
The Effects of Al and Ti Additions on the Structural Stability, Mechanical and Electronic Properties of D8m-Structured Ta5Si3

Abstract
:
1. Introduction
2. Calculation Details
3. Results and Discussion
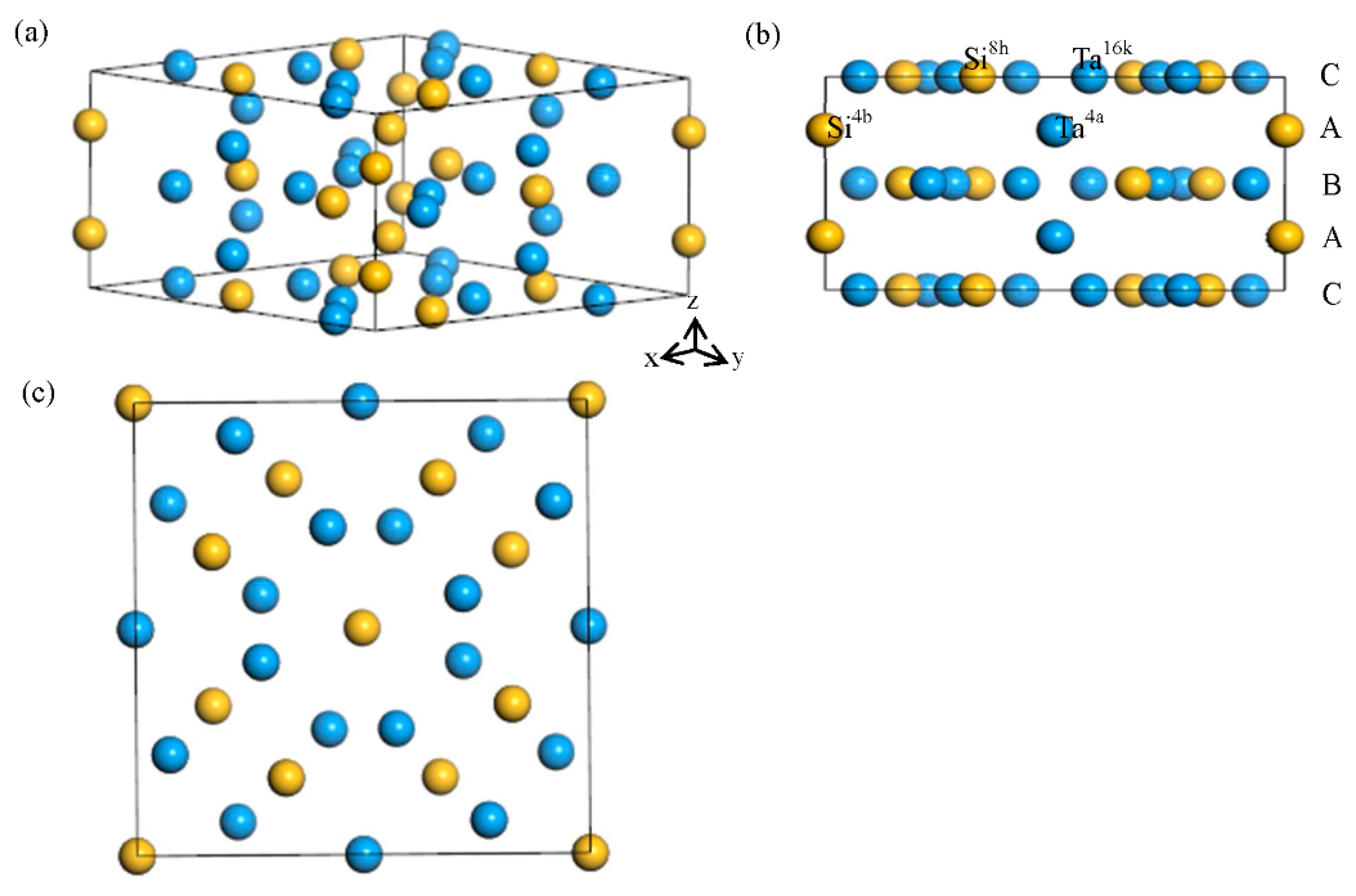
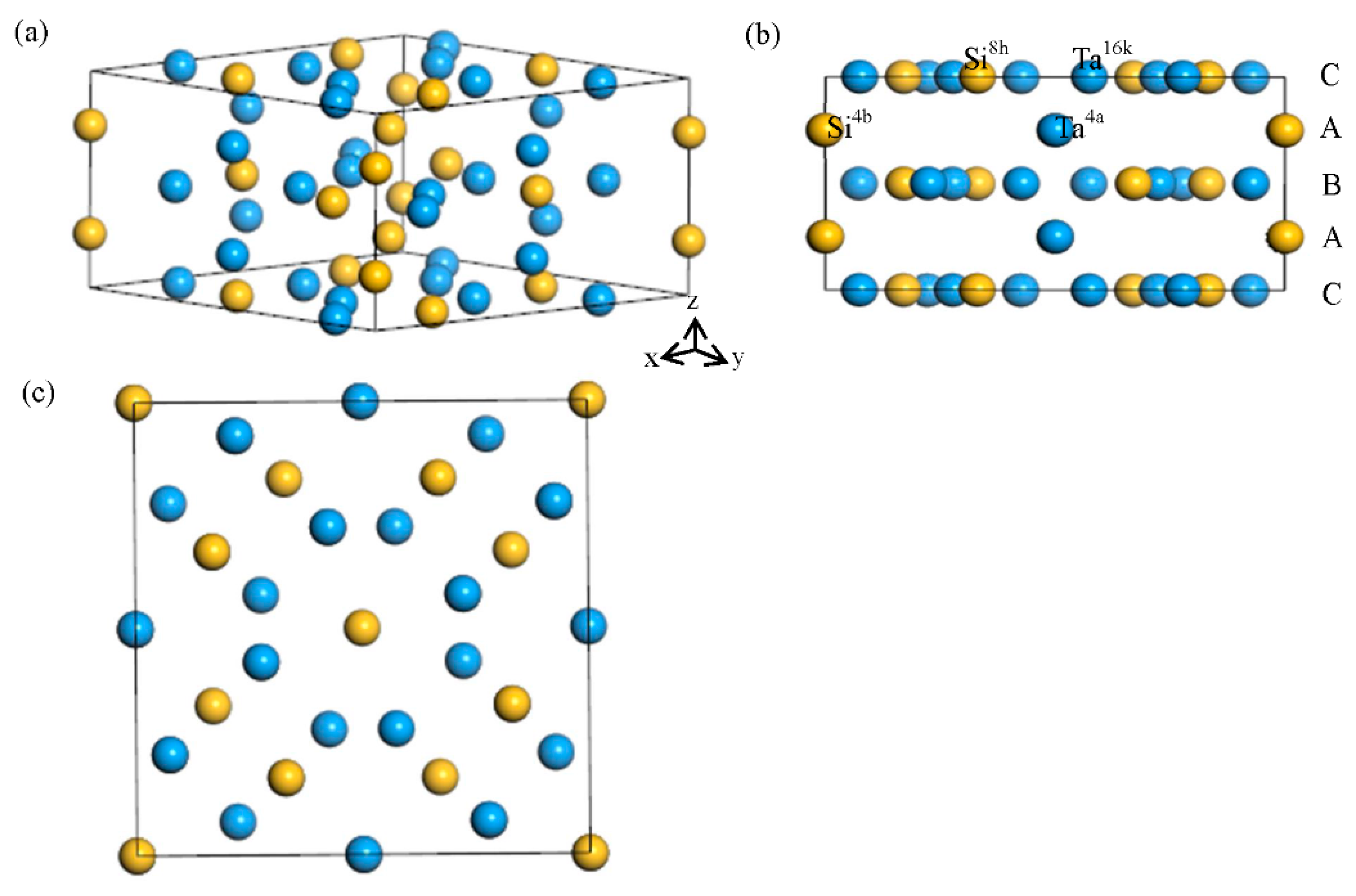
3.1. Lattice Parameter
3.2. Site Occupancy of Ternary Elements
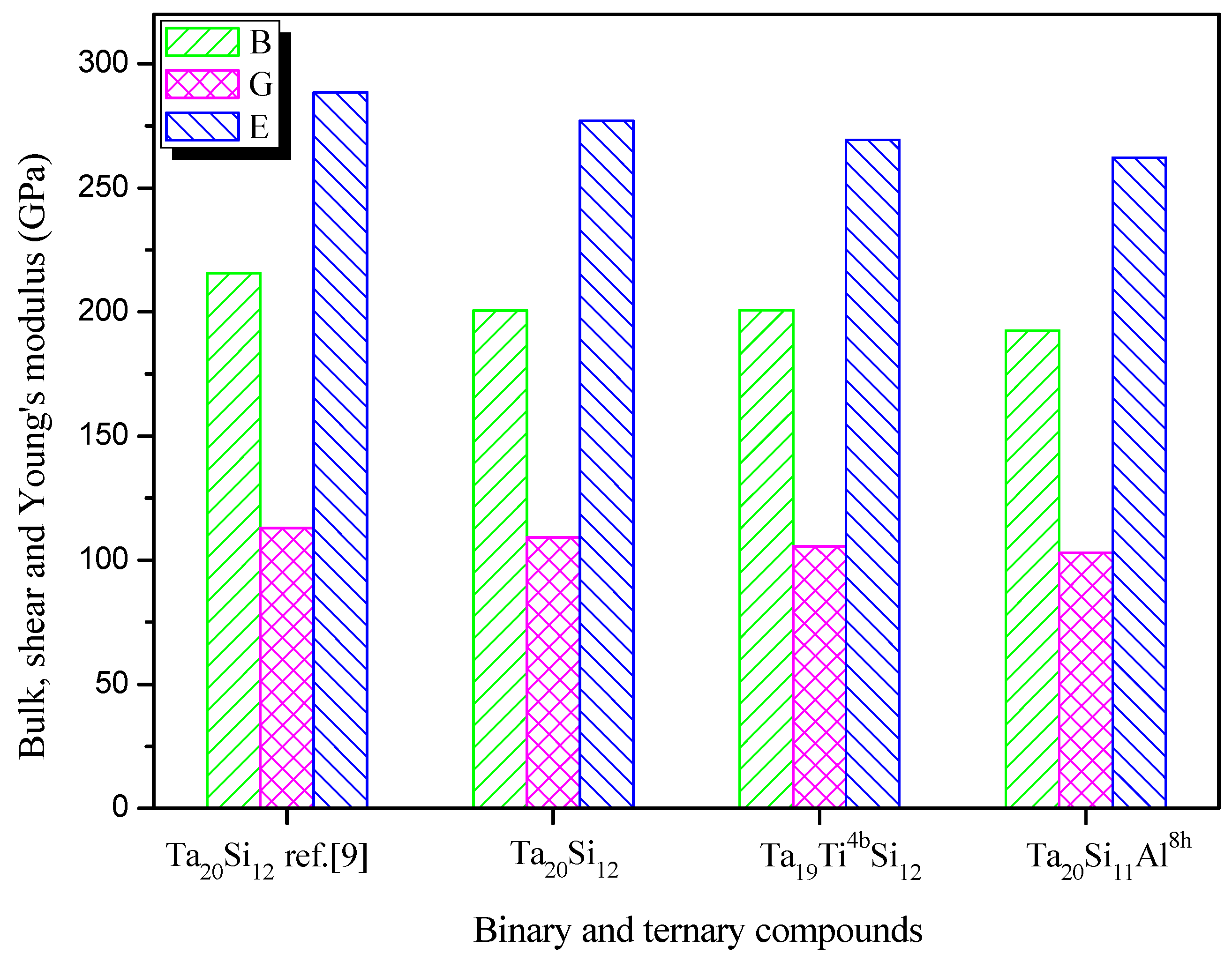
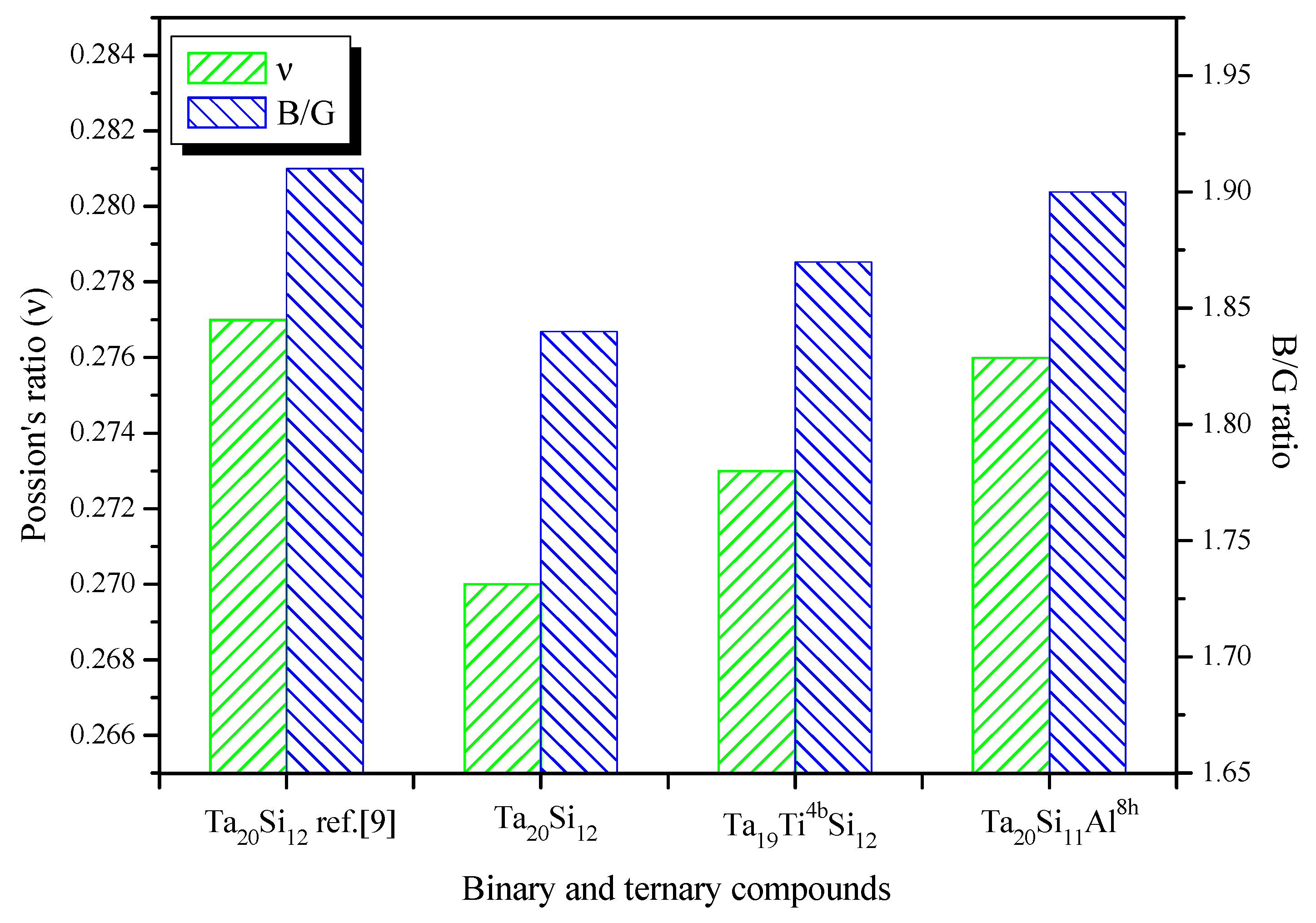
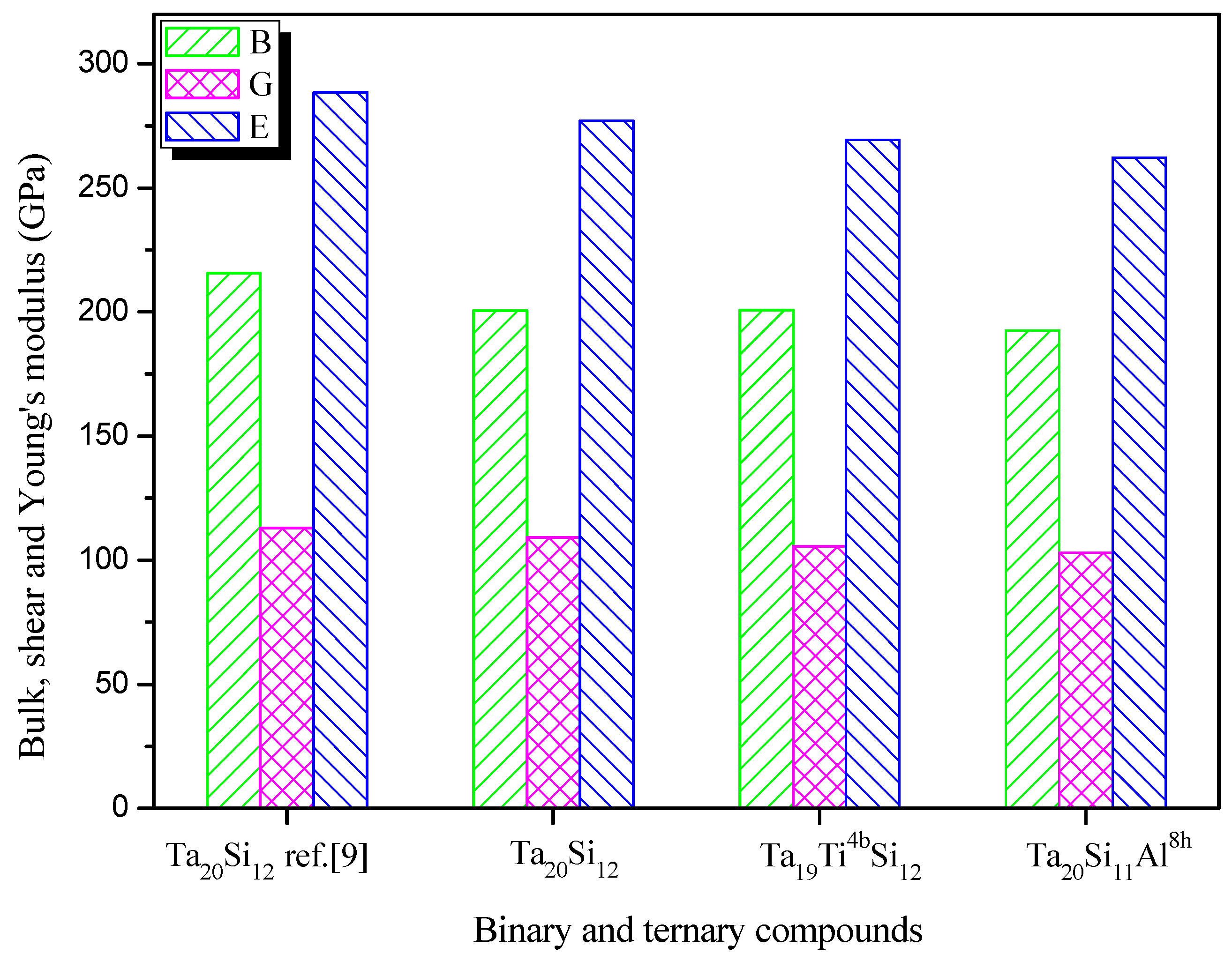
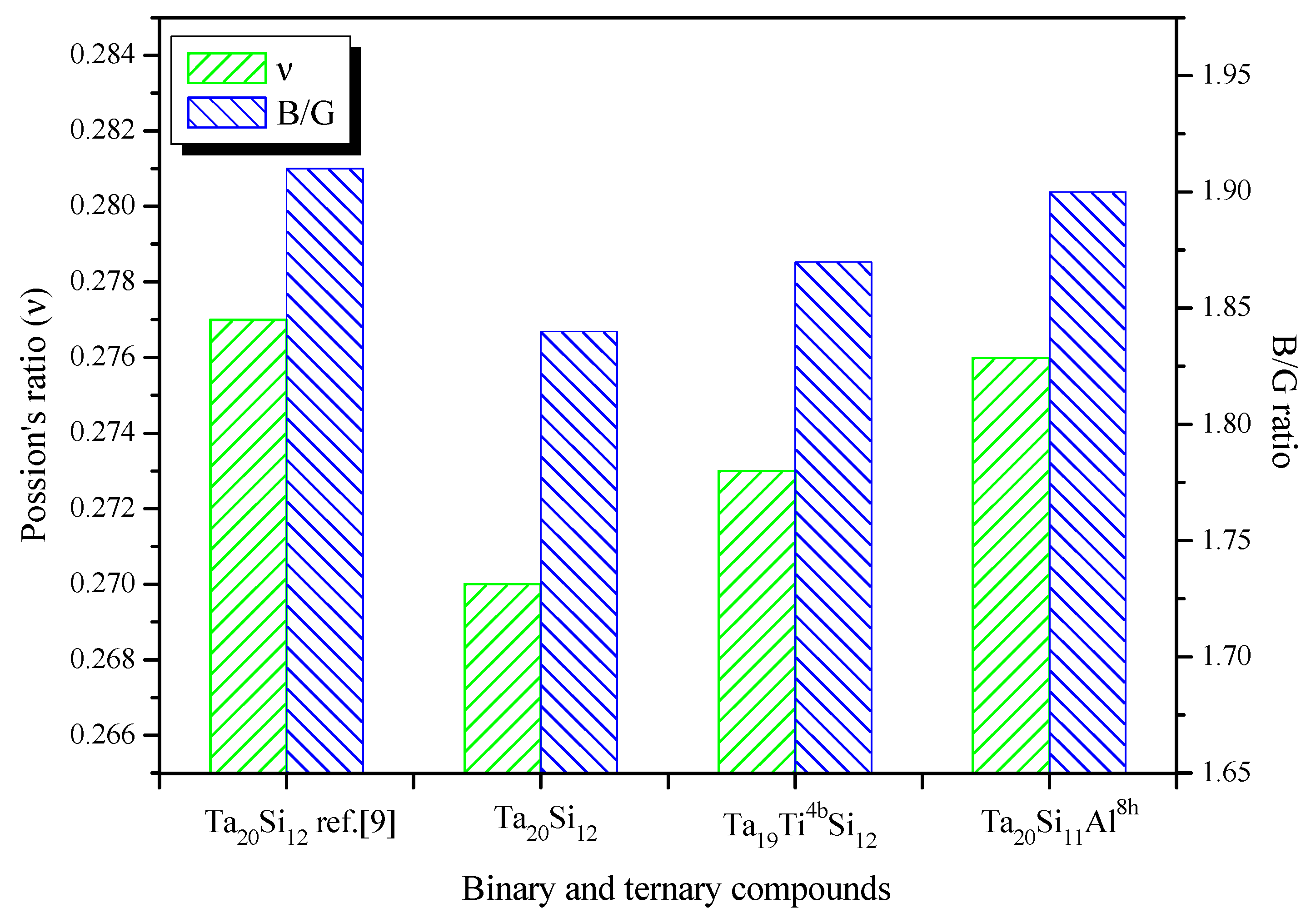
3.3. Elastic and Mechanical Properties
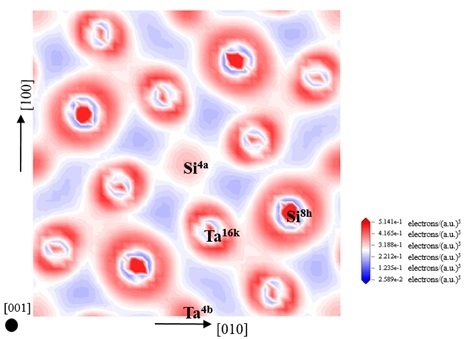
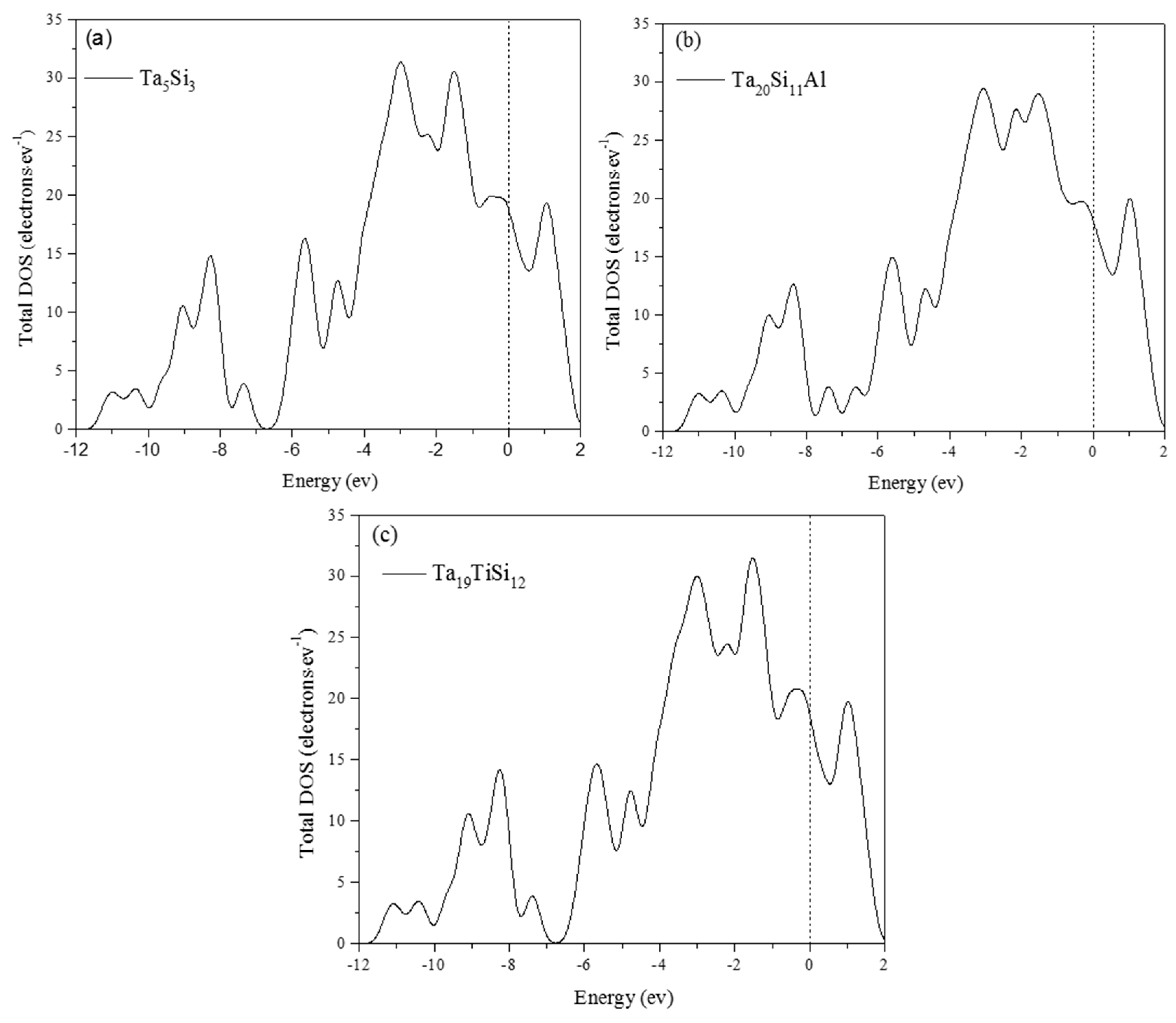
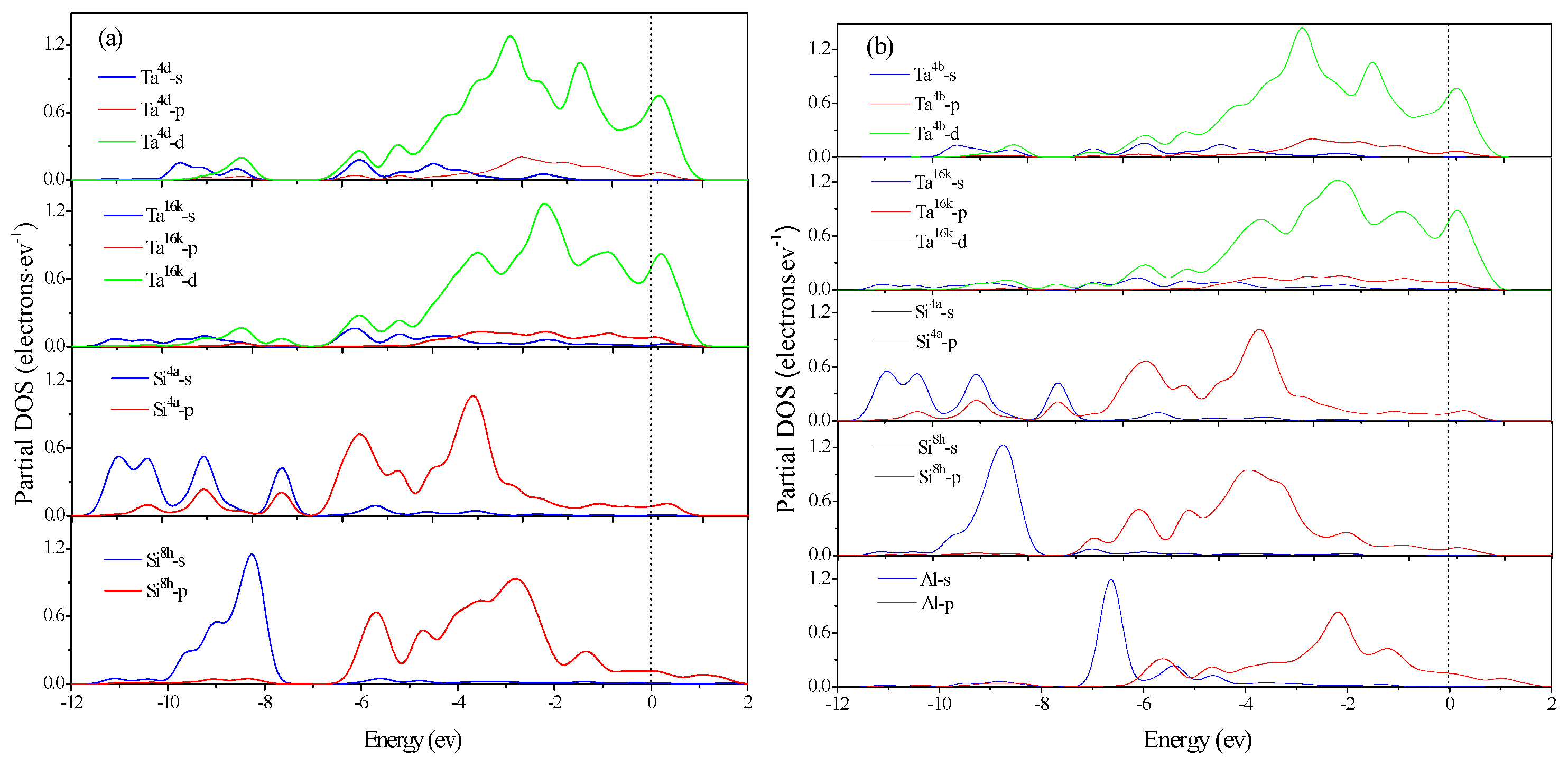
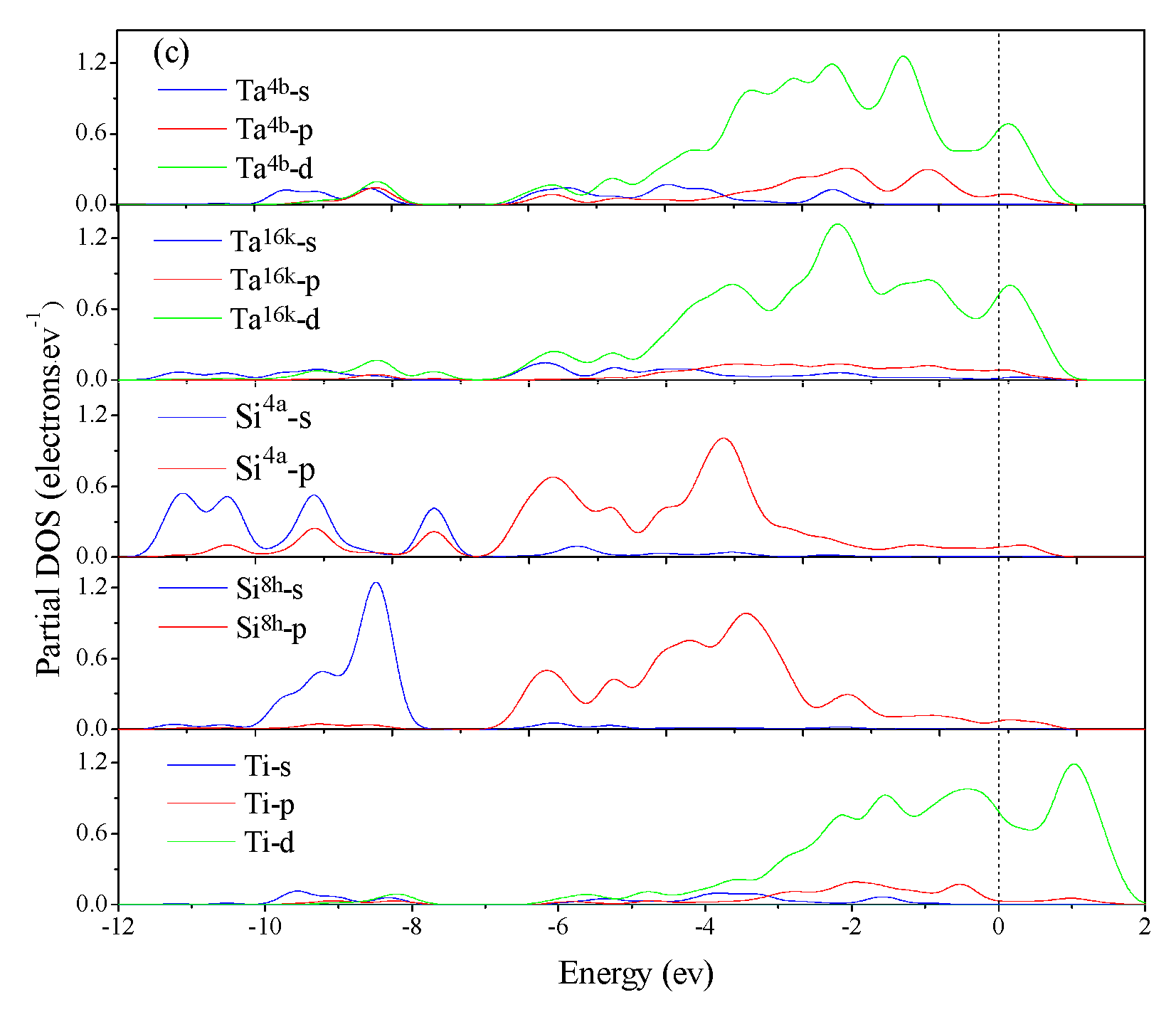
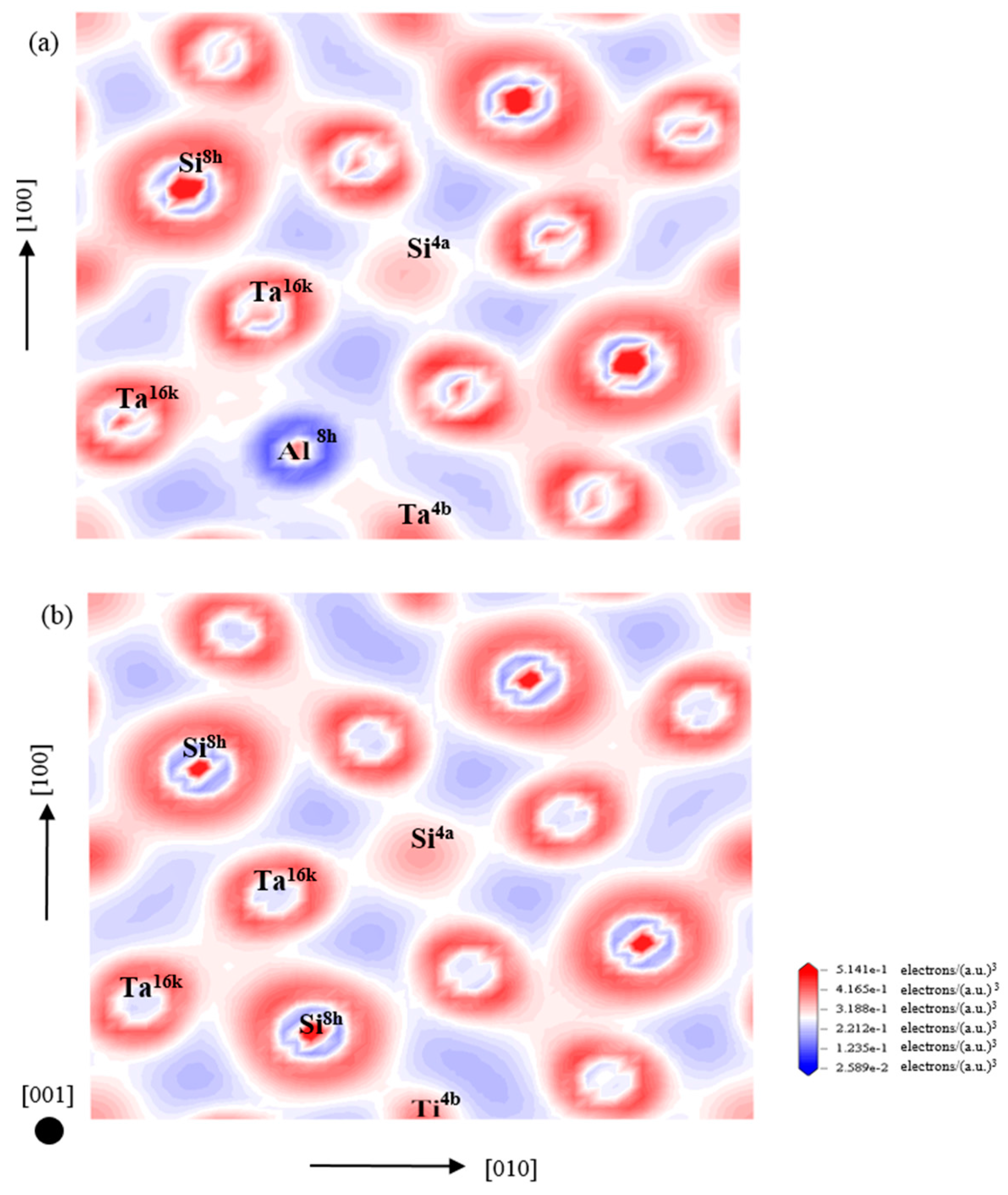
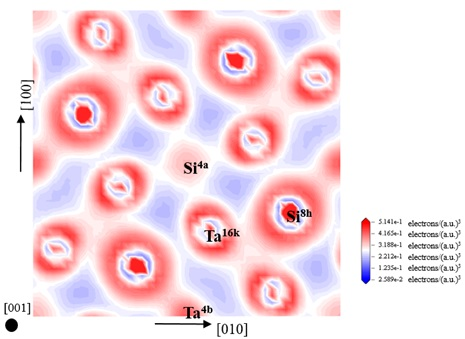
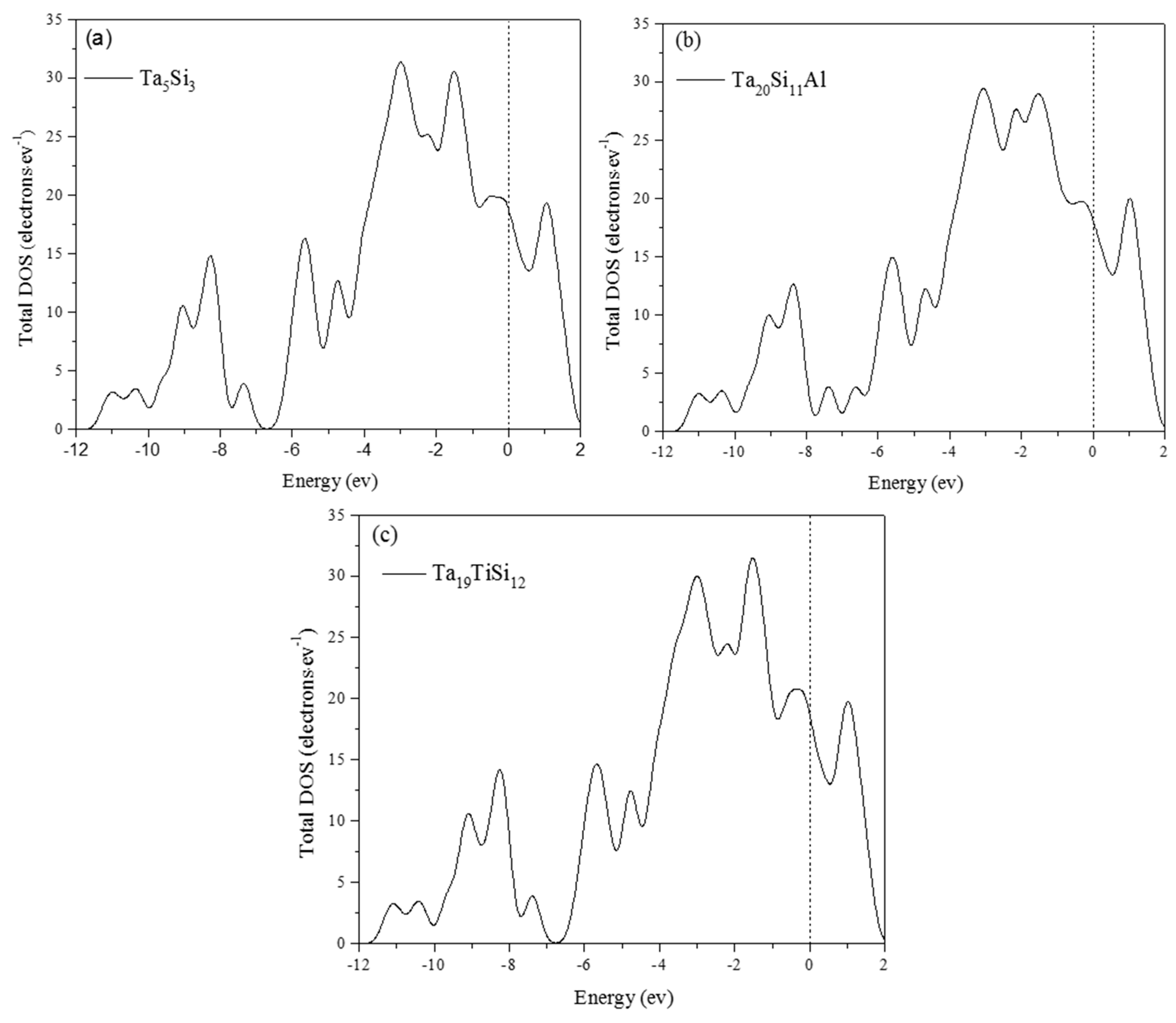
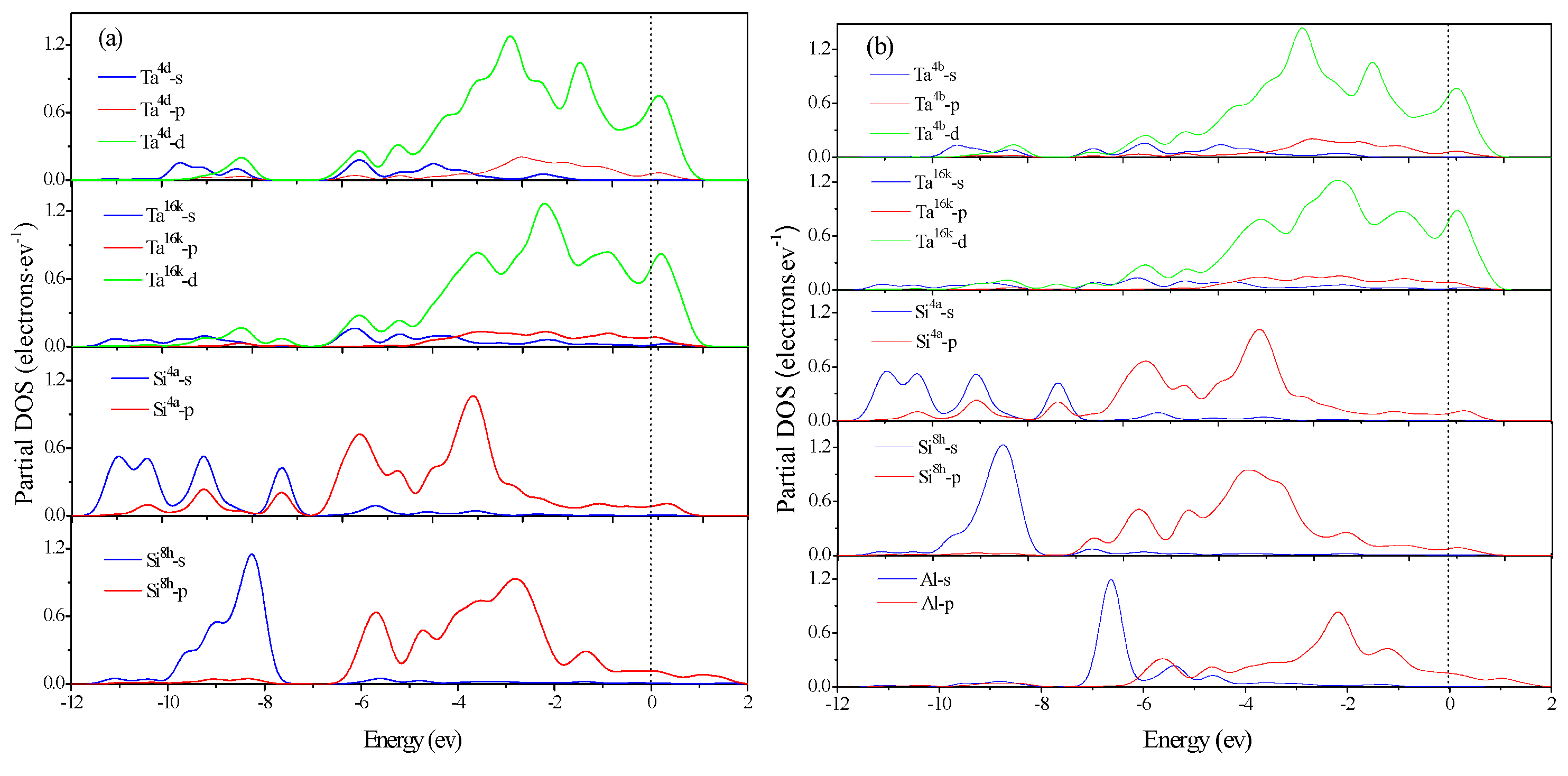
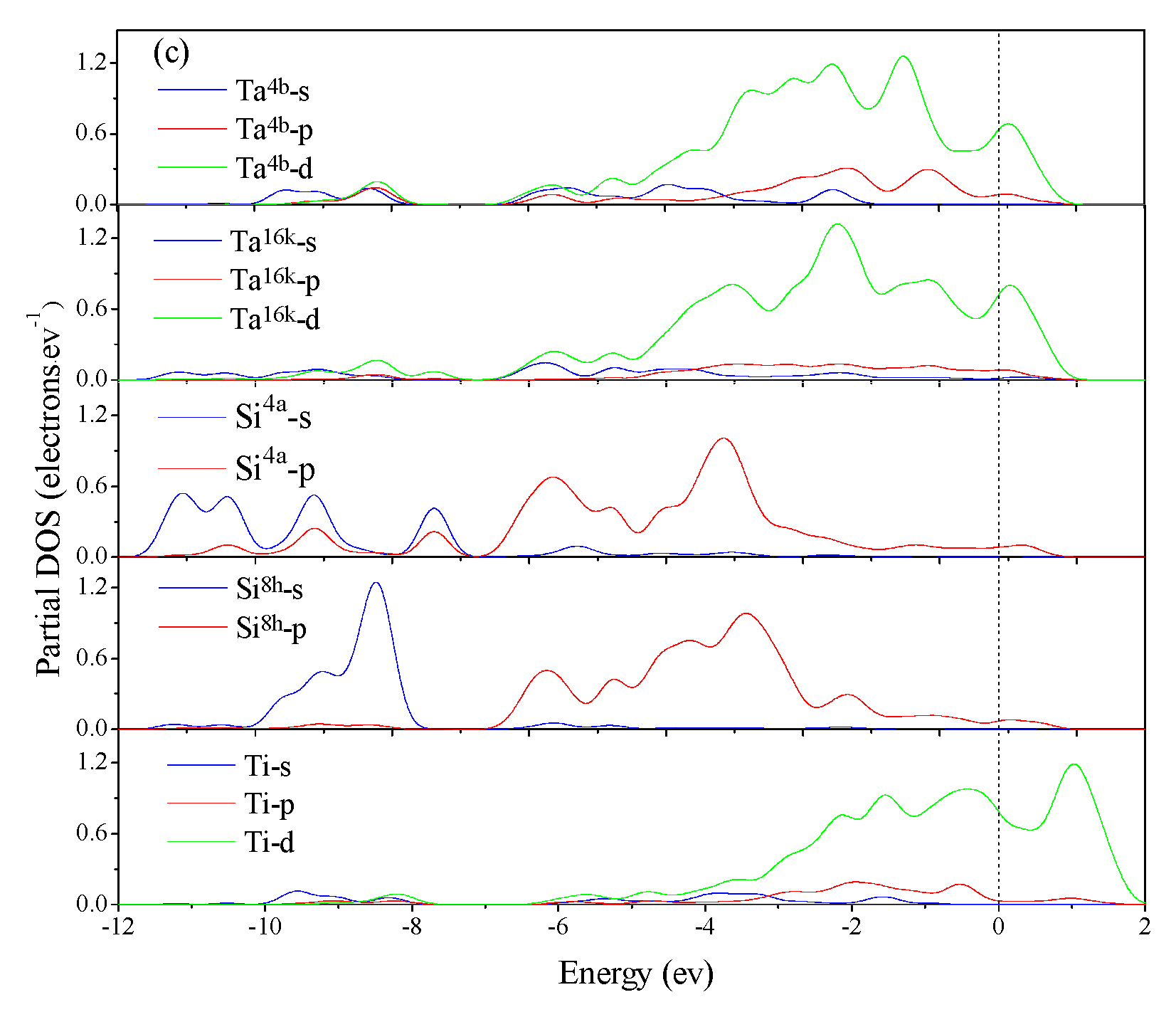
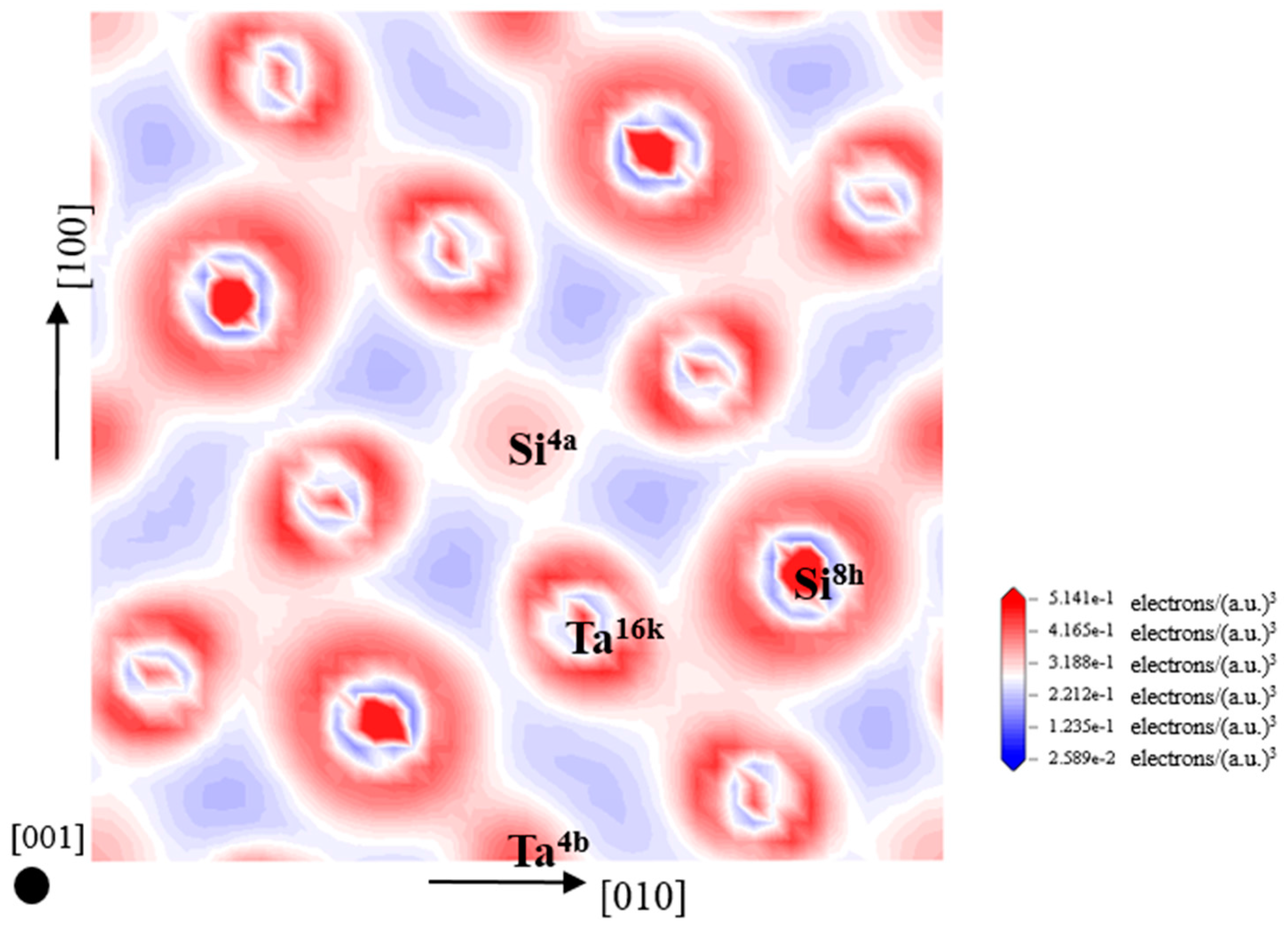
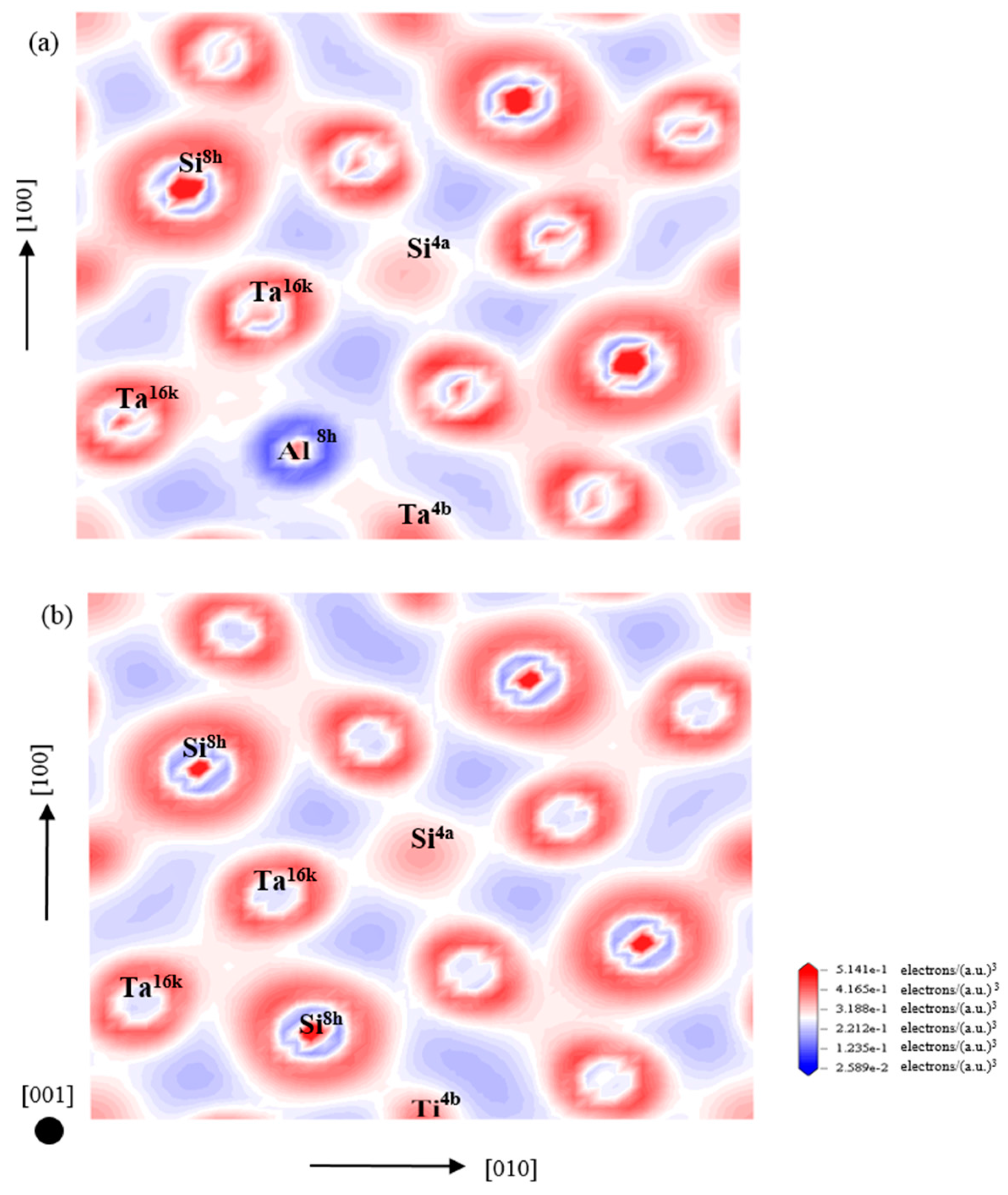
3.4. Electronic Structure and Population Analysis
3.5. Debye Temperature
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nakano, T.; Azuma, M.; Umakoshi, Y. Tensile deformation and fracture behaviour in NbSi2 and MoSi2 single crystals. Acta Mater. 2002, 14, 3731–3742. [Google Scholar] [CrossRef]
- Xu, J.; Leng, Y.; Li, H.; Zhang, H. Preparation and characterization of SiC/(Mo, W)Si2 composites from powders resulting from a SHS in a chemical oven. Int. J. Refract. Met. Hard Mater. 2009, 27, 74–77. [Google Scholar] [CrossRef]
- Datta, M.K.; Pabi, S.K.; Murty, B.S. Thermal stability of nanocrystalline Ni silicides synthesized by mechanical alloying. Mater. Sci. Eng. A 2000, 1, 219–225. [Google Scholar] [CrossRef]
