DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: I. Molecule–Surface and Cl–Surface Bonding


Abstract
1. Introduction
2. Technical Details and Definitions
2.1. Computational Details
2.2. CuO as a Model of Oxidized Copper Surface
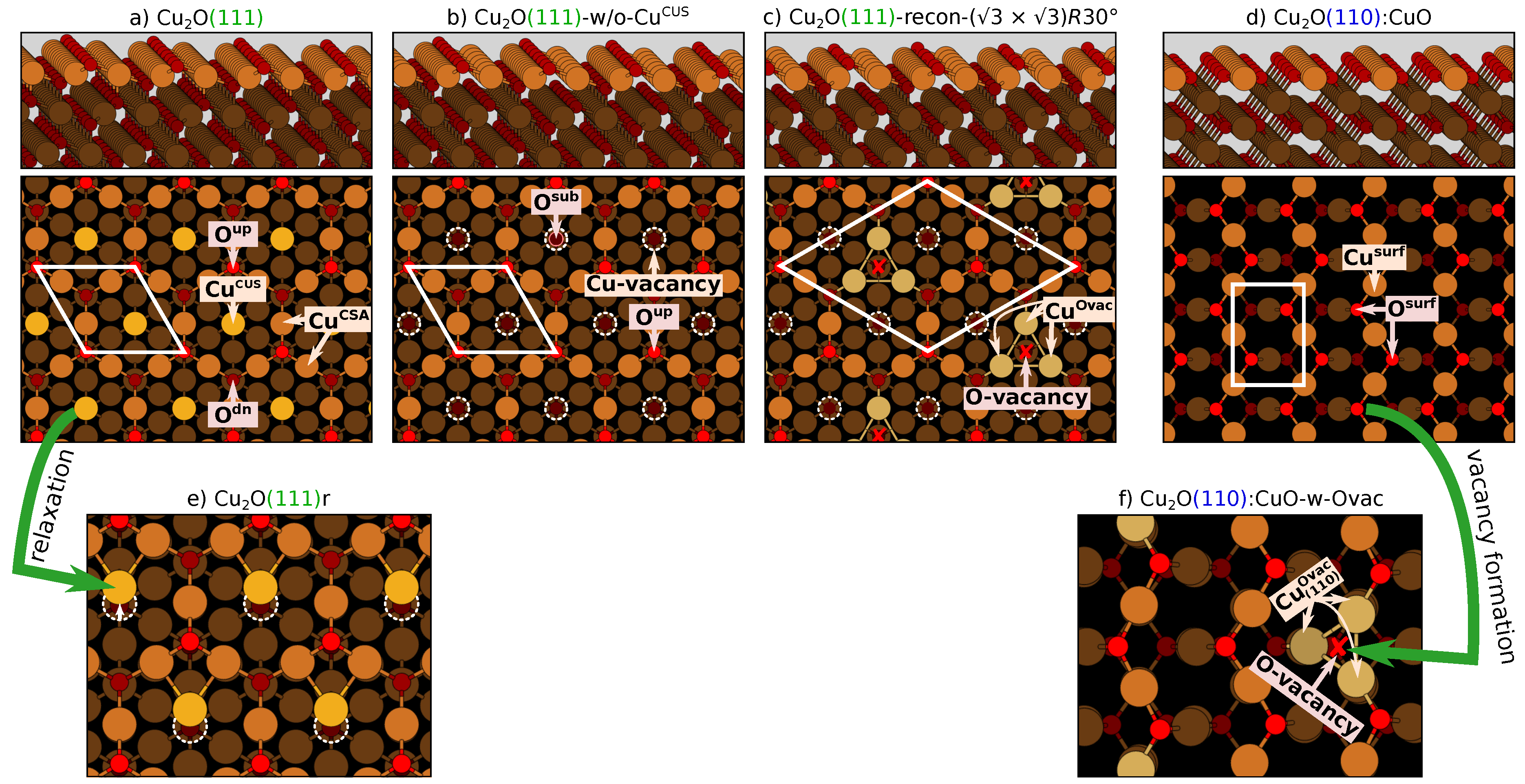
2.3. Description of Considered Surfaces
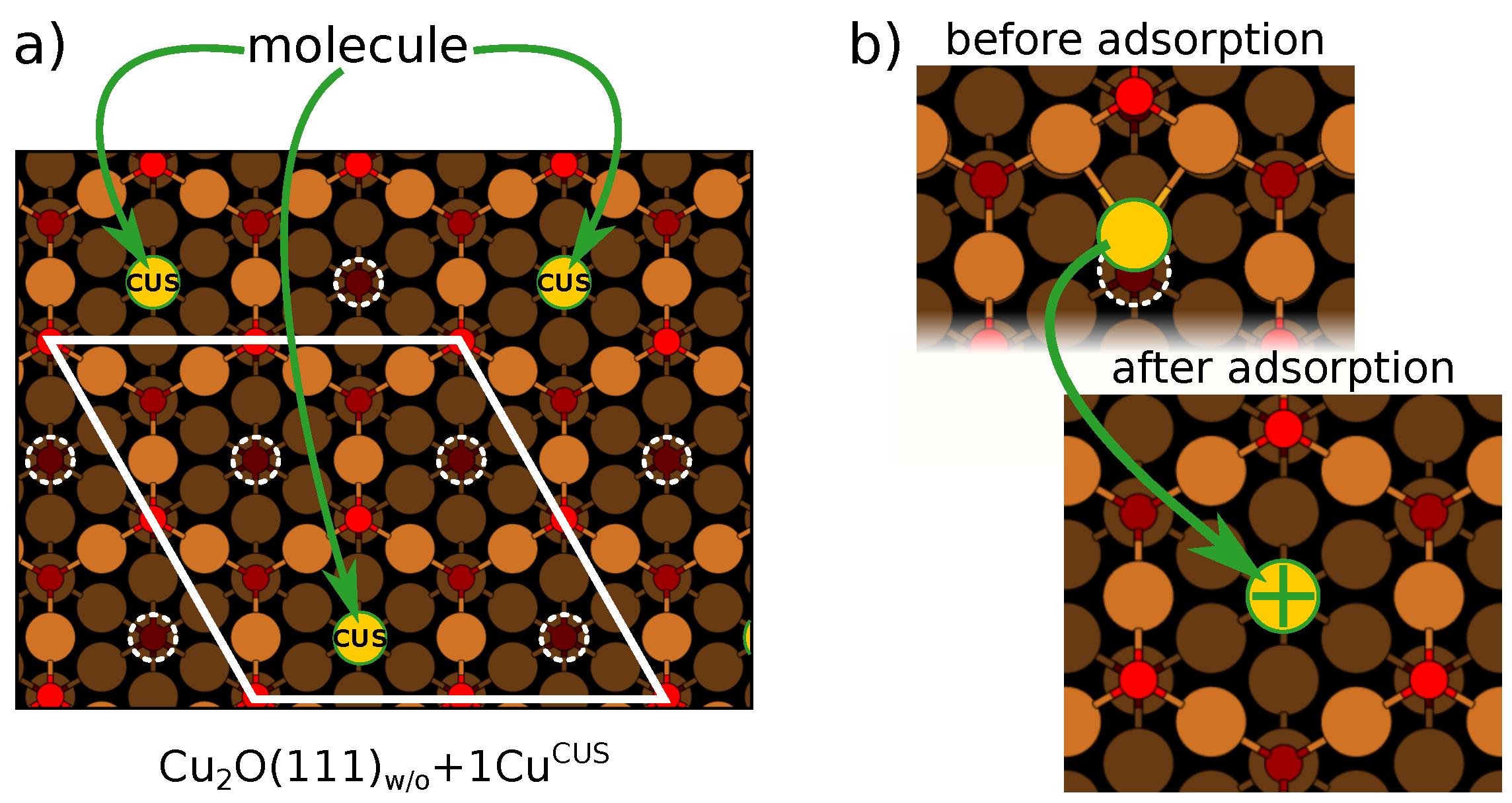
2.4. An Issue with Sites: A CuO(111)+1 Model
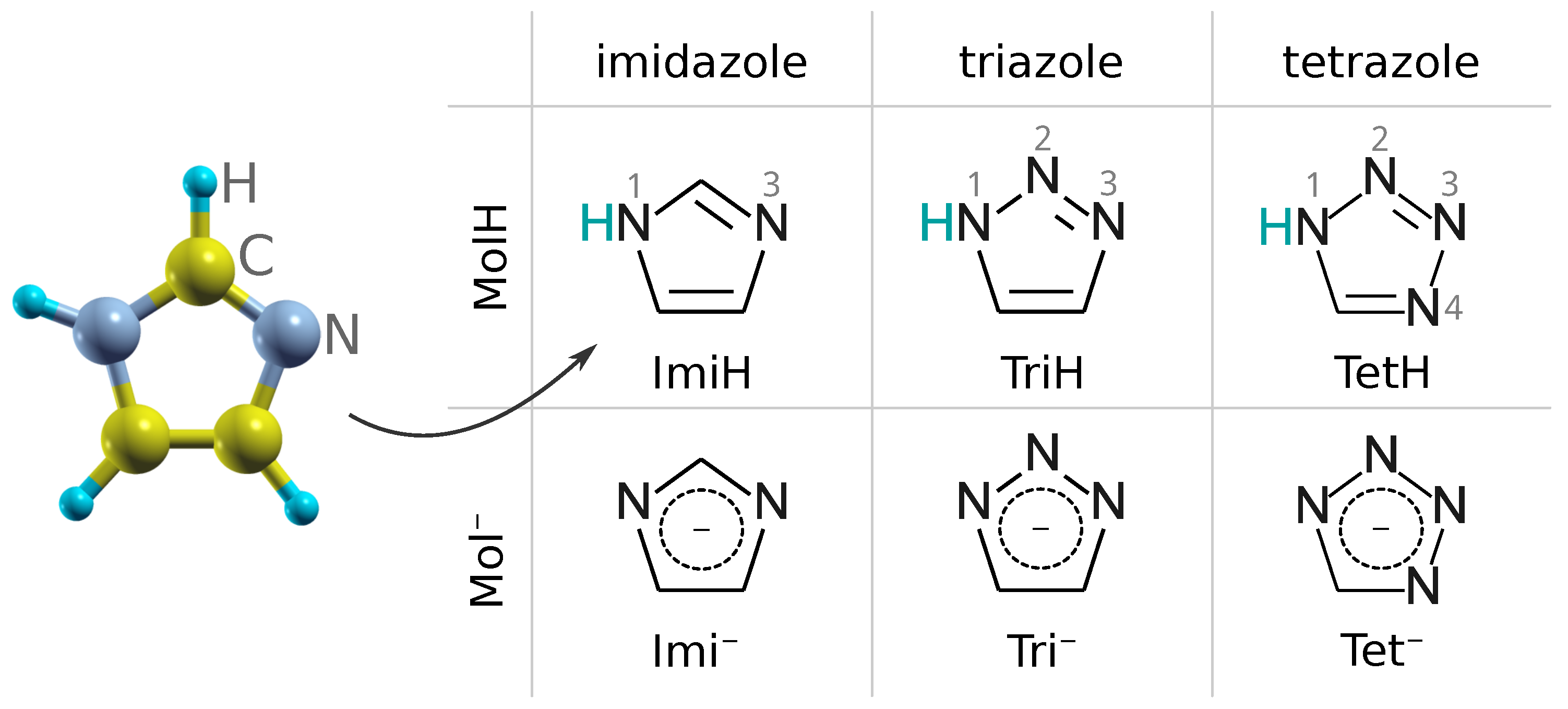
2.5. Molecular Labels
2.6. Adsorption Calculations
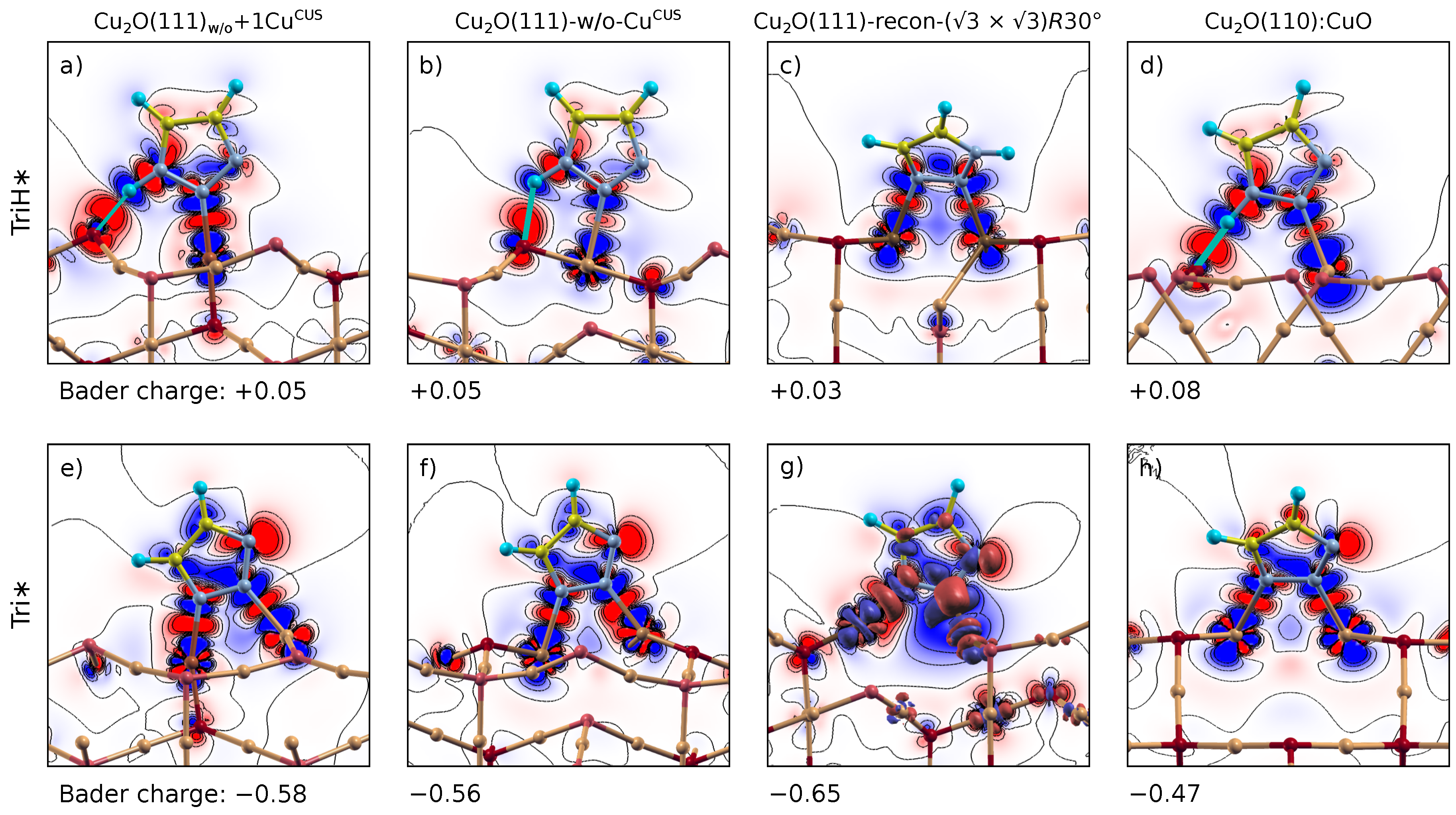
2.6.1. Charge Density Difference
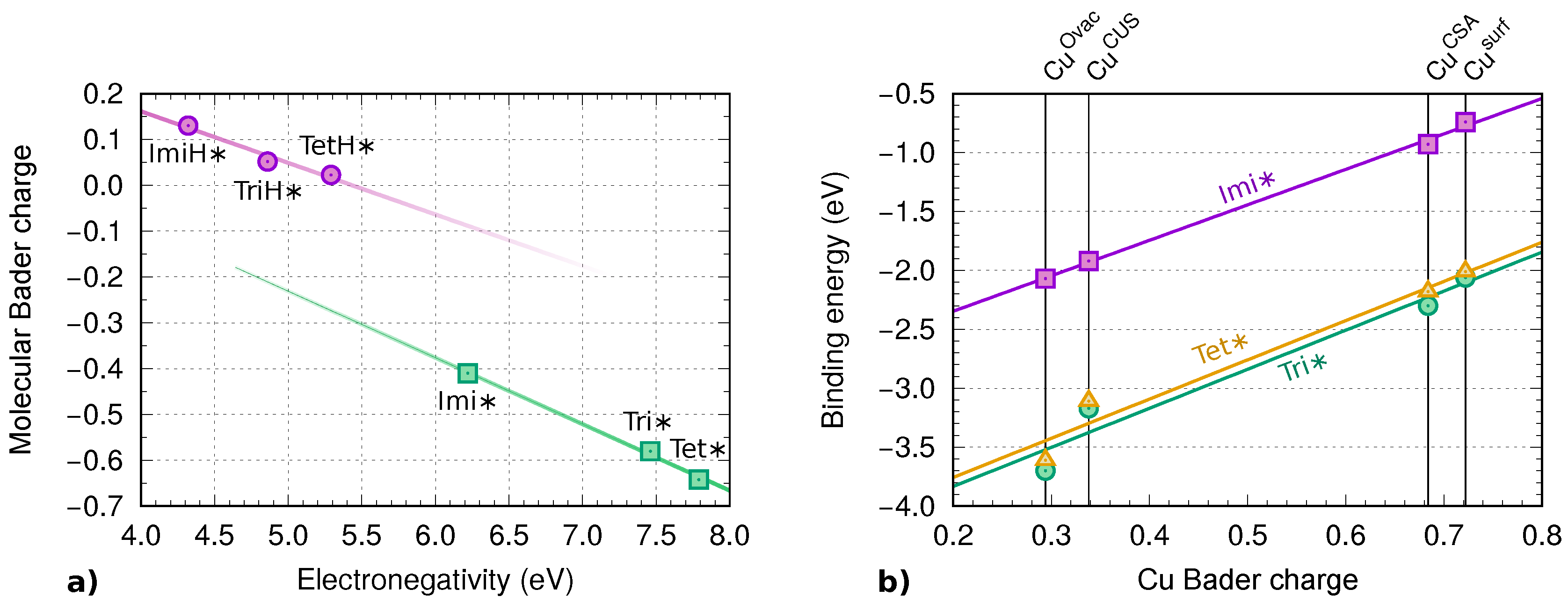
2.6.2. Bader Charge Analysis
2.6.3. Dissociation Activation Energies
3. Results
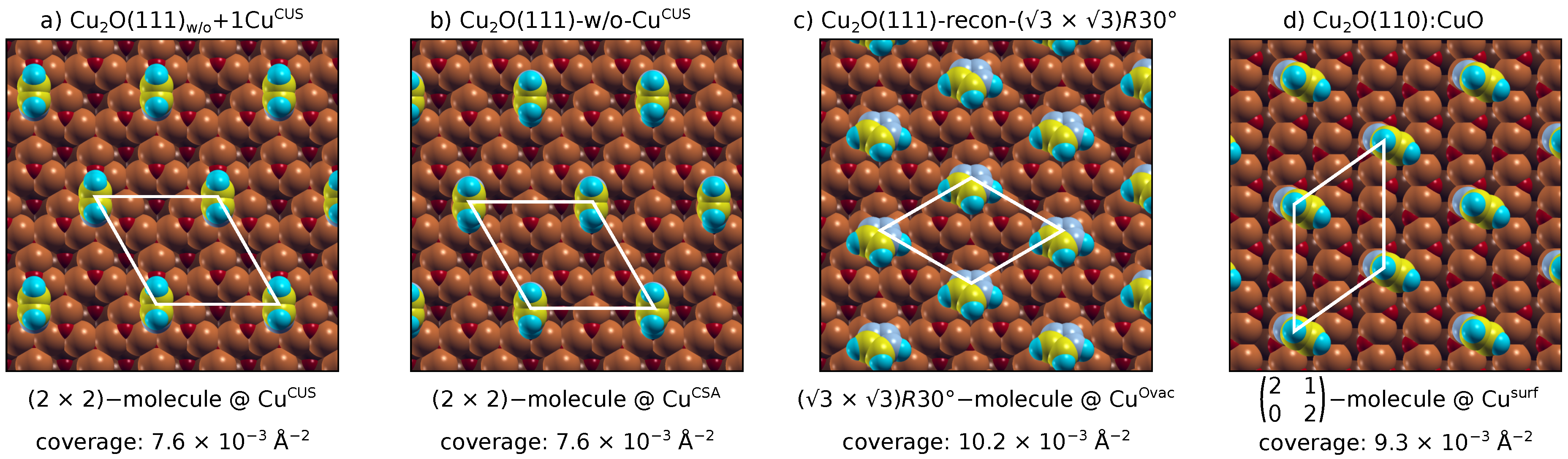
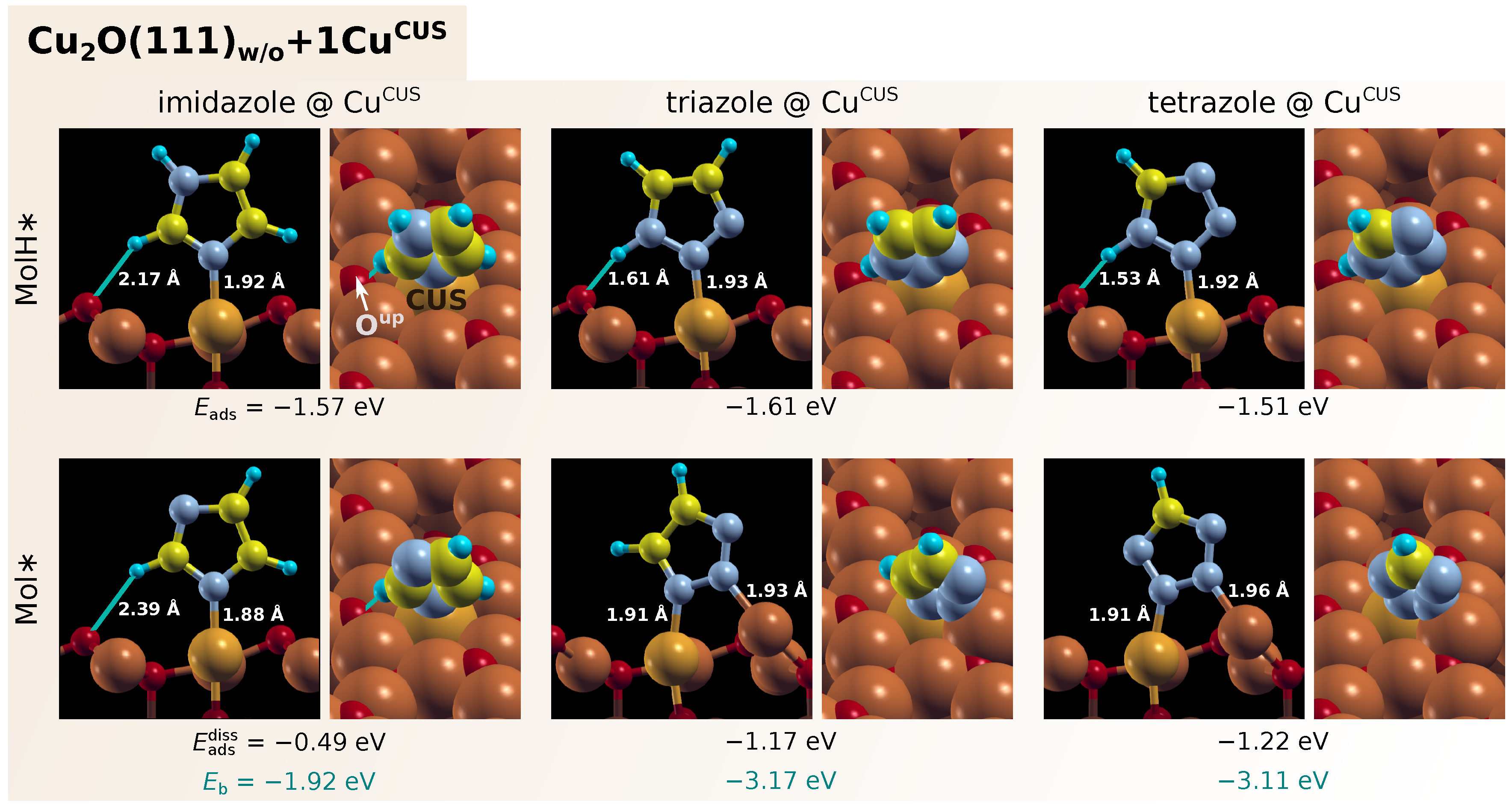
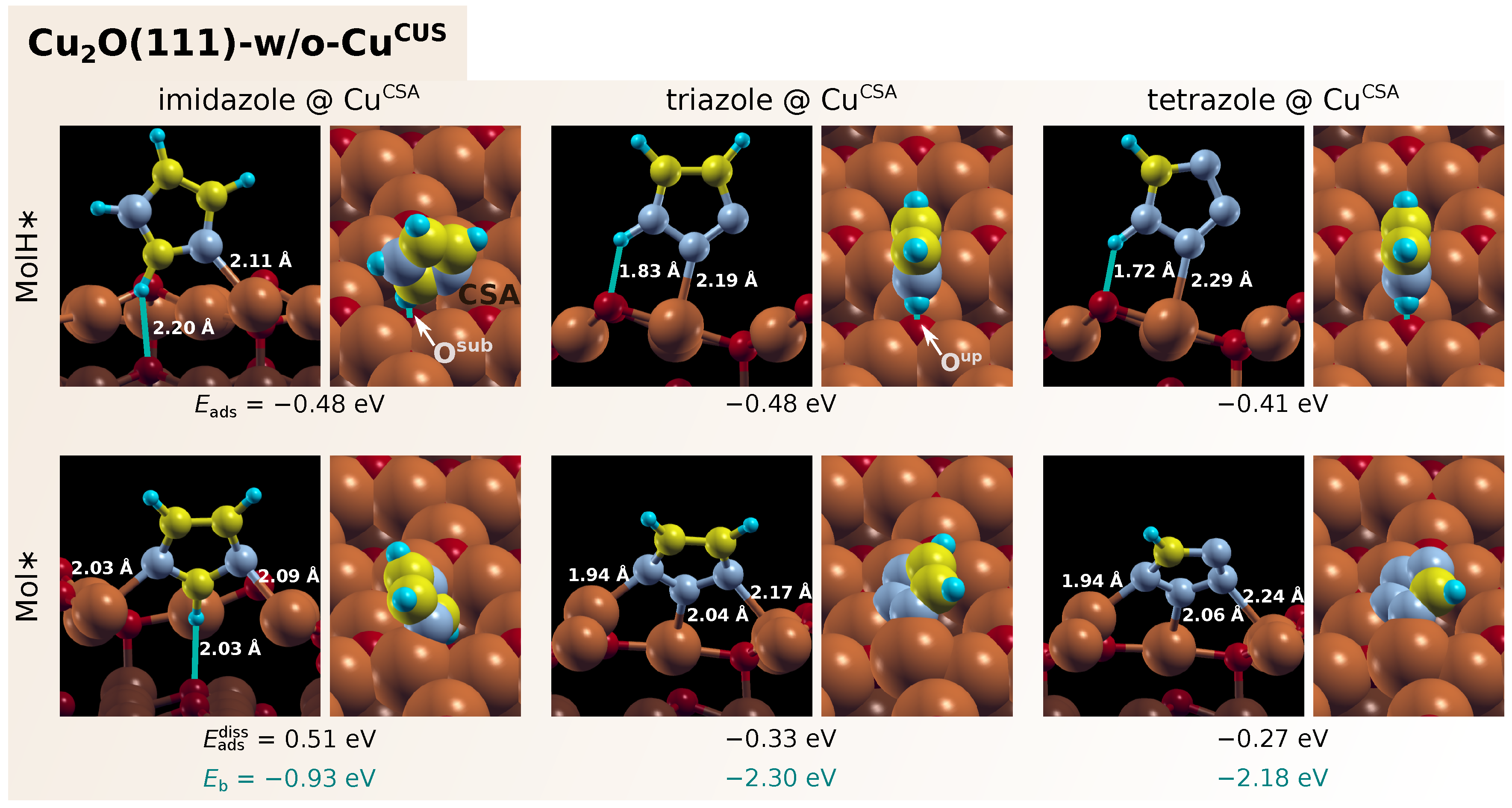
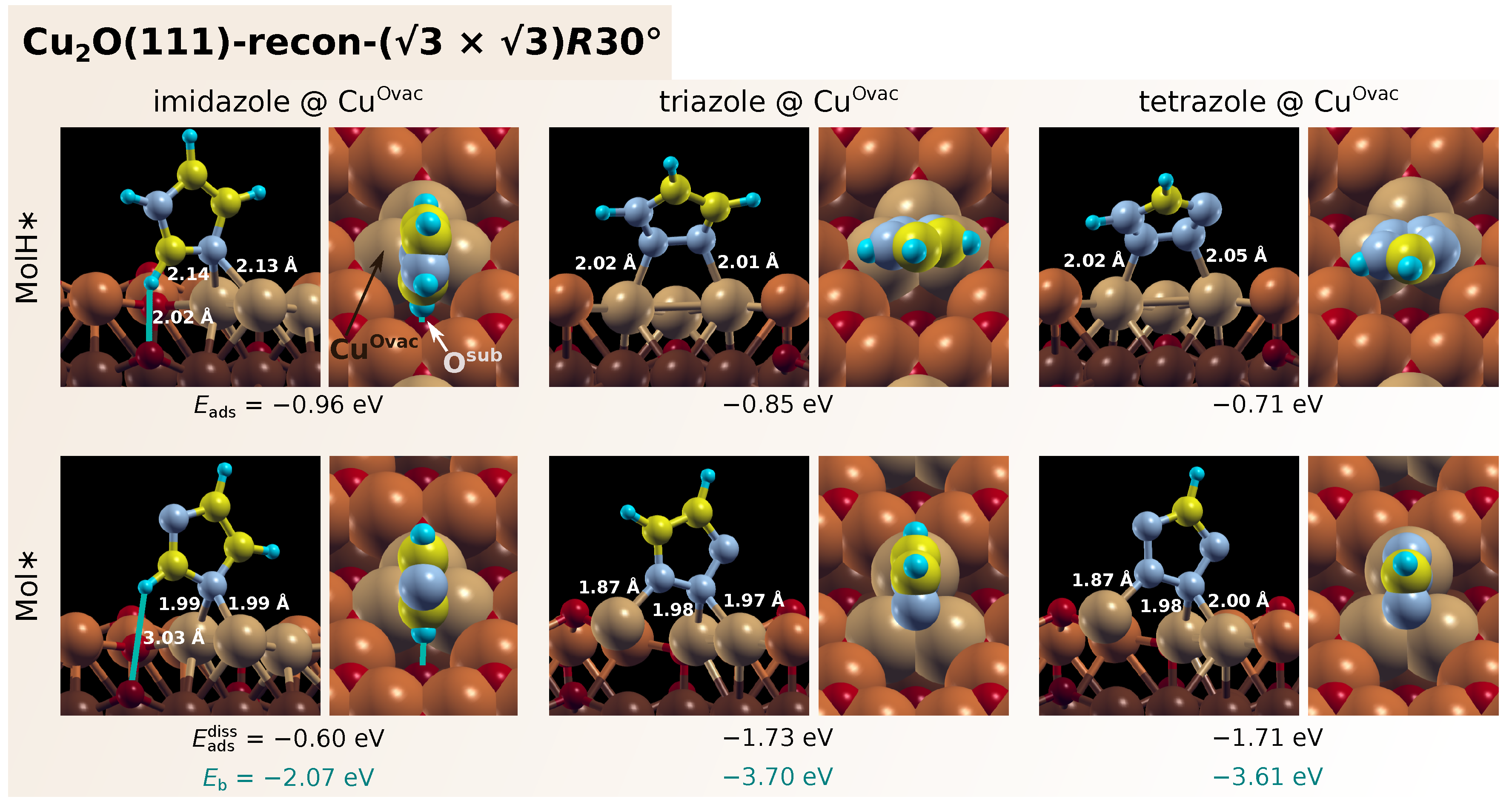
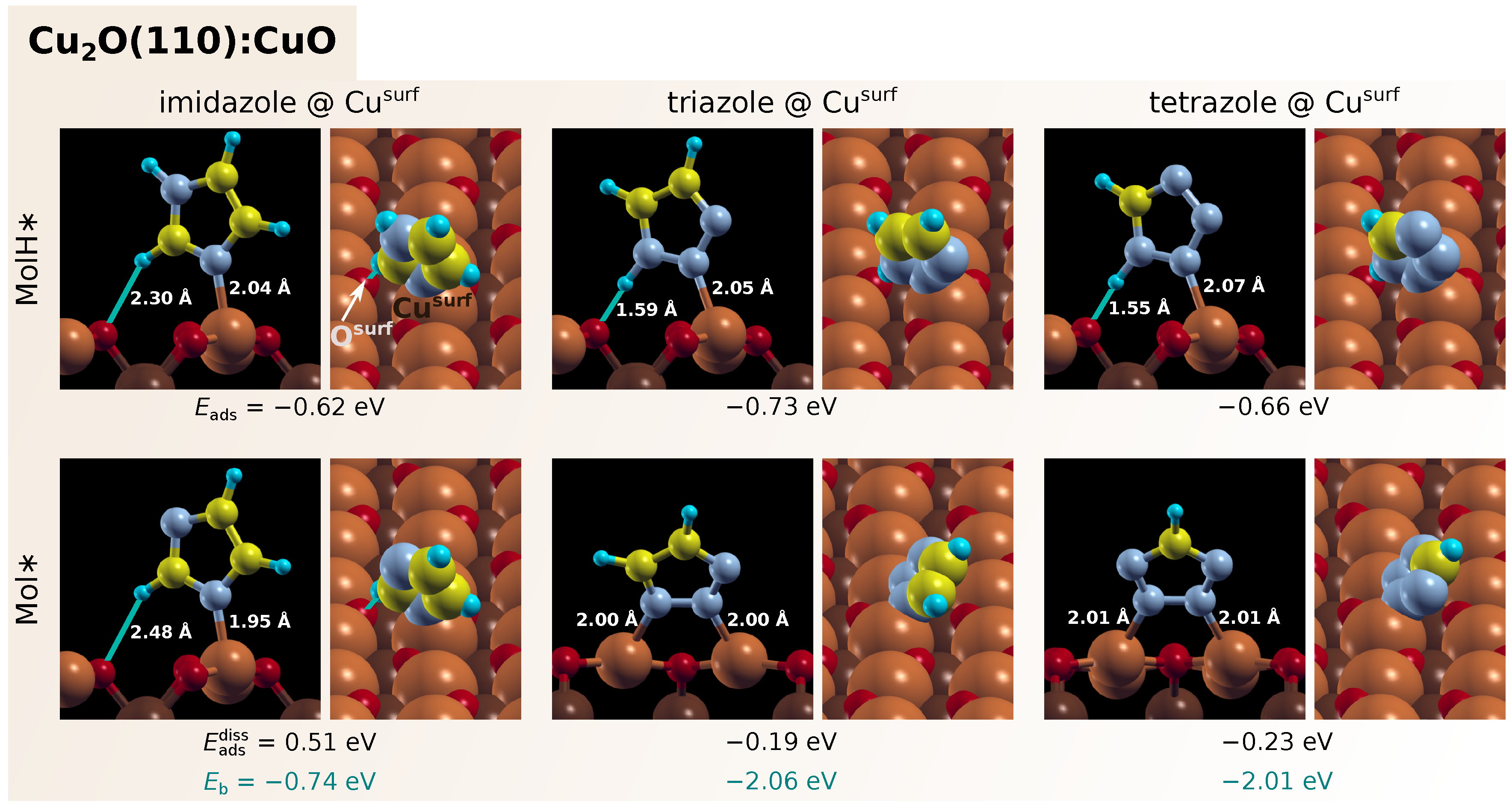
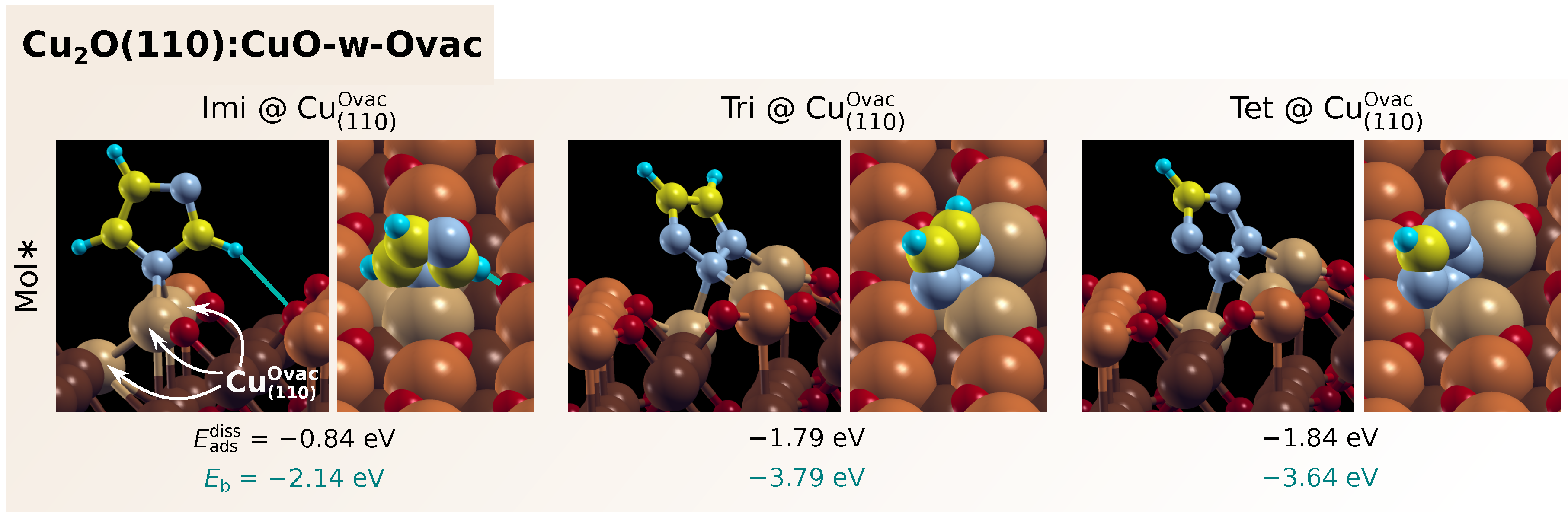
3.1. Adsorption of Standalone Molecules at Lower Coverage
3.2. Electronic Structure Analysis
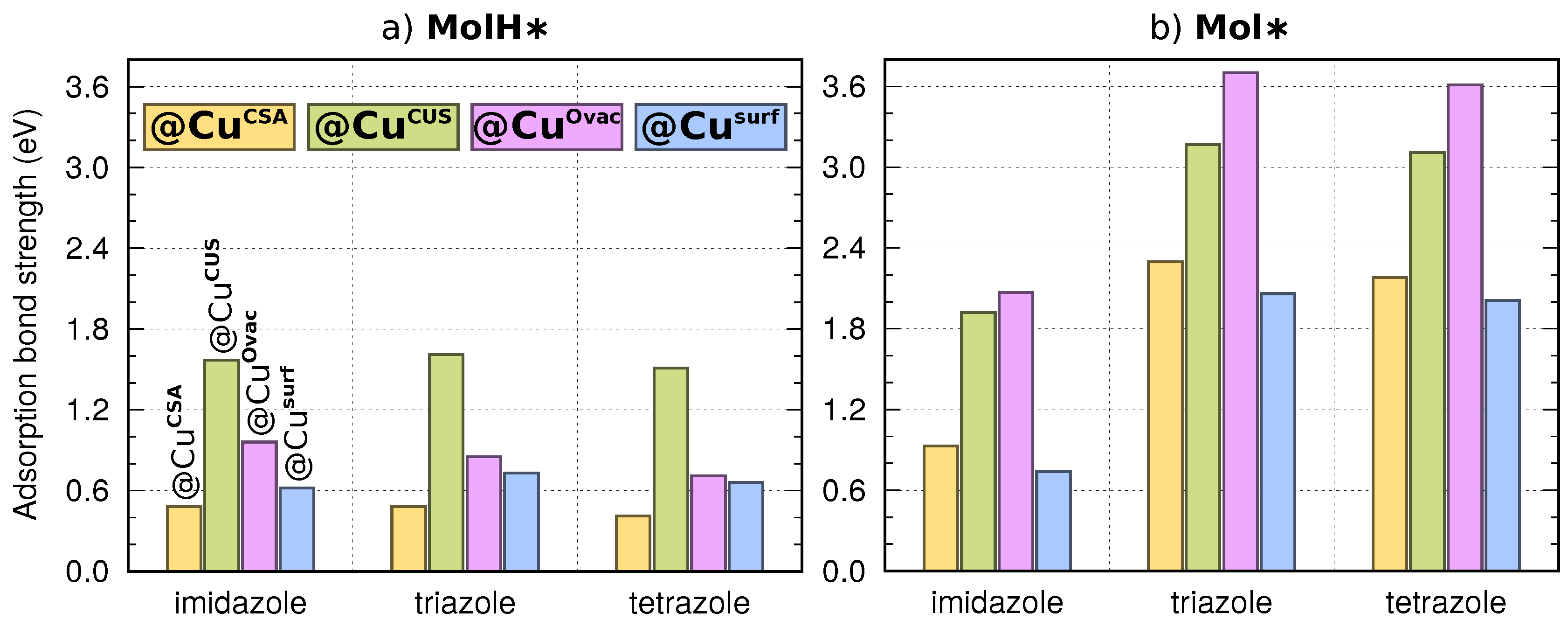
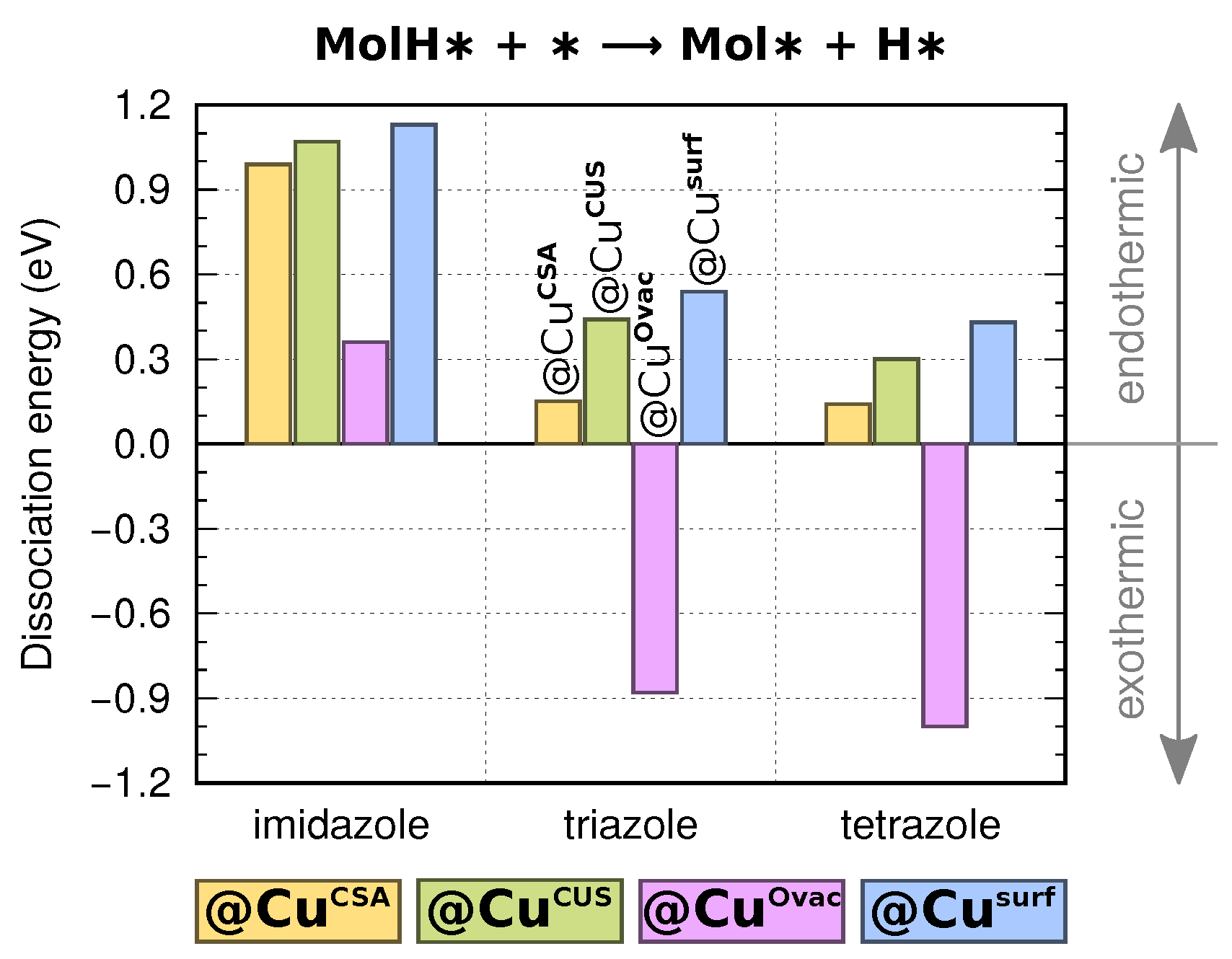
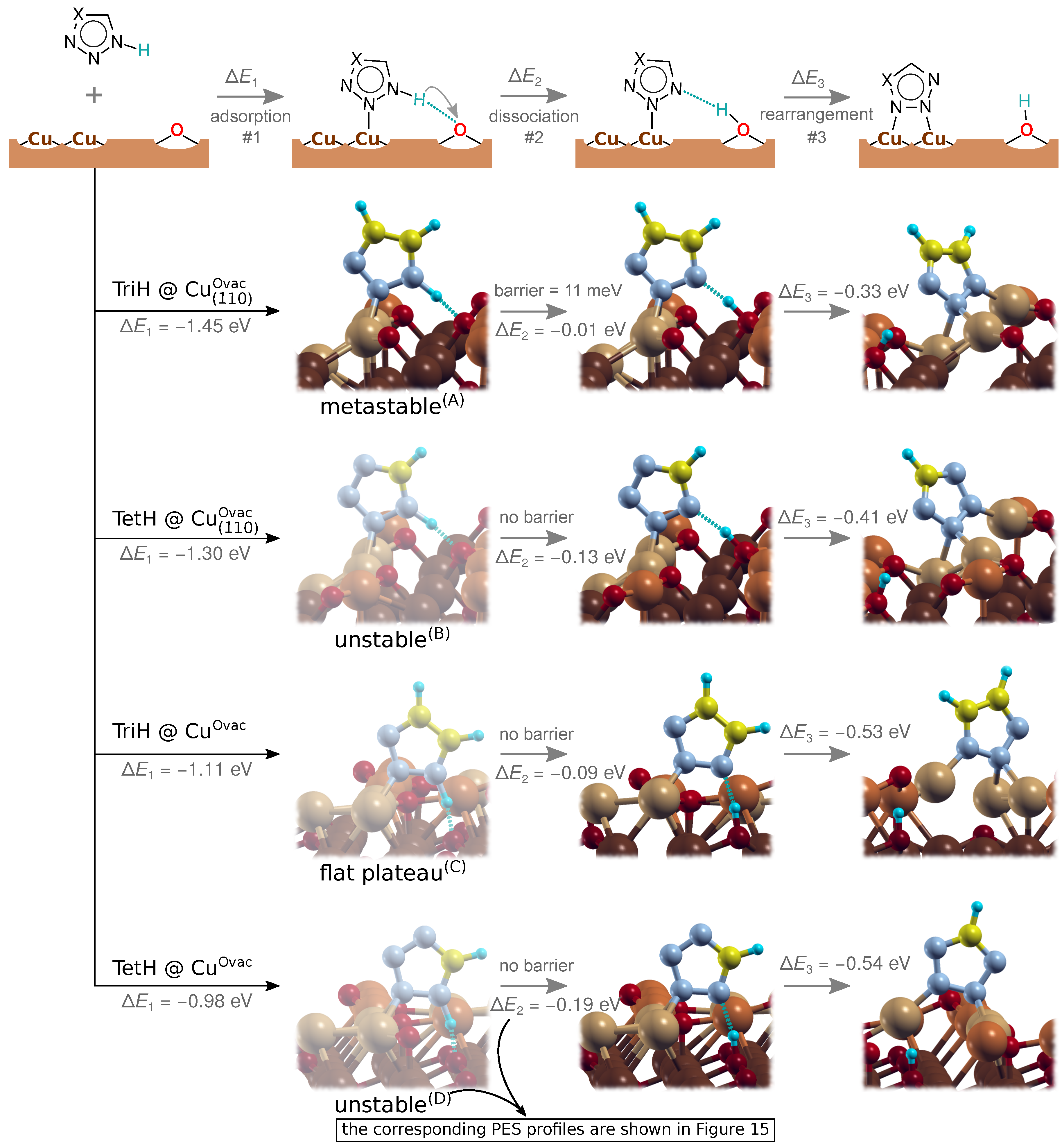
3.3. Dissociation of Adsorbed Azole Molecules, MolH∗ + ∗ → Mol∗ + H∗
