Differential Transcriptome Profile of Peripheral White Cells to Identify Biomarkers Involved in Oxaliplatin Induced Neuropathy



Abstract
:
1. Introduction
2. Results
2.1. Patients and Clinical Evaluation of Neurotoxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Anemia | Thrombopenia | Neutropenia | Neurotoxicity | Mucositis |
|---|---|---|---|---|---|
| ASC | Grade I | No | No | Grade III | No |
| LGM | Grade II | No | No | Grade II | No |
| SDB | No | Grade II | No | Grade III | No |
| ICHB | Grade II | Grade I | No | Grade I | Grade I |
| ASA | No | Grade I | No | Grade I | No |
| AGS | No | Grade I | No | Grade I | No |
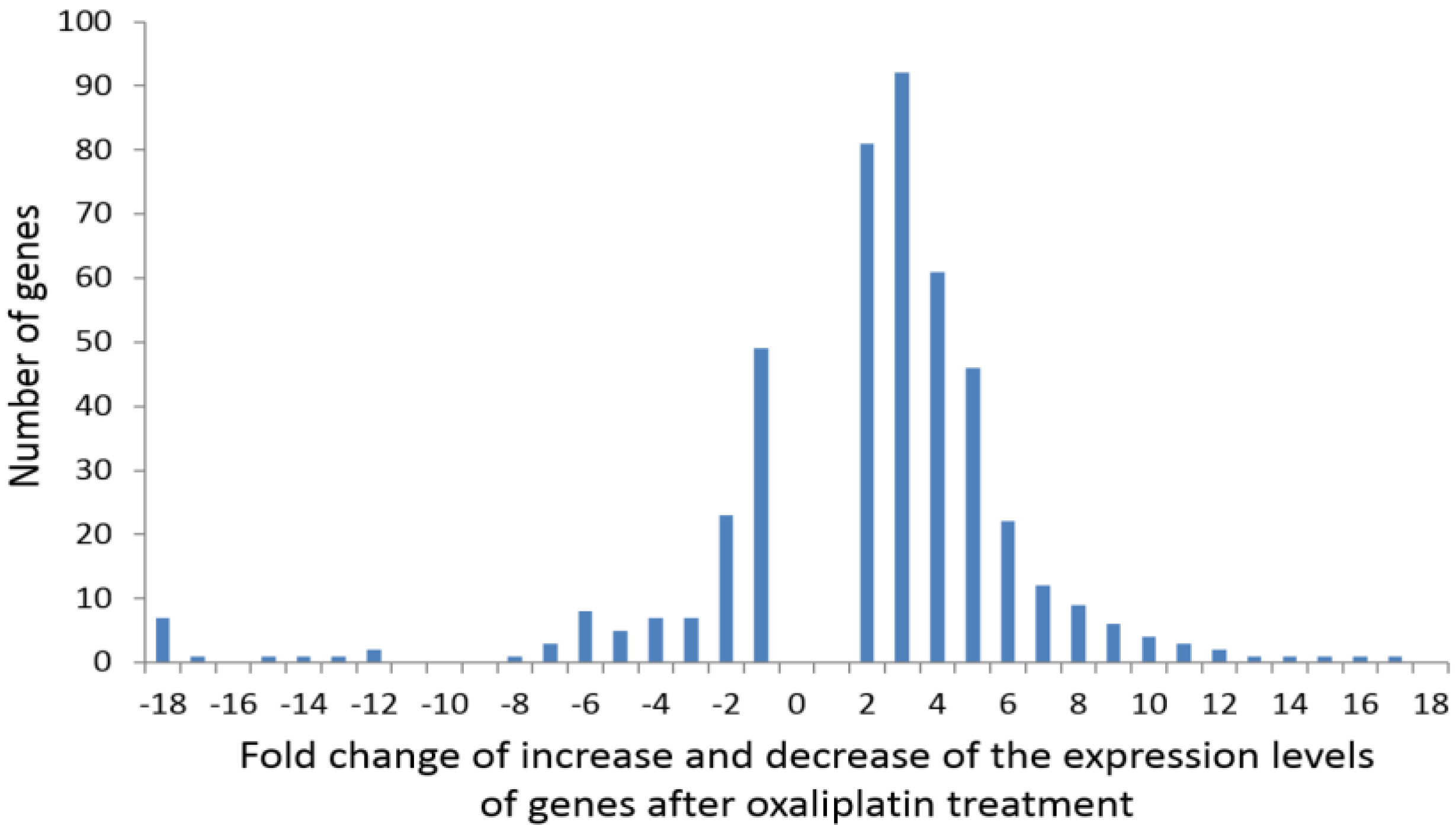
2.2. Analysis of Transcriptomics
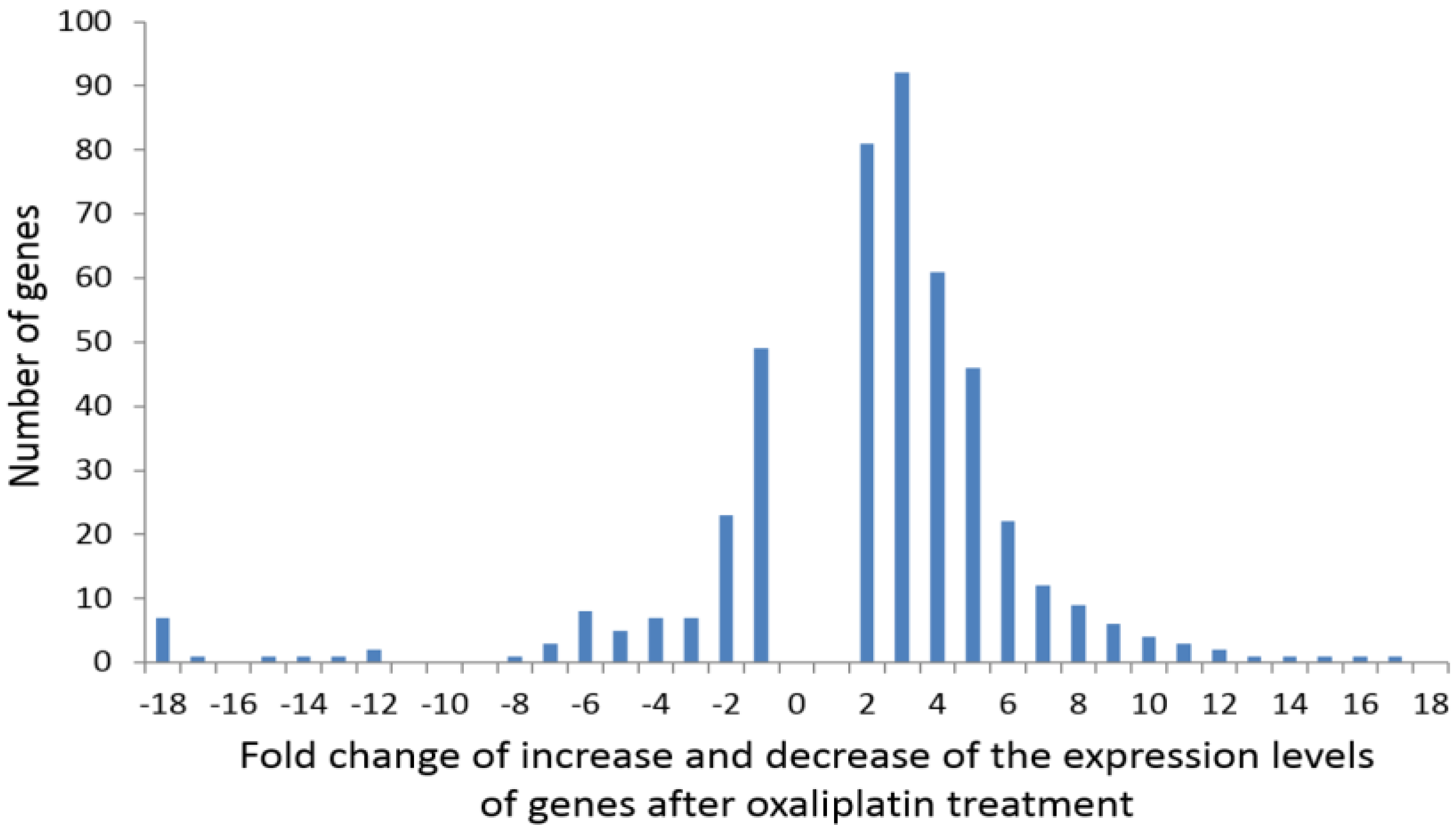
2.3. Characteristics of Genes with “On-Off” Differential Expression
| Gene | Genbank Acc. Number | PRE | POST |
|---|---|---|---|
| Gonadotropin-releasing hormone 2 (GNRH2) | NM_001501.1 | 0 | 5.8 |
| Cysteine and glycine-rich protein 2 (CSRP2) | NM_001321.1 | 0 | 7.8 |
| C5orf17 chromosome 5 open reading frame 17 (C5orf17) | NC_000005.9 | 0 | 8.8 |
| IQ motif containing GTPase activating protein 1 (IQGAP1) | NM_003870.3 | 0 | 229.5 |
| EF-hand calcium binding domain 8 (EFCAB8) | XM_006723897.1 | 4.1 | 0 |
| Chorionic somatomammotropin hormone-like 1 (CSHL1)- Growth hormone 1 (GH1) | NM_001318.2- NM_000515.3 | 7.2 | 0 |
| Small integral membrane protein 1 (SMIM1) | NM_001163724.2 | 8.2 | 0 |
| Potassium channel modulatory factor 1 (KCMF1) | NM_020122.4 | 12.3 | 0 |
| Interleukin 36, gamma (IL36G) | NM_001278568.1 | 13.4 | 0 |
| Lamin A/C (LMNA) | NM_001257374.2 | 325 | 1075.5 |
| Deleted in lymphocytic leukemia 7 (DLEU7) | NM_198989.2 | 25.7 | 3.9 |
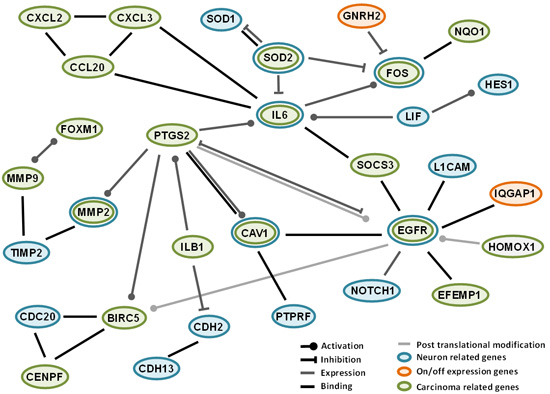
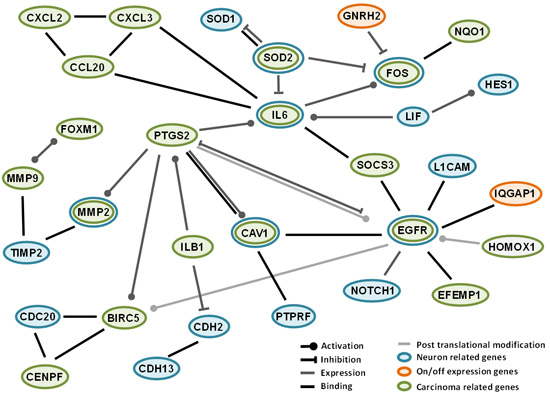
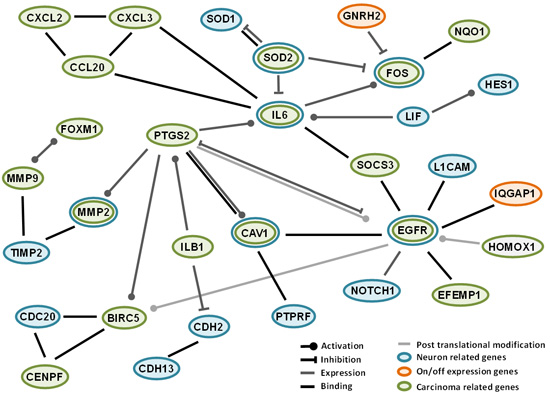
2.4. Interactions among Proteins Coded by Differentially Expressed Genes
3. Discussion
Some Characteristics and Comments on Genes with “On-Off” Variations in this Study