- Mason, K.N. Growth and characterization of transition metal silicides. Prog. Cryst. Growth Charact. Mater. 1981, 2, 269–307. [Google Scholar] [CrossRef]
- Fujiwara, H.; Ueda, Y. Thermodynamic properties of molybdenum silicides by molten electrolyte EMF measurements. J. Alloy. Compd. 2007, 441, 168–173. [Google Scholar] [CrossRef]
- Schlesinger, M.E. The Si-Ta (silicon-tantalum) system. J. Phase Equilib. 1994, 15, 90–95. [Google Scholar] [CrossRef]
- Matsuno, H.; Yokoyama, A.; Watari, F.; Uo, M.; Kawasaki, T. Biocompatibility and osteogenesis of refractory metal implants, titanium, hafnium, niobium, tantalum and rhenium. Biomaterials 2001, 22, 1253–1262. [Google Scholar] [CrossRef]
- Tillard, M. The mixed intermetallic silicide Nb5−xTaxSi3 (0 ≤ x ≤ 5). Crystal and electronic structure. J. Alloy. Compd. 2014, 584, 385–392. [Google Scholar] [CrossRef]
- Tao, X.; Jund, P.; Viennois, R.; Catherine, C.; Tedenacet, J.C. Physical properties of thallium–tellurium based thermoelectric compounds using first-principles simulations. J. Phys. Chem. A 2011, 31, 8761–8766. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R. Microstructure and mechanical behavior of reaction hot-pressed titanium silicide and titanium silicide-based alloys and composites. Metall. Mater. Trans. A 1998, 6, 1629–1641. [Google Scholar] [CrossRef]
- Rosenkranz, R.; Frommeyer, G.; Smarsly, W. Microstructures and properties of high melting point intermetallic Ti5Si3 and TiSi2 compounds. Mater. Sci. Eng. A 1992, 1, 288–294. [Google Scholar] [CrossRef]
- Korznikov, A.V.; Pakieła, Z.; Kurzydłowski, K.J. Influence of long-range ordering on mechanical properties of nanocrystalline Ni3Al. Scr. Mater. 2001, 45, 309–315. [Google Scholar] [CrossRef]
- Dasgupta, T.; Umarji, A.M. Thermal properties of MoSi2 with minor aluminum substitutions. Intermetallics 2007, 15, 128–132. [Google Scholar] [CrossRef]