3.3.1. Bonding of H to Various Sites on CuO Surfaces
3.3.2. Co-Adsorption of Mol and H
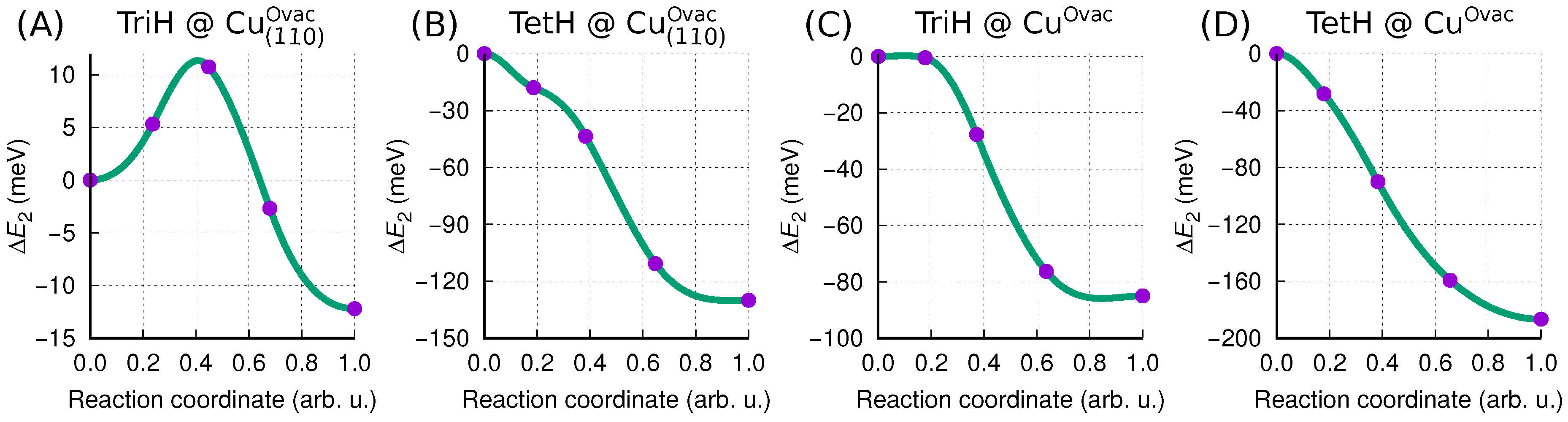
3.3.3. Mechanistic Insight into MolH∗ + O∗ → Mol∗ + OH∗ Dissociation
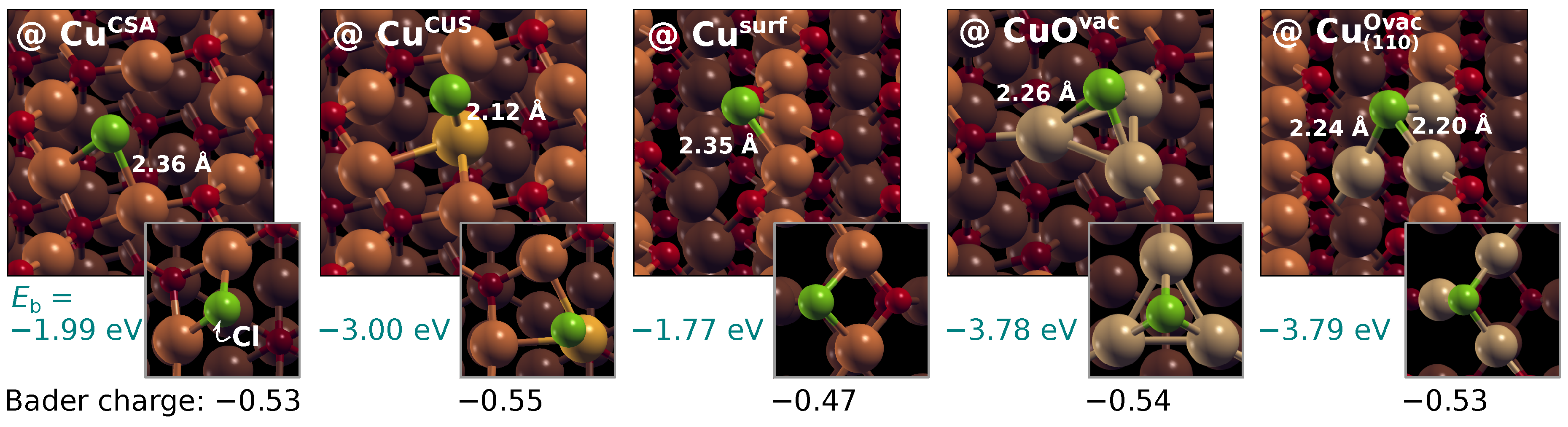
3.4. Comparison between Molecular and Chloride Adsorption
3.5. Adsorption of Molecules from Vacuum and Aqueous-Phase: Differences
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | density functional theory |
| GGA | generalized gradient approximation |
| GGA+U | +U corrected GGA, where U stands for screened on-site Coulomb interaction |
| PBE | Perdew–Burke–Ernzerhof |
| BZ | Brillouin zone |
| CSA | coordinatively saturated |
| CUS | coordinatively unsaturated |
| PES | potential energy surface |
| MEP | minimum energy path |
| CI-NEB | climbing-image nudged-elastic-band |
| US-PP | ultrasoft pseudopotentials |
| STM | scanning tunneling microscopy |
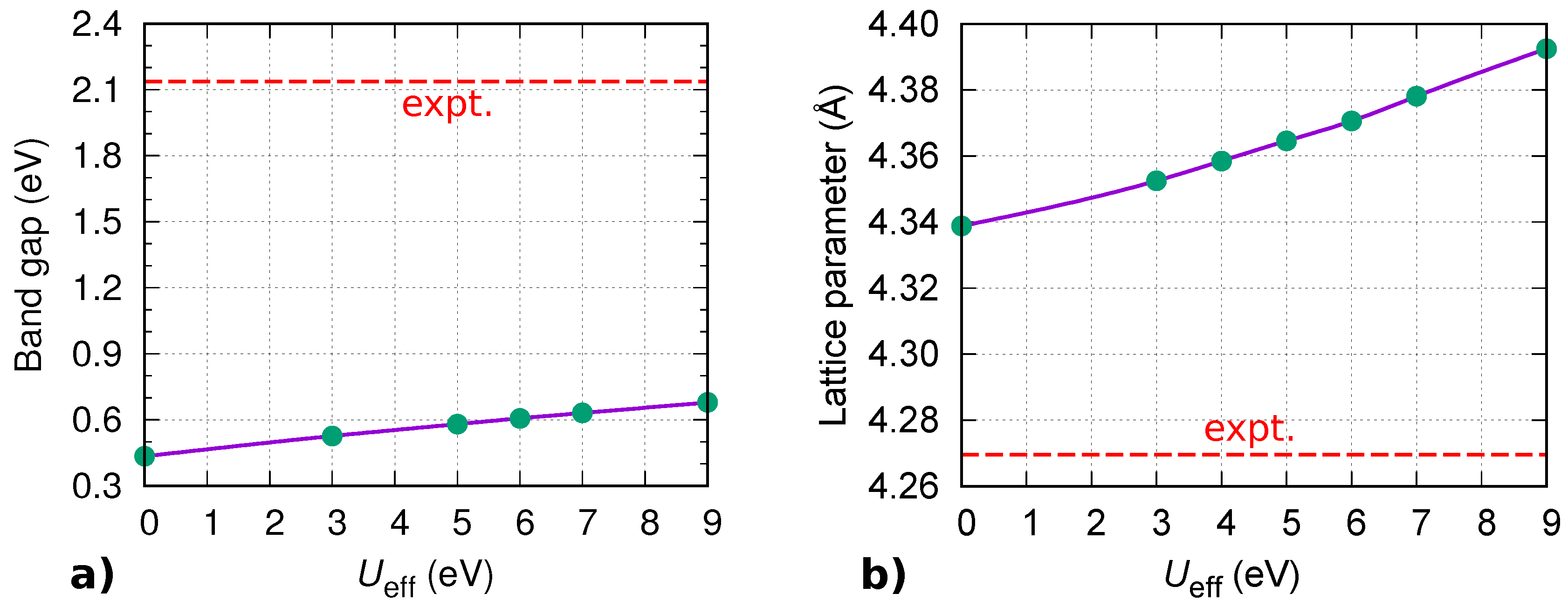
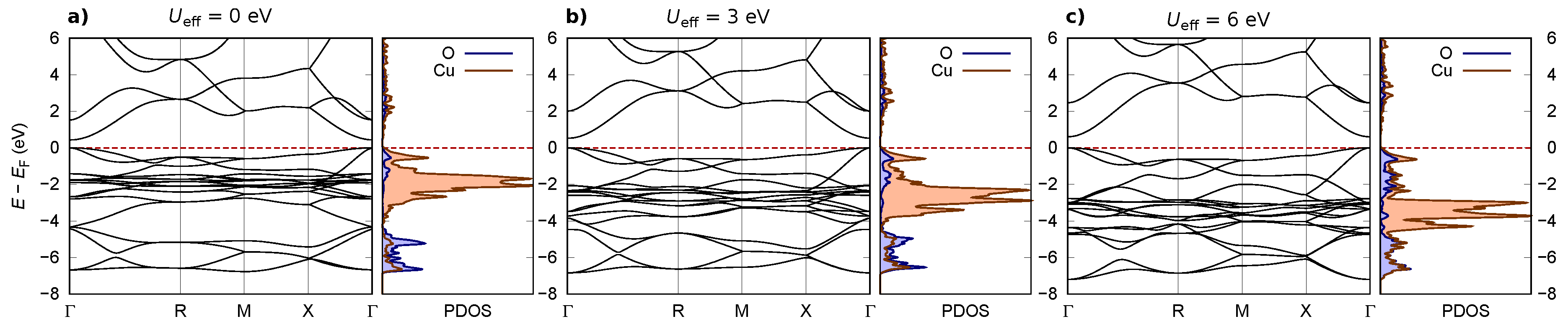
Appendix A. Effect of Hubbard U Parameter on Electronic and Adsorption Properties



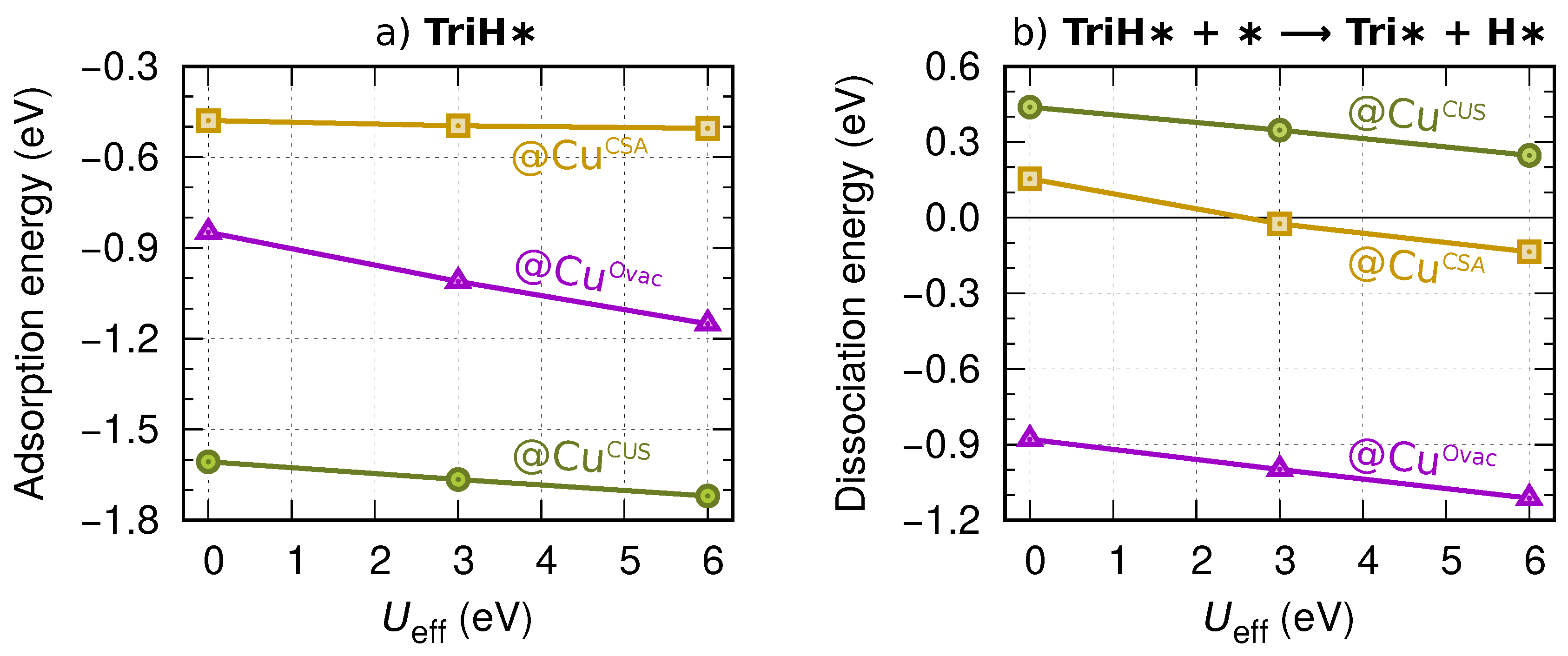
Appendix B. Binding Energies of Mol∗ and Cl∗
References
- Kuznetsov, Y.; Kazansky, L. Physicochemical aspects of metal protection by azoles as corrosion inhibitors. Russ. Chem. Rev. 2008, 77, 219–232. [Google Scholar] [CrossRef]