4. Experimental
4.1. Patients
4.2. Leukocytes Isolation
4.3. White Cell mRNA Extraction
4.4. RNA-Seq
4.5. Computational Analysis of RNA-Seq Data
4.6. Protein-Protein Interactions and Association Study
5. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pachman, D.R.; Loprinzi, C.L.; Grothey, A.; Ta, L.E. The search for treatments to reduce chemotherapy-induced peripheral neuropathy. J. Clin. Investig. 2014, 124, 72–74. [Google Scholar] [CrossRef]
- Andre, T.; Boni, C.; Mounedji-Boudiaf, L.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Zaninelli, M.; Clingan, P.; Bridgewater, J.; et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar] [CrossRef]
- O’Connor, E.S.; Greenblatt, D.Y.; LoConte, N.K.; Gangnon, R.E.; Liou, J.I.; Heise, C.P.; Smith, M.A. Adjuvant chemotherapy for stage II colon cancer with poor prognostic features. J. Clin. Oncol. 2011, 29, 3381–3388. [Google Scholar] [CrossRef]
- Gamelin, E.; Gamelin, L.; Bossi, L.; Quasthoff, S. Clinical aspects and molecular basis of oxaliplatin neurotoxicity: Current management and development of preventive measures. Semin. Oncol. 2002, 29, 21–33. [Google Scholar] [CrossRef]
- Grothey, A. Oxaliplatin-safety profile: Neurotoxicity. Semin. Oncol. 2003, 30, 5–13. [Google Scholar] [CrossRef]
- Raymond, E.; Faivre, S.; Woynarowski, J.M.; Chaney, S.G. Oxaliplatin: Mechanism of action and antineoplastic activity. Semin. Oncol. 1998, 25, 4–12. [Google Scholar]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D793–D800. [Google Scholar] [CrossRef]
- Kerrien, S.; Alam-Faruque, Y.; Aranda, B.; Bancarz, I.; Bridge, A.; Derow, C.; Dimmer, E.; Feuermann, M.; Friedrichsen, A.; Huntley, R.; et al. IntAct—Open source resource for molecular interaction data. Nucleic Acids Res. 2007, 35, D561–D565. [Google Scholar] [CrossRef]
- Podratz, J.L.; Knight, A.M.; Ta, L.E.; Staff, N.P.; Gass, J.M.; Genelin, K.; Schlattau, A.; Lathroum, L.; Windebank, A.J. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 2011, 41, 661–668. [Google Scholar]