- Gutmanas, E.Y.; Gotman, I. Reactive synthesis of ceramic matrix composites under pressure. Ceram. Int. 2000, 7, 699–707. [Google Scholar] [CrossRef]
- Williams, J.J.; Ye, Y.Y.; Kramer, M.J.; Ho, K.M.; Hong, L.; Fu, C.L.; Malik, S.K. Theoretical calculations and experimental measurements of the structure of Ti5Si3 with interstitial additions. Intermetallics 2000, 8, 937–943. [Google Scholar] [CrossRef]
- Du, W.; Zhang, L.; Ye, F.; Ni, X.; Lin, J. Intrinsic embrittlement of MoSi2 and alloying effect on ductility: Studied by first-principles. Phys. B Condens. Matter 2010, 7, 1695–1700. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 11. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 11. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. Special points for Brillouin-zone integrations—A reply. Phys. Rev. B 1977, 4. [Google Scholar] [CrossRef]
- Chu, F.; Thoma, D.J.; McClellan, K.J.; Peralta, P. Mo5Si3 single crystals: Physical properties and mechanical behavior. Mater. Sci. Eng. A 1999, 261, 44–52. [Google Scholar] [CrossRef]
- Kocherzhinskij, Y.A.; Kulik, O.G.; Shishkin, E.A. Phase diagram of the tantalum-silicon system. Dokl. Akad. Nauk SSSR 1981, 261, 106–108. [Google Scholar]
- Fu, C.L.; Wang, X.; Ye, Y.Y.; Ho, K.M. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principles calculation. Intermetallics 1999, 2, 179–184. [Google Scholar] [CrossRef]
- Voigt, W. A Determination of the elastic constants for beta-quartz lehrbuch de kristallphysik. Terubner Leipzig. 1928, 40, 2856–2860. [Google Scholar]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. ZAMM J. Appl. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Brauer, G.; Zapp, K.H. Die nitride des tantals. Anorg. Allg. Chem. 1954, 277, 129–139. [Google Scholar] [CrossRef]