- Antonijević, M.M.; Milić, S.M.; Petrović, M.B. Films formed on copper surface in chloride media in the presence of azoles. Corros. Sci. 2009, 51, 1228–1237. [Google Scholar] [CrossRef]
- Antonijević, M.M.; Petrović, M.B. Copper corrosion inhibitors. A review. Int. J. Electrochem. Sci. 2008, 3, 1–28. [Google Scholar]
- Mihajlović, M.B.P.; Antonijević, M.M. Copper Corrosion Inhibitors. Period 2008–2014. A Review. Int. J. Electrochem. Sci. 2015, 10, 1027–1053. [Google Scholar]
- Bockris, J.O.; Reddy, A.K.N. Modern Electrochemistry, 2nd ed.; Kluwer Academic/Plenum Publishers: New York/Boston, NY, USA, 2000; Volume 2B. [Google Scholar]
- Kokalj, A.; Peljhan, S.; Finšgar, M.; Milošev, I. What Determines the Inhibition Effectiveness of ATA, BTAH, and BTAOH Corrosion Inhibitors on Copper? J. Am. Chem. Soc. 2010, 132, 16657–16668. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.; Ribeiro, T.; Cornette, P.; Marcus, P. DFT Modeling of Corrosion Inhibition by Organic Molecules: Carboxylates as Inhibitors of Aluminum Corrosion. J. Phys. Chem. C 2016, 120, 28607–28616. [Google Scholar] [CrossRef]
