448. [Google Scholar] [CrossRef]
- Degrassi, G.; Devescovi, G.; Solis, R.; Steindler, L.; Venturi, V. Oryza sativa rice plants contain molecules that activate different quorum-sensing N-acyl homoserine lactone biosensors and are sensitive to the specific AiiA lactonase. FEMS Microbiol. Lett. 2007, 269, 213–220. [Google Scholar] [CrossRef]
- Gao, M.; Teplitski, M.; Robinson, J.B.; Bauer, W.D. Production of Substances by Medicago truncatula that Affect Bacterial Quorum Sensing. Mol. Plant-Microbe Interact. 2003, 16, 827–834. [Google Scholar] [CrossRef]
- Osman, S.F.; Fett, W.F.; Fishman, M.L. Exopolysaccharides of the phytopathogen Pseudomonas syringae pv. glycinea. J. Bacteriol. 1986, 166, 66–71. [Google Scholar]
- Franklin, M.J.; Nivens, D.E.; Weadge, J.T.; Howell, P.L. Biosynthesis of the Pseudomonas aeruginosa Extracellular Polysaccharides, Alginate, Pel, and Psl. Front. Microbiol. 2011, 2, 167. [Google Scholar]
- Li, H.; Ullrich, M.S. Characterization and mutational analysis of three allelic lsc genes encoding levansucrase in Pseudomonas syringae. J. Bacteriol. 2001, 183, 3282–3292. [Google Scholar] [CrossRef]
- Penaloza-Vazquez, A.; Kidambi, S.; Chakrabarty, A.; Bender, C. Characterization of the alginate biosynthetic gene cluster in Pseudomonas syringae pv. syringae. J. Bacteriol. 1997, 179, 4464–4472. [Google Scholar]
- Laue, H.; Schenk, A.; Li, H.; Lambertsen, L.; Neu, T.R.; Molin, S.; Ullrich, M.S. Contribution of alginate and levan production to biofilm formation by Pseudomonas syringae. Microbiology 2006, 152, 2909–2918. [Google Scholar] [CrossRef]
- Byrd, M.S.; Sadovskaya, I.; Vinogradov, E.; Lu, H.; Sprinkle, A.B.; Richardson, S.H.; Ma, L.; Ralston, B.; Parsek, M.R.; Anderson, E.M.; et al. Genetic and biochemical analyses of the Pseudomonas aeruginosa Psl exopolysaccharide reveal overlapping roles for polysaccharide synthesis enzymes in Psl and LPS production. Mol. Microbiol. 2009, 73, 622–638. [Google Scholar] [CrossRef]
- Jackson, K.D.; Starkey, M.; Kremer, S.; Parsek, M.R.; Wozniak, D.J. Identification of psl, a locus encoding a potential exopolysaccharide that is essential for Pseudomonas aeruginosa PAO1 biofilm formation. J. Bacteriol. 2004, 186, 4466–4475. [Google Scholar] [CrossRef]
- Hockett, K.L.; Burch, A.Y.; Lindow, S.E. Thermo-regulation of genes mediating motility and plant interactions in Pseudomonas syringae. PLoS One 2013, 8, e59850. [Google Scholar] [CrossRef]
- Shrivastava, S.; Mande, S.S. Identification and functional characterization of gene components of Type VI Secretion system in bacterial genomes. PLoS One 2008, 3, e2955. [Google Scholar] [CrossRef]
- Kapitein, N.; Mogk, A. Deadly syringes: type VI secretion system activities in pathogenicity and interbacterial competition. Curr. Opin. Microbiol. 2013, 16, 52–58. [Google Scholar] [CrossRef]
- Barret, M.; Egan, F.; Fargier, E.; Morrissey, J.P.; O’Gara, F. Genomic analysis of the type VI secretion systems in Pseudomonas spp.: Novel clusters and putative effectors uncovered. Microbiology 2011, 157, 1726–1739. [Google Scholar] [CrossRef]
- Hachani, A.; Lossi, N.S.; Hamilton, A.; Jones, C.; Bleves, S.; Albesa-Jové, D.; Filloux, A. Type VI secretion system in Pseudomonas aeruginosa: Secretion and multimerization of VgrG proteins. J. Biol. Chem. 2011, 286, 12317–12327. [Google Scholar]
- Burtnick, M.N.; Brett, P.J.; Harding, S.V.; Ngugi, S.A.; Ribot, W.J.; Chantratita, N.; Scorpio, A.; Milne, T.S.; Dean, R.E.; Fritz, D.L.; et al. The cluster 1 type VI secretion system is a major virulence determinant in Burkholderia pseudomallei. Infect. Immun. 2011, 79, 1512–1525. [Google Scholar] [CrossRef]
- Sarris, P.F.; Skandalis, N.; Kokkinidis, M.; Panopoulos, N.J. In silico analysis reveals multiple putative type VI secretion systems and effector proteins in Pseudomonas syringae pathovars. Mol. Plant Pathol. 2010, 11, 795–804. [Google Scholar]
- Sarris, P.F.; Trantas, E.A.; Skandalis, N.; Tampakaki, A.P.; Kapanidou, M.; Kokkinidis, M.; Panopoulos, N.J. Phytobacterial type VI secretion system—Gene distribution, phylogeny, structure and biological functions. In InTech Plant Pathology; Cumagun, C.J.R., Ed.; InTech: Rijeca, Croatia, 2012; pp. 53–84. [Google Scholar]