- Wang, F.E.; Buehler, W.J.; Pickart, S.J. Crystal structure and a unique “Martensitic” transition of TiNi. J. Appl. Phys. 1965, 36, 3232–3239. [Google Scholar] [CrossRef]
- Nowotny, H.; Laube, E. The thermal expansion of high-melting phases. Plansee. Pulver. 1961, 9, 85–92. [Google Scholar]
- Jette, E.R.; Foote, F. Precision determination of lattice constants. J. Chem. Phys. 1935, 3, 605–616. [Google Scholar] [CrossRef]
- Sakakibara, N.; Takahashi, Y.; Okumura, K.; Hattori, K.H.; Yaita, T.; Suzuki, K.; Shimizu, H. Speciation of osmium in an iron meteorite and a platinum ore specimen based on X-ray absorption fine-structure spectroscopy. Geochem. J. 2005, 39, 383–389. [Google Scholar] [CrossRef]
- Zhang, C.; Han, P.; Li, J.; Chi, M.; Yan, L.; Liu, Y.; Liu, X.; Xu, B. First-principles study of the mechanical properties of NiAl microalloyed by M (Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd). J. Phys. D Appl. Phys. 2008, 41. [Google Scholar] [CrossRef]
- Featherston, F.H.; Neighbours, J.R. Elastic constants of tantalum, tungsten, and molybdenum. Phys. Rev. 1963, 130. [Google Scholar] [CrossRef]
- McSkimin, H.J.; Andreatch, P., Jr. Elastic moduli of silicon vs. hydrostatic pressure at 25.0 °C and −195.8 °C. J. Appl. Phys. 1964, 35, 2161–2165. [Google Scholar] [CrossRef]
- Kang, L.U.O.; Bing, Z.; Shang, F.U.; Jiang, Y.; Yi, D.Q. Stress/strain aging mechanisms in Al alloys from first principles. Trans. Nonferr. Met. Soc. China 2014, 24, 2130–2137. [Google Scholar]
- Chu, F.; Lei, M.; Maloy, S.A.; Petrovic, J.J.; Mitchell, T.E. Elastic properties of C40 transition metal disilicides. Acta Mater. 1996, 44, 3035–3048. [Google Scholar] [CrossRef]