- Costa, D.; Marcus, P. Adsorption of Organic Inhibitor Molecules on Metal and Oxidized Surfaces Studied by Atomistic Theoretical Methods. In Molecular Modeling of Corrosion Processes; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 125–156. [Google Scholar]
- Kokalj, A.; Costa, D. Molecular Modeling of Corrosion Inhibitors. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Encyclopedia of Interfacial Chemistry: Surface Science and Electrochemistry; Elsevier: Amsterdam, The Netherlands, 2018; Volume 6.1, pp. 332–345. Available online: https://www.sciencedirect.com/science/article/pii/B9780124095472134444 (accessed on 25 April 2018).
- Taylor, C.D.; Chandra, A.; Vera, J.; Sridhar, N. Multiphysics modelling, quantum chemistry and risk analysis for corrosion inhibitor design and lifetime prediction. Faraday Discuss. 2015, 180, 459–477. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.A. Predicting the Performance of Organic Corrosion Inhibitors. Metals 2017, 7, 553. [Google Scholar] [CrossRef]
- Winkler, D.A.; Breedon, M.; Hughes, A.E.; Burden, F.R.; Barnard, A.S.; Harvey, T.G.; Cole, I. Towards chromate-free corrosion inhibitors: Structure-property models for organic alternatives. Green Chem. 2014, 16, 3349–3357. [Google Scholar] [CrossRef]