- Casciato, D.; Barry, B. Lowitz Manual of Clinical Oncology; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004; p. 5. [Google Scholar]
- Chaouch, M.; Allal, Y.; de Sandre-Giovannoli, A.; Vallat, J.M.; Amer-el-Khedoud, A.; Kassouri, N.; Chaouch, A.; Sindou, P.; Hammadouche, T.; Tazir, M.; et al. The phenotypic manifestations of autosomal recessive axonal Charcot-Marie-Tooth due to a mutation in Lamin A/C gene. Neuromuscul. Disord. 2003, 13, 60–67. [Google Scholar] [CrossRef]
- Custodio, A.; Moreno-Rubio, J.; Aparicio, J.; Gallego-Plazas, J.; Yaya, R.; Maurel, J.; Higuera, O.; Burgos, E.; Ramos, D.; Calatrava, A.; et al. Pharmacogenetic predictors of severe peripheral neuropathy in colon cancer patients treated with oxaliplatin-based adjuvant chemotherapy: A GEMCAD group study. Ann. Oncol. 2014, 25, 398–403. [Google Scholar] [CrossRef]
- Kumamoto, K.; Ishibashi, K.; Okada, N.; Tajima, Y.; Kuwabara, K.; Kumagai, Y.; Baba, H.; Haga, N.; Ishida, H. Polymorphisms of GSTP1, ERCC2 and TS-3'UTR are associated with the clinical outcome of mFOLFOX6 in colorectal cancer patients. Oncol. Lett. 2013, 6, 648–654. [Google Scholar]
- Won, H.H.; Lee, J.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, J.W.; Lee, S.Y.; Park, S.H. Polymorphic markers associated with severe oxaliplatin-induced, chronic peripheral neuropathy in colon cancer patients. Cancer 2012, 118, 2828–2836. [Google Scholar] [CrossRef]
- McWhinney, S.R.; Goldberg, R.M.; McLeod, H.L. Platinum neurotoxicity pharmacogenetics. Mol. Cancer Ther. 2009, 8, 10–16. [Google Scholar] [CrossRef]
- Wu, Z.S.; Yang, K.; Wan, Y.; Qian, P.X.; Perry, J.K.; Chiesa, J.; Mertani, H.C.; Zhu, T.; Lobie, P.E. Tumor expression of human growth hormone and human prolactin predict a worse survival outcome in patients with mammary or endometrial carcinoma. J. Clin. Endocrinol. Metab. 2011, 96, E1619–E1629. [Google Scholar] [CrossRef]
- Jang, J.H. FIGC, a novel FGF-induced ubiquitin-protein ligase in gastric cancers. FEBS Lett. 2004, 578, 21–25. [Google Scholar] [CrossRef]