- Tao, X.; Chen, H.; Tong, X.; Ouyang, Y.; Jund, P.; Tedenac, J.C. Structural, electronic and elastic properties of V5Si3 phases from first-principles calculations. Comput. Mater. Sci. 2012, 53, 169–174. [Google Scholar] [CrossRef]
- Ström, E.; Eriksson, S.; Rundlöf, H.; Zhang, J. Effect of site occupation on thermal and mechanical properties of ternary alloyed Mo5Si3. Acta Mater. 2005, 53, 357–365. [Google Scholar] [CrossRef]
- Pan, Y.; Lin, Y.; Xue, Q.; Ren, C.; Wang, H. Relationship between Si concentration and mechanical properties of Nb–Si compounds: A first-principles study. Mater. Des. 2016, 89, 676–683. [Google Scholar]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Chumakov, A.I.; Monaco, G.; Fontana, A.; Bosak, A.; Hermann, R.P.; Bessas, D.; Wehinger, B.; Crichton, W.A.; Krisch, M.; Baldi, G.; et al. Role of disorder in the thermodynamics and atomic dynamics of glasses. Phys. Rev. Lett. 2014, 112. [Google Scholar] [CrossRef] [PubMed]
- Pettifor, D.G. Theoretical predictions of structure and related properties of intermetallics. Mater. Sci. Technol. 1992, 8, 345–349. [Google Scholar] [CrossRef]
- Mukhanov, V.A.; Kurakevych, O.O.; Solozhenko, V.L. Thermodynamic aspects of materials’ hardness: Prediction of novel superhard high-pressure phases. High Press. Res. 2008, 28, 531–537. [Google Scholar] [CrossRef]
- Gilman, J.J. Hardness—A strength microprobe. In The Science of Hardness Testing and Its Research Applications; Westbrook, J.H., Conrad, H., Eds.; American Society of Metal: Metal Park, OH, USA, 1973. [Google Scholar]
- Liu, A.Y.; Cohen, M.L. Structural properties and electronic structure of low-compressibility materials: β-Si3N4 and hypothetical β-C3N4. Phys. Rev. B 1990, 41, 10727–10734. [Google Scholar] [CrossRef]
- Teter, D.M.; Bull, M.R.S. Hardness and fracture toughness of brittle materials. Phys. Rev. B 1998, 23. [Google Scholar] [CrossRef]