- Winkler, D.A.; Breedon, M.; White, P.; Hughes, A.E.; Sapper, E.D.; Cole, I. Using high throughput experimental data and in silico models to discover alternatives to toxic chromate corrosion inhibitors. Corros. Sci. 2016, 106, 229–235. [Google Scholar] [CrossRef]
- Gustinčič, D.; Kokalj, A. DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: II. Lateral Interactions and Thermodynamic Stability. Metals 2018, 8, 311. [Google Scholar] [CrossRef]
- Kovačević, N.; Kokalj, A. DFT Study of Interaction of Azoles with Cu(111) and Al(111) Surfaces: Role of Azole Nitrogen Atoms and Dipole–Dipole Interactions. J. Phys. Chem. C 2011, 115, 24189–24197. [Google Scholar] [CrossRef]
- Kovačević, N.; Kokalj, A. The relation between adsorption bonding and corrosion inhibition of azole molecules on copper. Corros. Sci. 2013, 73, 7–17. [Google Scholar] [CrossRef]
- Gustinčič, D.; Kokalj, A. A DFT study of adsorption of imidazole, triazole, and tetrazole on oxidized copper surfaces: Cu2O(111) and Cu2O(111)-w/o-CuCUS. Phys. Chem. Chem. Phys. 2015, 17, 28602–28615. [Google Scholar] [CrossRef] [PubMed]
- Kovačević, N.; Milošev, I.; Kokalj, A. How relevant is the adsorption bonding of imidazoles and triazoles for their corrosion inhibition of copper? Corros. Sci. 2017, 124, 25–34. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd ed.; NACE, Cebelcor: Houston, TX, USA, 1974. [Google Scholar]