- Beilke, S.; Oswald, F.; Genze, F.; Wirth, T.; Adler, G.; Wagner, M. The zinc-finger protein KCMF1 is overexpressed during pancreatic cancer development and downregulation of KCMF1 inhibits pancreatic cancer development in mice. Oncogene 2010, 29, 4058–4067. [Google Scholar] [CrossRef]
- Kreppel, M.; Aryee, D.N.; Schaefer, K.L.; Amann, G.; Kofler, R.; Poremba, C.; Kovar, H. Suppression of KCMF1 by constitutive high CD99 expression is involved in the migratory ability of Ewing’s sarcoma cells. Oncogene 2006, 25, 2795–2800. [Google Scholar] [CrossRef]
- Zou, J.; Mi, L.; Yu, X.F.; Dong, J. Interaction of 14–3-3sigma with KCMF1 suppresses the proliferation and colony formation of human colon cancer stem cells. World J. Gastroenterol. 2013, 19, 3770–3780. [Google Scholar] [CrossRef]
- Deloukas, P.; Matthews, L.H.; Ashurst, J.; Burton, J.; Gilbert, J.G.; Jones, M.; Stavrides, G.; Almeida, J.P.; Babbage, A.K.; Bagguley, C.L.; et al. The DNA sequence and comparative analysis of human chromosome 20. Nature 2001, 414, 865–871. [Google Scholar] [CrossRef]
- Ebejer, J.L.; Duffy, D.L.; van der Werf, J.; Wright, M.J.; Montgomery, G.; Gillespie, N.A; Hickie, I.B.; Martin, N.G.; Medland, S.E. Genome-wide association study of inattention and hyperactivity-impulsivity measured as quantitative traits. Twin Res. Hum. Genet. 2013, 16, 560–574. [Google Scholar] [CrossRef]
- Sandholm, N.; Salem, R.M.; McKnight, A.J.; Brennan, E.P.; Forsblom, C.; Isakova, T.; McKay, G.J.; Williams, W.W.; Sadlier, D.M.; Makinen, V.P.; et al. New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet. 2012, 8, e1002921. [Google Scholar] [CrossRef] [Green Version]
- Gresnigt, M.S.; van de Veerdonk, F.L. Biology of IL-36 cytokines and their role in disease. Semin. Immunol. 2013, 25, 458–465. [Google Scholar] [CrossRef]
- Bachmann, M.; Scheiermann, P.; Hardle, L.; Pfeilschifter, J.; Muhl, H. IL-36gamma/IL-1F9, an innate T-bet target in myeloid cells. J. Biol. Chem. 2012, 287, 41684–41696. [Google Scholar] [CrossRef]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar]