- Chen, X.Q.; Niu, H.; Franchini, C.; Li, D.; Li, Y. Hardness of T-carbon: Density functional theory calculations. Phys. Rev. B 2011, 84. [Google Scholar] [CrossRef]
- Tao, X.; Jund, P.; Colinet, C.; Tédenac, J.C. First-principles study of the structural, electronic and elastic properties of W5Si3. Intermetallics 2010, 18, 688–693. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Cao, Y.; Zhu, P.; Zhu, J.; Liu, Y. First-principles study of NiAl alloyed with Co. Comput. Mater. Sci. 2016, 111, 34–40. [Google Scholar] [CrossRef]
- Wang, H.Z.; Zhan, Y.Z.; Pang, M.J. The structure, elastic, electronic properties and Debye temperature of M2AlC (M = V, Nb and Ta) under pressure from first principles. Comput. Mater. Sci. 2012, 54, 16–22. [Google Scholar] [CrossRef]
- Shi, S.; Zhu, L.; Jia, L.; Zhang, H.; Sun, Z. Ab-initio study of alloying effects on structure stability and mechanical properties of α-Nb5Si3. Comput. Mater. Sci. 2015, 108, 121–127. [Google Scholar] [CrossRef]
- Chen, Q.; Sundman, B. Calculation of Debye temperature for crystalline structures—A case study on Ti, Zr, and Hf. Acta Mater. 2001, 49, 947–961. [Google Scholar] [CrossRef]
- Du, L.; Wang, L.; Zheng, B.; Du, H. Numerical simulation of phase separation in Fe–Cr–Mo ternary alloys. J. Alloy. Compd. 2016, 663, 243–248. [Google Scholar] [CrossRef]
- Cuenya, B.R.; Frenkel, A.I.; Mostafa, S.; Behafarid, F.; Croy, J.R.; Ono, L.K.; Wang, Q. Anomalous lattice dynamics and thermal properties of supported size-and shape-selected Pt nanoparticles. Phys. Rev. B 2010, 82. [Google Scholar] [CrossRef]
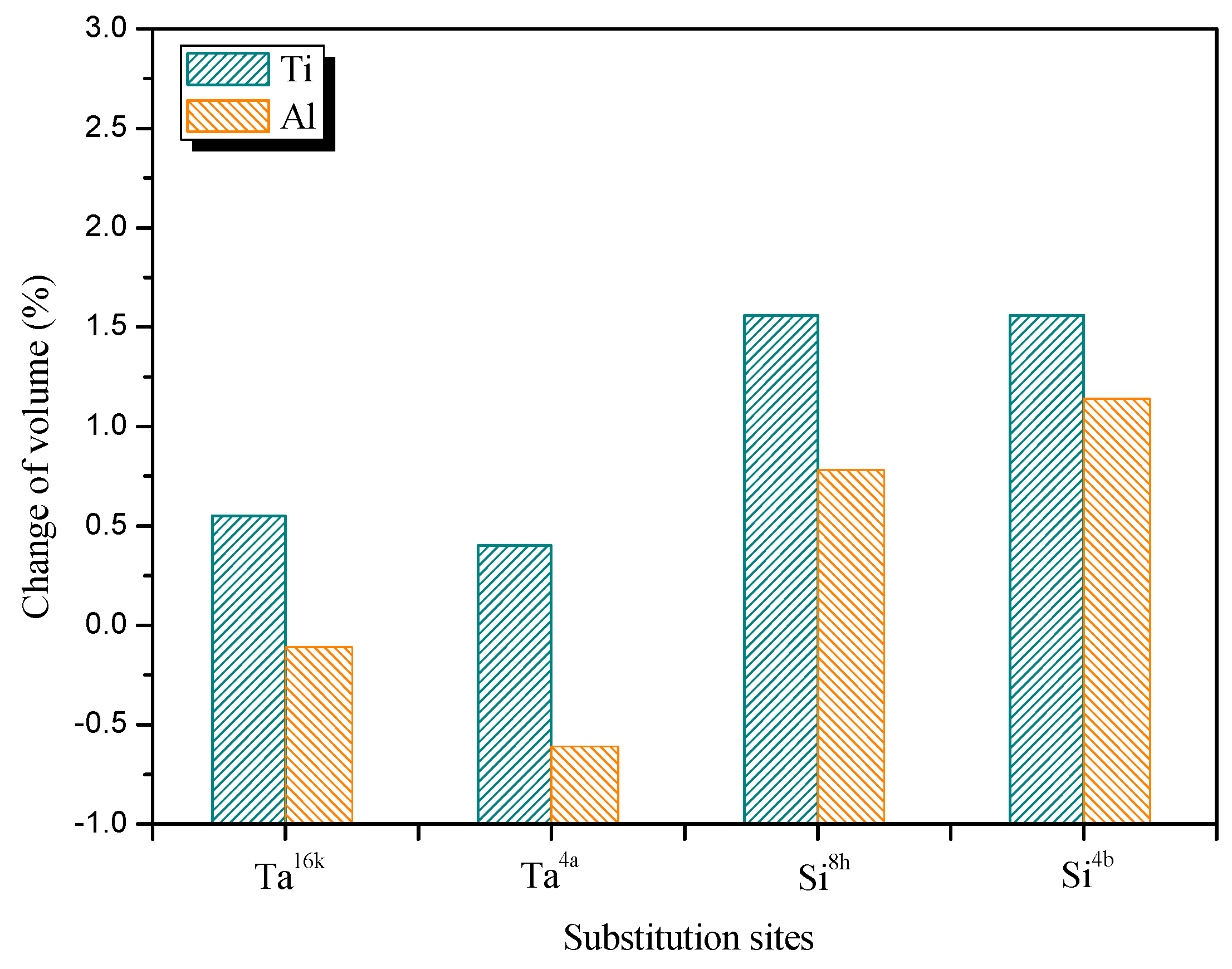
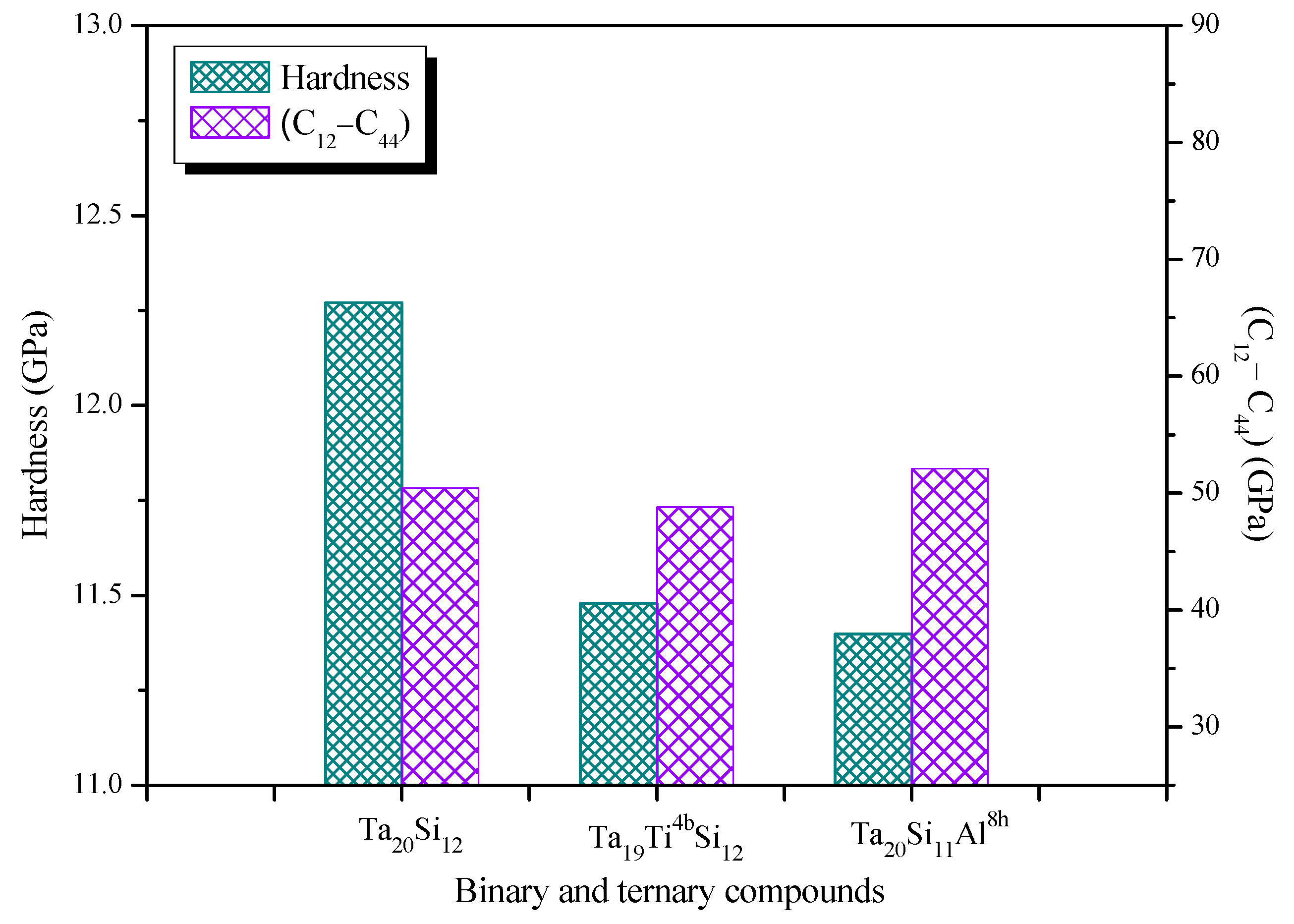


{kind=link}
{kind=link}
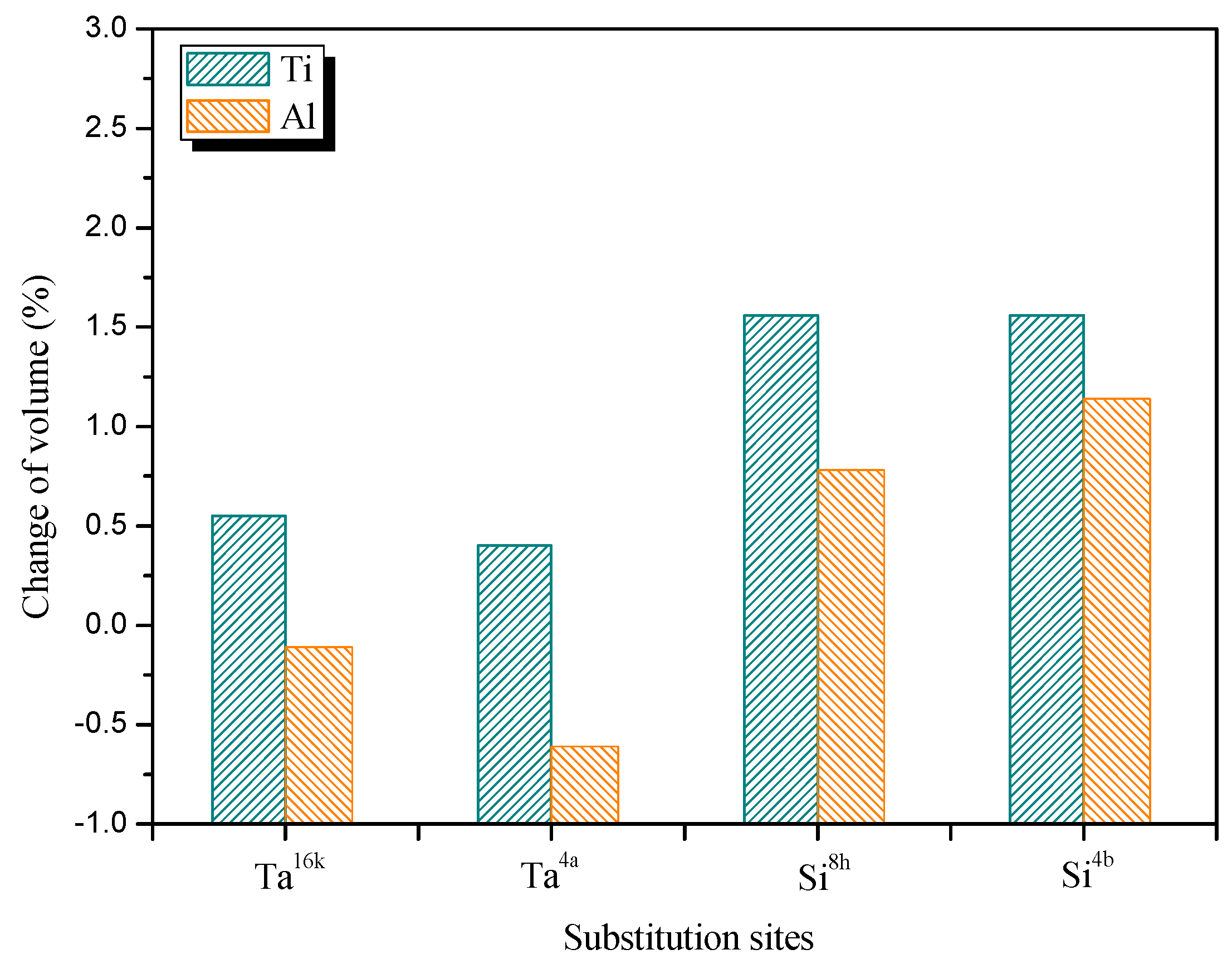{kind=link}