- Pukatzki, S.; Ma, A.T.; Revel, A.T.; Sturtevant, D.; Mekalanos, J.J. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl. Acad. Sci. USA 2007, 104, 15508–15513. [Google Scholar]
- Russell, A.B.; LeRoux, M.; Hathazi, K.; Agnello, D.M.; Ishikawa, T.; Wiggins, P.A.; Wai, S.N.; Mougous, J.D. Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 2013, 496, 508–512. [Google Scholar] [CrossRef]
- Hood, R.D.; Singh, P.; Hsu, F.; Güvener, T.; Carl, M.A.; Trinidad, R.R.S.; Silverman, J.M.; Ohlson, B.B.; Hicks, K.G.; Plemel, R.L.; et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 2010, 7, 25–37. [Google Scholar] [CrossRef]
- Lindeberg, M.; Myers, C.R.; Collmer, A.; Schneider, D.J. Roadmap to new virulence determinants in Pseudomonas syringae: Insights from comparative genomics and genome organization. Mol. Plant. Microbe. Interact. 2008, 21, 685–700. [Google Scholar] [CrossRef]
- Greenberg, J.T.; Vinatzer, B.A. Identifying type III effectors of plant pathogens and analyzing their interaction with plant cells. Curr. Opin. Microbiol. 2003, 6, 20–28. [Google Scholar] [CrossRef]
- Swingle, B.; Bao, Z.; Markel, E.; Chambers, A.; Cartinhour, S. Recombineering using RecTE from Pseudomonas syringae. Appl. Environ. Microbiol. 2010, 76, 4960–4968. [Google Scholar] [CrossRef]
- Juhas, M.; van der Meer, J.R.; Gaillard, M.; Harding, R.M.; Hood, D.W.; Crook, D.W. Genomic islands: Tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 2009, 33, 376–393. [Google Scholar] [CrossRef]
- Gaillard, M.; Vallaeys, T.; Vorhölter, F.J.; Minoia, M.; Werlen, C.; Sentchilo, V.; Pühler, A.; van der Meer, J.R. The clc element of Pseudomonas sp. strain B13, a genomic island with various catabolic properties. J. Bacteriol. 2006, 188, 1999–2013. [Google Scholar] [CrossRef]
- Wozniak, R.A.F.; Waldor, M.K. Integrative and conjugative elements: Mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 2010, 8, 552–563. [Google Scholar] [CrossRef]
- Rodríguez-Blanco, A.; Lemos, M.L.; Osorio, C.R. Integrating conjugative elements as vectors of antibiotic, mercury, and quaternary ammonium compound resistance in marine aquaculture environments. Antimicrob. Agents Chemother. 2012, 56, 2619–2626. [Google Scholar] [CrossRef]
- Pitman, A.R.; Jackson, R.W.; Mansfield, J.W.; Kaitell, V.; Thwaites, R.; Arnold, D.L. Exposure to host resistance mechanisms drives evolution of bacterial virulence in plants. Curr. Biol. 2005, 15, 2230–2235. [Google Scholar] [CrossRef]
- Zhao, Y.; Ma, Z.; Sundin, G.W. Comparative genomic analysis of the pPT23A plasmid family of Pseudomonas syringae. J. Bacteriol. 2005, 187, 2113–2126. [Google Scholar] [CrossRef]
- Bender, C.; Palmer, D.; Peñaloza-Vázquez, A.; Rangaswamy, V.; Ullrich, M. Biosynthesis of coronatine, a thermoregulated phytotoxin produced by the phytopathogen Pseudomonas syringae. Arch. Microbiol. 1996, 166, 71–75. [Google Scholar] [CrossRef]
- Pérez-Martínez, I.; Zhao, Y.; Murillo, J.; Sundin, G.W.; Ramos, C. Global genomic analysis of Pseudomonas savastanoi pv. savastanoi plasmids. J. Bacteriol. 2008, 190, 625–635. [Google Scholar] [CrossRef]
- Juhas, M.; Crook, D.W.; Hood, D.W. Type IV secretion systems: tools of bacterial horizontal gene transfer and virulence. Cell. Microbiol. 2008, 10, 2377–2386. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 2013, 41, 4360–4377. [Google Scholar] [CrossRef]
- Toussaint, A.; Prangishvili, D.; Molineux, I.; Fineran, P.C.; Charpentier, E. Memory of viral infections by CRISPR-Cas adaptive immune systems: Acquisition of new information. Virology 2012, 434, 202–209. [Google Scholar] [CrossRef]
- Haft, D.H.; Selengut, J.; Mongodin, E.F.; Nelson, K.E. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005, 1, e60. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; van der Oost, J.; Koonin, E. V Prokaryotic homologs of Argonaute proteins are predicted to function as key components of a novel system of defense against mobile genetic elements. Biol. Direct 2009, 4, 29. [Google Scholar] [CrossRef]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins—A viable alternative to antibiotics? Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef]