- Weiskirchen, R.; Moser, M.; Weiskirchen, S.; Erdel, M.; Dahmen, S.; Buettner, R.; Gressner, A.M. LIM-domain protein cysteine- and glycine-rich protein 2 (CRP2) is a novel marker of hepatic stellate cells and binding partner of the protein inhibitor of activated STAT1. Biochem. J. 2001, 359, 485–496. [Google Scholar] [CrossRef]
- Midorikawa, Y.; Tsutsumi, S.; Taniguchi, H.; Ishii, M.; Kobune, Y.; Kodama, T.; Makuuchi, M.; Aburatani, H. Identification of genes associated with dedifferentiation of hepatocellular carcinoma with expression profiling analysis. Jpn. J. Cancer Res. 2002, 93, 636–643. [Google Scholar] [CrossRef]
- Johnson, M.; Sharma, M.; Henderson, B.R. IQGAP1 regulation and roles in cancer. Cell Signal. 2009, 21, 1471–1478. [Google Scholar] [CrossRef]
- White, C.D.; Brown, M.D.; Sacks, D.B. IQGAPs in cancer: A family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009, 583, 1817–1824. [Google Scholar] [CrossRef]
- Erdemir, H.H.; Li, Z.; Sacks, D.B. IQGAP1 binds to estrogen receptor-alpha and modulates its function. J. Biol. Chem. 2014, 289, 9100–9112. [Google Scholar] [CrossRef]
- Tamm-Rosenstein, K.; Simm, J.; Suhorutshenko, M.; Salumets, A.; Metsis, M. Changes in the transcriptome of the human endometrial Ishikawa cancer cell line induced by estrogen, progesterone, tamoxifen, and mifepristone (RU486) as detected by RNA-sequencing. PLoS One 2013, 8, e68907. [Google Scholar]
- Jameson, K.L.; Mazur, P.K.; Zehnder, A.M.; Zhang, J.; Zarnegar, B.; Sage, J.; Khavari, P.A. IQGAP1 scaffold-kinase interaction blockade selectively targets RAS-MAP kinase-driven tumors. Nat. Med. 2013, 19, 626–630. [Google Scholar]
- Sanchez-Laorden, B.; Viros, A.; Marais, R. Mind the IQGAP. Cancer Cell 2013, 23, 715–717. [Google Scholar] [CrossRef]
- Stuart, D.D.; Sellers, W.R. Targeting RAF-MEK-ERK kinase-scaffold interactions in cancer. Nat. Med. 2013, 19, 538–540. [Google Scholar] [CrossRef]