- Soon, A.; Todorova, M.; Delley, B.; Stampfl, C. Thermodynamic stability and structure of copper oxide surfaces: A first-principles investigation. Phys. Rev. B 2007, 75, 125420–125429. [Google Scholar] [CrossRef]
- Soon, A.; Todorova, M.; Delley, B.; Stampfl, C. Erratum: Thermodynamic stability and structure of copper oxide surfaces: A first-principles investigation [Phys. Rev. B 75, 125420 (2007)]. Phys. Rev. B 2007, 76, 129902. [Google Scholar] [CrossRef]
- Önsten, A.; Göthelid, M.; Karlsson, U.O. Atomic structure of Cu2O(111). Surf. Sci. 2009, 603, 257–264. [Google Scholar] [CrossRef]
- Besharat, Z.; Stenlid, J.H.; Soldemo, M.; Marks, K.; Önsten, A.; Johnson, M.; Öström, H.; Weissenrieder, J.; Brinck, T.; Göthelid, M. Dehydrogenation of methanol on Cu2O(100) and (111). J. Chem. Phys. 2017, 146, 244702. [Google Scholar] [CrossRef] [PubMed]
- Soldemo, M.; Stenlid, J.H.; Besharat, Z.; Johansson, N.; Önsten, A.; Knudsen, J.; Schnadt, J.; Göthelid, M.; Brinck, T.; Weissenrieder, J. Interaction of Sulfur Dioxide and Near-Ambient Pressures of Water Vapor with Cuprous Oxide Surfaces. J. Phys. Chem. C 2017, 121, 24011–24024. [Google Scholar] [CrossRef]