{kind=link}
{kind=link}
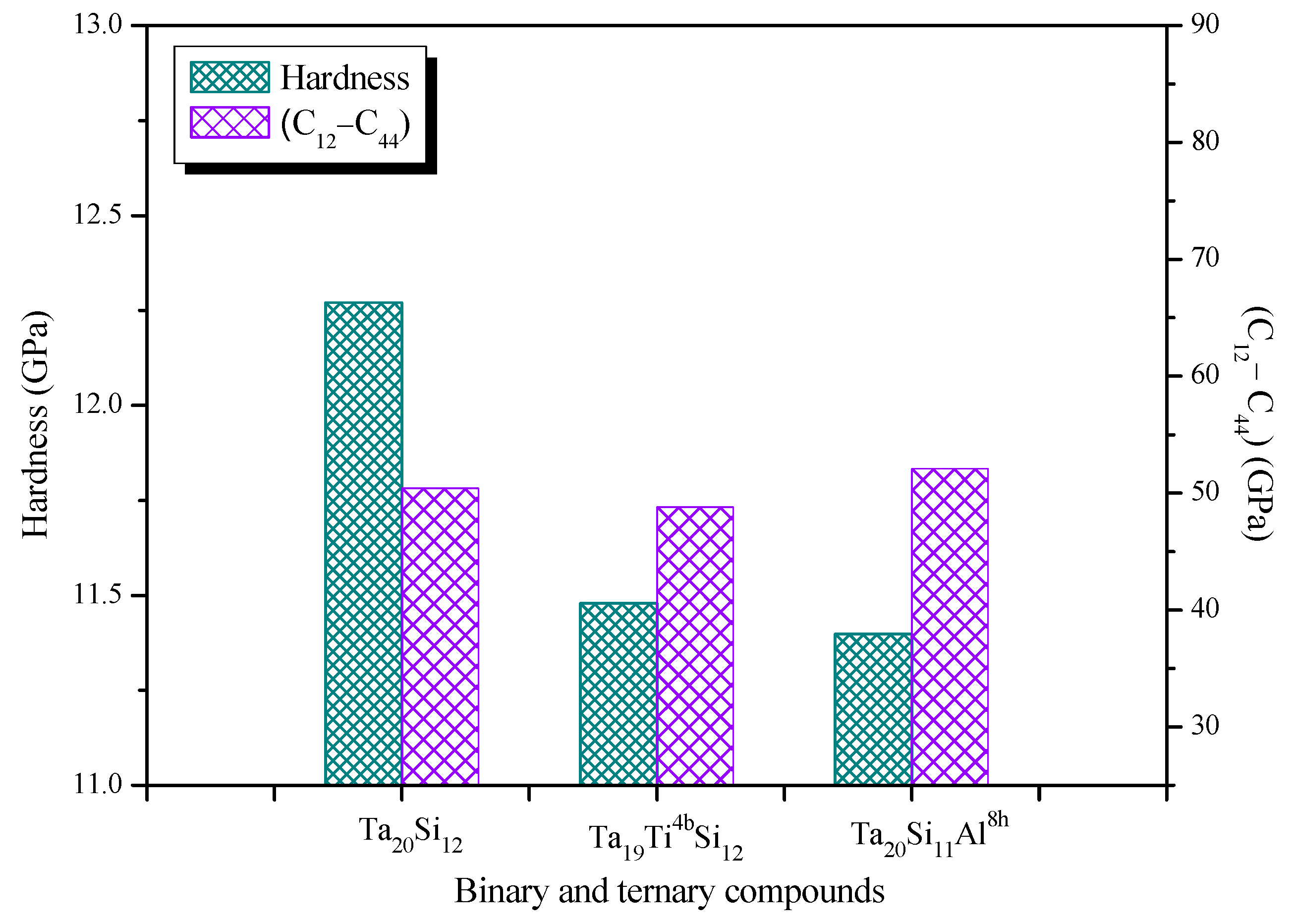{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Type | Substitution Sites | Lattice Parameters | Enthalpy of Formation (eV/Atom) | ||
|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | ||||
| Ta | W(A2) | - | 3.309 3.306 a | - | - | - |
| Ti | - | - | 2.981 2.954 b | - | - | - |
| D8m Ta20Si12 | W5Si3 | - | 10.08 10.01 c 9.862 d | - | 5.183 5.106 c 5.05 d | −0.619 −0.544 c |
| Ta19TiSi12 | - | Ta16k | 10.09 | 10.09 | 5.199 | −0.634 |
| - | W5Si3 | Ta4b | 10.084 | - | 5.197 | −0.639 |
| Ta20Si11Ti | - | Si4a | 10.12 | 10.12 | 5.221 | −0.554 |
| - | - | Si8h | 10.15 | 10.11 | 5.210 | −0.557 |
| Ta19AlSi12 | - | Ta16k | 10.10 | 10.06 | 5.175 | −0.591 |
| - | - | Ta4b | 10.09 | 10.10 | 5.137 | −0.602 |
| Ta20Si11Al | - | Si4a | 10.08 | 10.15 | 5.199 | −0.605 |
| - | W5Si3 | Si8h | 10.12 | - | 5.180 | −0.605 |
| Si | Si(A4) | - | 5.462 5.433 e | - | - | - |
| Al | - | - | 4.071 4.052 f | - | - | - |
| Structural | Ta (BCC) | Si (Diamond) | Al (FCC) | Ti (Hexagonal) | Ta20Si12 | Ta19Ti4bSi12 | Ta20Si11Al8h |
|---|---|---|---|---|---|---|---|
| C11 (GPa) | 260 266 a | 160 168 b | 102 97 c | 196 | 386.80 410.19 d | 365.51 | 378.39 |
| C12 (GPa) | 149 158 a | 70 65 b | 70 62 c | 57 | 137.12 145.66 d | 133.36 | 137.31 |
| C13 (GPa) | - | - | - | 69.59 | 106.55 125.45 d | 112.33 | 116.75 |
| C33 (GPa) | - | - | - | 200.12 | 339.40 338.89 d | 298.14 | 317.29 |
| C44 (GPa) | 85 87 a | 75 80 b | 22 21 c | 45.98 | 86.68 92.94 d | 84.55 | 85.22 |
| C66 (GPa) | - | - | - | 69.86 | 128.03 132.91 d | 130.85 | 131.50 |
| Bond | Ta20Si12 | Ta20Si11Al8h | Ta19Ti4bSi12 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| P | L | N | P | L | N | P | L | N | |