- Hassan, M.; Kjos, M.; Nes, I.F.; Diep, D.B.; Lotfipour, F. Natural antimicrobial peptides from bacteria: Characteristics and potential applications to fight against antibiotic resistance. J. Appl. Microbiol. 2012, 113, 723–736. [Google Scholar] [CrossRef]
- Grinter, R.; Roszak, A.W.; Cogdell, R.J.; Milner, J.J.; Walker, D. The crystal structure of the lipid II-degrading bacteriocin syringacin M suggests unexpected evolutionary relationships between colicin M-like bacteriocins. J. Biol. Chem. 2012, 287, 38876–38888. [Google Scholar]
- Nikolouli, K.; Mossialos, D. Bioactive compounds synthesized by non-ribosomal peptide synthetases and type-I polyketide synthases discovered through genome-mining and metagenomics. Biotechnol. Lett. 2012, 34, 1393–1403. [Google Scholar] [CrossRef]
- Ramel, C.; Tobler, M.; Meyer, M.; Bigler, L.; Ebert, M.-O.; Schellenberg, B.; Dudler, R. Biosynthesis of the proteasome inhibitor syringolin A: The ureido group joining two amino acids originates from bicarbonate. BMC Biochem. 2009, 10, 26. [Google Scholar] [CrossRef]
- Arrebola, E.; Carrión, V.J.; Cazorla, F.M.; Pérez-García, A.; Murillo, J.; de Vicente, A. Characterisation of the mgo operon in Pseudomonas syringae pv. syringae UMAF0158 that is required for mangotoxin production. BMC Microbiol. 2012, 12, 10. [Google Scholar] [CrossRef]
- Yu, X.; Lund, S.P.; Scott, R.A.; Greenwald, J.W.; Records, A.H.; Nettleton, D.; Lindow, S.E.; Gross, D.C.; Beattie, G.A. Transcriptional responses of Pseudomonas syringae to growth in epiphytic versus apoplastic leaf sites. Proc. Natl. Acad. Sci. USA 2013, 110, E425–E434. [Google Scholar] [CrossRef]
- NCBI FTP Server. Available online: Available online: ftp://ftp.ncbi.nlm.nih.gov/genomes/ (accessed on 4 September 2013).
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 2008, 9, 75. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Prokaryotic Genome Annotation System. Available online: http://vicbioinformatics.com/ (Accessed on 10 September 2013).
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. PGAP: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Pseudomonas syringae Genome Resources Home Page. Available online: http://www.pseudomonas-syringae.org/ (accessed on 20 January 2014).
- Mucyn, T.S.; Yourstone, S.; Lind, A.L.; Biswas, S.; Nishimura, M.T.; Baltrus, D.A.; Cumbie, J.S.; Chang, J.H.; Jones, C.D.; Dangl, J.L.; et al. Variable suites of non-effector genes are co-regulated in the type III secretion virulence regulon across the Pseudomonas syringae phylogeny. PLoS Pathog. 2014, 10, e1003807. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, V.M.; Mavromatis, K.; Ivanova, N.N.; Chen, I.-M.A.; Chu, K.; Kyrpides, N.C. IMG ER: A system for microbial genome annotation expert review and curation. Bioinformatics 2009, 25, 2271–2278. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef]
- De Jong, A.; van Heel, A.J.; Kok, J.; Kuipers, O.P. BAGEL2: Mining for bacteriocins in genomic data. Nucleic Acids Res. 2010, 38, W647–W651. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dudnik, A.; Dudler, R. Genomics-Based Exploration of Virulence Determinants and Host-Specific Adaptations of Pseudomonas syringae Strains Isolated from Grasses. Pathogens 2014, 3, 121-148. https://doi.org/10.3390/pathogens3010121
Dudnik A, Dudler R. Genomics-Based Exploration of Virulence Determinants and Host-Specific Adaptations of Pseudomonas syringae Strains Isolated from Grasses. Pathogens. 2014; 3(1):121-148. https://doi.org/10.3390/pathogens3010121
Chicago/Turabian StyleDudnik, Alexey, and Robert Dudler. 2014. "Genomics-Based Exploration of Virulence Determinants and Host-Specific Adaptations of Pseudomonas syringae Strains Isolated from Grasses" Pathogens 3, no. 1: 121-148. https://doi.org/10.3390/pathogens3010121
APA StyleDudnik, A., & Dudler, R. (2014). Genomics-Based Exploration of Virulence Determinants and Host-Specific Adaptations of Pseudomonas syringae Strains Isolated from Grasses. Pathogens, 3(1), 121-148. https://doi.org/10.3390/pathogens3010121