- Nilius, N.; Fedderwitz, H.; Groß, B.; Noguera, C.; Goniakowski, J. Incorrect DFT-GGA predictions of the stability of non-stoichiometric/polar dielectric surfaces: The case of Cu2O(111). Phys. Chem. Chem. Phys. 2016, 18, 6729–6733. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, D.O.; Morgan, B.J.; Watson, G.W. Modeling the polaronic nature of p-type defects in Cu2O: The failure of GGA and GGA+U. J. Chem. Phys. 2009, 131, 124703. [Google Scholar] [CrossRef] [PubMed]
- Kokalj, A.; Peljhan, S. Density Functional Theory Study of Adsorption of Benzotriazole on Cu2O Surfaces. J. Phys. Chem. C 2015, 119, 11625–11635. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Isseroff, L.Y.; Carter, E.A. Importance of reference Hamiltonians containing exact exchange for accurate one-shot GW calculations of Cu2O. Phys. Rev. B 2012, 85, 235142. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Ultrasoft Pseudopotentials for H, C, N, O, and Cu Were Taken from the Quantum Espresso Pseudopotential. (files: H.pbe-rrkjus.UPF, C.pbe-rrkjus.UPF, N.pbe-rrkjus.UPF, O.pbe-rrkjus.UPF, Cl.pbe-n-van.UPF, and Cu.pbe-d-rrkjus.UPF). 2017. Available online: http://www.quantum-espresso.org/pseudopotentials (accessed on 21 August 2017).