- Ballif, B.A.; Helias, V.; Peyrard, T.; Menanteau, C.; Saison, C.; Lucien, N.; Bourgouin, S.; Le, G.M.; Cartron, J.P.; Arnaud, L. Disruption of SMIM1 causes the Vel-blood type. EMBO Mol. Med. 2013, 5, 751–761. [Google Scholar] [CrossRef]
- Cvejic, A.; Haer-Wigman, L.; Stephens, J.C.; Kostadima, M.; Smethurst, P.A.; Frontini, M.; van den Akker, E.; Bertone, P.; Bielczyk-Maczynska, E.; Farrow, S.; et al. SMIM1 underlies the Vel blood group and influences red blood cell traits. Nat. Genet. 2013, 45, 542–545. [Google Scholar] [CrossRef]
- Storry, J.R.; Joud, M.; Christophersen, M.K.; Thuresson, B.; Akerstrom, B.; Sojka, B.N.; Nilsson, B.; Olsson, M.L. Homozygosity for a null allele of SMIM1 defines the Vel-negative blood group phenotype. Nat. Genet. 2013, 45, 537–541. [Google Scholar] [CrossRef]
- Shamay, M.; Liu, J.; Li, R.; Liao, G.; Shen, L.; Greenway, M.; Hu, S.; Zhu, J.; Xie, Z.; Ambinder, R.F.; et al. A protein array screen for Kaposi’s sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity, and telomere shortening. J. Virol. 2012, 86, 5179–5191. [Google Scholar] [CrossRef]
- McNulty, D.E.; Li, Z.; White, C.D.; Sacks, D.B.; Annan, R.S. MAPK scaffold IQGAP1 binds the EGF receptor and modulates its activation. J. Biol. Chem. 2011, 286, 15010–15021. [Google Scholar] [CrossRef]
- Wu, H.M.; Wang, H.S.; Huang, H.Y.; Soong, Y.K.; MacCalman, C.D.; Leung, P.C. GnRH signaling in intrauterine tissues. Reproduction 2009, 137, 769–777. [Google Scholar] [CrossRef]
- Rauscher, F.J., III; Sambucetti, L.C.; Curran, T.; Distel, R.J.; Spiegelman, B.M. Common DNA binding site for Fos protein complexes and transcription factor AP-1. Cell 1988, 52, 471–480. [Google Scholar] [CrossRef]
- Herdegen, T.; Leah, J.D. Inducible and constitutive transcription factors in the mammalian nervous system: Control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res. Brain Res. Rev. 1998, 28, 370–490. [Google Scholar] [CrossRef]
- Kaczmarek, L. Molecular biology of vertebrate learning: Is c-fos a new beginning? J. Neurosci. Res. 1993, 34, 377–381. [Google Scholar] [CrossRef]