| Ta4b-Ta4b | −0.08 | 2.592 | 2 | 0.01 | 2.597 | 2 | 0.01 | 2.599 | 1 |
| Ta16k-Ta16k | 0.46 | 2.918 | 8 | 0.45 | 2.903 | 8 | 0.34 | 2.965 | 12 |
| Si4a-Si4a | 0.41 | 2.592 | 2 | 0.40 | 2.598 | 2 | 0.40 | 2.599 | 2 |
| Si4a-Ta16k | 0.19 | 2.677 | 32 | 0.19 | 2.677 | 32 | 0.19 | 2.682 | 32 |
| Ta4b-Si8h | 0.31 | 2.677 | 16 | 0.31 | 2.69 | 14 | 0.35 | 2.696 | 12 |
| Si8h-Ta16k | 0.35 | 2.833 | 48 | 0.33 | 2.832 | 43 | 0.40 | 2.78 | 43 |
| Al8h-Ta4b | - - - - | 0.30 | 2.735 | 2 | - | ||||
| Al8h-Ta16k | 0.41 | 2.930 | 6 | - | |||||
| Ti4b-Si8h | - | 0.13 | 2.678 | 4 | |||||
| Ti4b-Ta4b | - | −0.24 | 2.599 | 1 | |||||
| Phase | Ta | Si | Al | Ti | Ta20Si12 | Ta20Si11Al8h | Ta19Ti4bSi12 |
|---|---|---|---|---|---|---|---|
| ρ | 16.58 | 2.286 | 2.66 | 4.56 | 12.5 | 12.5 | 12.1 |
| νl | 5827 | 8906 | 6474 | 6398 | 5262 | 5227 | 5221 |
| νt | 2940 | 5166 | 2980 | 3561 | 2954 | 2906 | 2918 |
| νm | 3296 | 5733 | 3357 | 3965 | 3287 | 3236 | 3248 |
| Θ | 374 253 a | 626 635 a | 390 428 b | 450 374 b | 385 388 c | 380 | 381 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Cheng, J.; Xu, J.; Munroe, P.; Xie, Z.-H. The Effects of Al and Ti Additions on the Structural Stability, Mechanical and Electronic Properties of D8m-Structured Ta5Si3. Metals 2016, 6, 127. https://doi.org/10.3390/met6060127
Liu L, Cheng J, Xu J, Munroe P, Xie Z-H. The Effects of Al and Ti Additions on the Structural Stability, Mechanical and Electronic Properties of D8m-Structured Ta5Si3. Metals. 2016; 6(6):127. https://doi.org/10.3390/met6060127
Chicago/Turabian StyleLiu, Linlin, Jian Cheng, Jiang Xu, Paul Munroe, and Zong-Han Xie. 2016. "The Effects of Al and Ti Additions on the Structural Stability, Mechanical and Electronic Properties of D8m-Structured Ta5Si3" Metals 6, no. 6: 127. https://doi.org/10.3390/met6060127