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Marzari, N.; Vanderbilt, D.; De Vita, A.; Payne, M.C. Thermal Contraction and Disordering of the Al(110) Surface. Phys. Rev. Lett. 1999, 82, 3296–3299. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- Kokalj, A. XCrySDen-a new program for displaying crystalline structures and electron densities. J. Mol. Graph. Modell. 1999, 17, 176–179. [Google Scholar] [CrossRef]
- Costa, D.; Ribeiro, T.; Mercuri, F.; Pacchioni, G.; Marcus, P. Atomistic Modeling of Corrosion Resistance: A First Principles Study of O2 Reduction on the Al(111) Surface Covered with a Thin Hydroxylated Alumina Film. Adv. Mater. Interfaces 2014, 1, 1300072. [Google Scholar] [CrossRef]
- Finšgar, M.; Peljhan, S.; Kokalj, A.; Kovač, J.; Milošev, I. Determination of the Cu2O Thickness on BTAH-Inhibited Copper by Reconstruction of Auger Electron Spectra. J. Electrochem. Soc. 2010, 157, C295–C301. [Google Scholar] [CrossRef]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 85th ed.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Li, C.; Wang, F.; Li, S.; Sun, Q.; Jia, Y. Stability and electronic properties of the O-terminated Cu2O(111) surfaces: First-principles investigation. Phys. Lett. A 2010, 374, 2994–2998. [Google Scholar] [CrossRef]
- Bengtsson, L. Dipole correction for surface supercell calculations. Phys. Rev. B 1999, 59, 12301–12304. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: a Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Arnaldsson, A.; Tang, W.; Henkelman, G. Computer Program for Bader Charge Analysis. Available online: http://theory.cm.utexas.edu/henkelman/code/bader/ (accessed on 21 August 2017).
- Henkelman, G.; Jonsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Kokalj, A. Electrostatic model for treating long-range lateral interactions between polar molecules adsorbed on metal surfaces. Phys. Rev. B 2011, 84, 045418. [Google Scholar] [CrossRef]
- Stenlid, J.H.; Johansson, A.J.; Brinck, T. Local Lewis Acidity of (TiO2)n (n = 7–10) Nanoparticles Characterized by DFT-Based Descriptors: Tools for Catalyst Design. J. Phys. Chem. C 2017, 121, 27483–27492. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, X.; Wang, H.; Wang, Z.; Feng, G. High-Coverage H2 Adsorption on the Reconstructed Cu2O(111) Surface. J. Phys. Chem. C 2017, 121, 22081–22091. [Google Scholar] [CrossRef]
- Kokalj, A.; Peljhan, S.; Koller, J. The Effect of Surface Geometry of Copper on Dehydrogenation of Benzotriazole. Part II. J. Phys. Chem. C 2014, 118, 944–954. [Google Scholar] [CrossRef]
- Kokalj, A. Ab initio modeling of the bonding of benzotriazole corrosion inhibitor to reduced and oxidized copper surfaces. Faraday Discuss. 2015, 180, 415–438. [Google Scholar] [CrossRef] [PubMed]
- Peljhan, S.; Koller, J.; Kokalj, A. The Effect of Surface Geometry of Copper on Adsorption of Benzotriazole and Cl. Part I. J. Phys. Chem. C 2014, 118, 933–943. [Google Scholar] [CrossRef]
- Marcus, P.; Maurice, V.; Strehblow, H.H. Localized corrosion (pitting): A model of passivity breakdown including the role of the oxide layer nanostructure. Corros. Sci. 2008, 50, 2698–2704. [Google Scholar] [CrossRef]
- Maurice, V.; Marcus, P. Passive films at the nanoscale. Electrochim. Acta 2012, 84, 129–138. [Google Scholar] [CrossRef]
- Walba, H.; Isensee, R.W. Acidity Constants of Some Arylimidazoles and Their Cations. J. Org. Chem. 1961, 26, 2789–2791. [Google Scholar] [CrossRef]
- Yet, L. Five-Membered Heterocycles with Three Heteroatoms: Triazoles. In Modern Heterocyclic Chemistry; Alvarez-Builla, J., Vaquero, J.J., Barluenga, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 989–1045. [Google Scholar]
- Boraei, A.A.A. Acidity Constants of Some Tetrazole Compounds in Various Aqueous–Organic Solvent Media. J. Chem. Eng. Data 2001, 46, 939–943. [Google Scholar] [CrossRef]
- Cococcioni, M.; de Gironcoli, S. Linear response approach to the calculation of the effective interaction parameters in the LDA + U method. Phys. Rev. B 2005, 71, 035105. [Google Scholar] [CrossRef]
- Yu, K.; Carter, E.A. Communication: Comparing ab initio methods of obtaining effective U parameters for closed-shell materials. J. Chem. Phys. 2014, 140, 121105. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Roldan, A.; de Leeuw, N.H. CuO surfaces and CO2 activation: A dispersion-corrected DFT+U study. J. Phys. Chem. C 2016, 120, 2198–2214. [Google Scholar] [CrossRef]
- Meyer, B.K.; Polity, A.; Reppin, D.; Becker, M.; Hering, P.; Klar, P.J.; Sander, T.; Reindl, C.; Benz, J.; Eickhoff, M.; et al. Binary copper oxide semiconductors: From materials towards devices. Phys. Status Solidi B 2012, 249, 1487–1509. [Google Scholar] [CrossRef]
- Caballero-Briones, F.; Artes, J.M.; Diez-Perez, I.; Gorostiza, P.; Sanz, F. Direct Observation of the Valence Band Edge by in Situ ECSTM-ECTS in p-Type Cu2O Layers Prepared by Copper Anodization. J. Phys. Chem. C 2009, 113, 1028–1036. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbate | ||||
|---|---|---|---|---|
| ImiH∗ | +0.13 | |||
| TriH∗ | +0.05 | +0.05 | +0.03 | +0.08 |
| TetH∗ | +0.02 | |||
| Imi∗ | −0.41 | |||
| Tri∗ | −0.58 | −0.56 | −0.65 | −0.47 |
| Tet∗ | −0.64 |
| Surface-Site | ||||
|---|---|---|---|---|
| (Ion, Symmetry) | (eV) | (e) | (Å) | (Å) |
| O, top | −1.88 | +0.62 | 0.97 | |
| O, top | −2.89 | +0.63 | 0.98 | |
| O, top | −3.02 | +0.62 | 0.98 | |
| O, tilted-top | −3.16 | +0.64 | 0.98 | |
| Cu, tilted-top | −1.03 | −0.06 | 1.51 | |
| Cu, top | −1.22 | −0.02 | 1.49 | |
| Cu, top | −1.97 | −0.22 | 1.49 | |
| Cu, hollow | −3.27 | −0.34 | 1.67 | |
| Cu, hollow | −3.34 | −0.33 | 1.66, 1.68 |
| Surface-Site | (eV) | |||
|---|---|---|---|---|
| Cl∗ | Imi∗ | Tri∗ | Tet∗ | |
| (111)-type surfaces | ||||
| Cu | −1.99 | −2.30 | −2.18 | |
| Cu | −3.00 | −3.17 | −3.11 | |
| Cu | −3.78 | −3.70 | −3.61 | |
| (110)-type surfaces | ||||
| Cu | −1.77 | −2.06 | −2.01 | |
| Cu | −3.79 | −3.79 | −3.61 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gustinčič, D.; Kokalj, A. DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: I. Molecule–Surface and Cl–Surface Bonding. Metals 2018, 8, 310. https://doi.org/10.3390/met8050310
Gustinčič D, Kokalj A. DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: I. Molecule–Surface and Cl–Surface Bonding. Metals. 2018; 8(5):310. https://doi.org/10.3390/met8050310
Chicago/Turabian StyleGustinčič, Dunja, and Anton Kokalj. 2018. "DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: I. Molecule–Surface and Cl–Surface Bonding" Metals 8, no. 5: 310. https://doi.org/10.3390/met8050310
APA StyleGustinčič, D., & Kokalj, A. (2018). DFT Study of Azole Corrosion Inhibitors on Cu2O Model of Oxidized Copper Surfaces: I. Molecule–Surface and Cl–Surface Bonding. Metals, 8(5), 310. https://doi.org/10.3390/met8050310