- Lo, H.W.; Hung, M.C. Nuclear EGFR signalling network in cancers: Linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br. J. Cancer 2006, 94, 184–188. [Google Scholar] [CrossRef]
- Lo, H.W. EGFR-targeted therapy in malignant glioma: Novel aspects and mechanisms of drug resistance. Curr. Mol. Pharmacol. 2010, 3, 37–52. [Google Scholar] [CrossRef]
- Lo, H.W. Nuclear mode of the EGFR signaling network: Biology, prognostic value, and therapeutic implications. Discov. Med. 2010, 10, 44–51. [Google Scholar]
- Aguirre, A.; Rubio, M.E.; Gallo, V. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature 2010, 467, 323–327. [Google Scholar] [CrossRef]
- Ayuso-Sacido, A.; Moliterno, J.A.; Kratovac, S.; Kapoor, G.S.; O’Rourke, D.M.; Holland, E.C.; Garcia-Verdugo, J.M.; Roy, N.S.; Boockvar, J.A. Activated EGFR signaling increases proliferation, survival, and migration and blocks neuronal differentiation in post-natal neural stem cells. J. Neurooncol. 2010, 97, 323–337. [Google Scholar] [CrossRef]
- Boockvar, J.A.; Kapitonov, D.; Kapoor, G.; Schouten, J.; Counelis, G.J.; Bogler, O.; Snyder, E.Y.; McIntosh, T.K.; O’Rourke, D.M. Constitutive EGFR signaling confers a motile phenotype to neural stem cells. Mol. Cell Neurosci. 2003, 24, 1116–1130. [Google Scholar] [CrossRef]
- Schafer, M.K.; Altevogt, P. L1CAM malfunction in the nervous system and human carcinomas. Cell. Mol. Life Sci. 2010, 67, 2425–2437. [Google Scholar] [CrossRef]
- Schafer, M.K.; Frotscher, M. Role of L1CAM for axon sprouting and branching. Cell Tissue Res. 2012, 349, 39–48. [Google Scholar] [CrossRef]
- Cheung, C.H.; Huang, C.C.; Tsai, F.Y.; Lee, J.Y.; Cheng, S.M.; Chang, Y.C.; Huang, Y.C.; Chen, S.H.; Chang, J.Y. Survivin—Biology and potential as a therapeutic target in oncology. Oncol. Targets Ther. 2013, 6, 1453–1462. [Google Scholar]
- Wang, Q.; Greene, M.I. EGFR enhances Survivin expression through the phosphoinositide 3 (PI-3) kinase signaling pathway. Exp. Mol. Pathol. 2005, 79, 100–107. [Google Scholar] [CrossRef]
- Camaj, P.; Seeliger, H.; Ischenko, I.; Krebs, S.; Blum, H.; de Toni, E.N.; Faktorova, D.; Jauch, K.W.; Bruns, C.J. EFEMP1 binds the EGF receptor and activates MAPK and Akt pathways in pancreatic carcinoma cells. Biol. Chem. 2009, 390, 1293–1302. [Google Scholar]
- Chen, J.; Wei, D.; Zhao, Y.; Liu, X.; Zhang, J. Overexpression of EFEMP1 correlates with tumor progression and poor prognosis in human ovarian carcinoma. PLoS One 2013, 8, e78783. [Google Scholar]
- Song, E.L.; Hou, Y.P.; Yu, S.P.; Chen, S.G.; Huang, J.T.; Luo, T.; Kong, L.P.; Xu, J.; Wang, H.Q. EFEMP1 expression promotes angiogenesis and accelerates the growth of cervical cancer in vivo. Gynecol. Oncol. 2011, 121, 174–180. [Google Scholar] [CrossRef]
- Sakarya, O.; Breu, H.; Radovich, M.; Chen, Y.; Wang, Y.N.; Barbacioru, C.; Utiramerur, S.; Whitley, P.P.; Brockman, J.P.; Vatta, P. RNA-Seq mapping and detection of gene fusions with a suffix array algorithm. PLoS Comput. Biol. 2012, 8, e1002464. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Morales, M.; Ávila, J.; González-Fernández, R.; Boronat, L.; Soriano, M.L.; Martín-Vasallo, P. Differential Transcriptome Profile of Peripheral White Cells to Identify Biomarkers Involved in Oxaliplatin Induced Neuropathy. J. Pers. Med. 2014, 4, 282-296. https://doi.org/10.3390/jpm4020282
Morales M, Ávila J, González-Fernández R, Boronat L, Soriano ML, Martín-Vasallo P. Differential Transcriptome Profile of Peripheral White Cells to Identify Biomarkers Involved in Oxaliplatin Induced Neuropathy. Journal of Personalized Medicine. 2014; 4(2):282-296. https://doi.org/10.3390/jpm4020282
Chicago/Turabian StyleMorales, Manuel, Julio Ávila, Rebeca González-Fernández, Laia Boronat, María Luisa Soriano, and Pablo Martín-Vasallo. 2014. "Differential Transcriptome Profile of Peripheral White Cells to Identify Biomarkers Involved in Oxaliplatin Induced Neuropathy" Journal of Personalized Medicine 4, no. 2: 282-296. https://doi.org/10.3390/jpm4020282