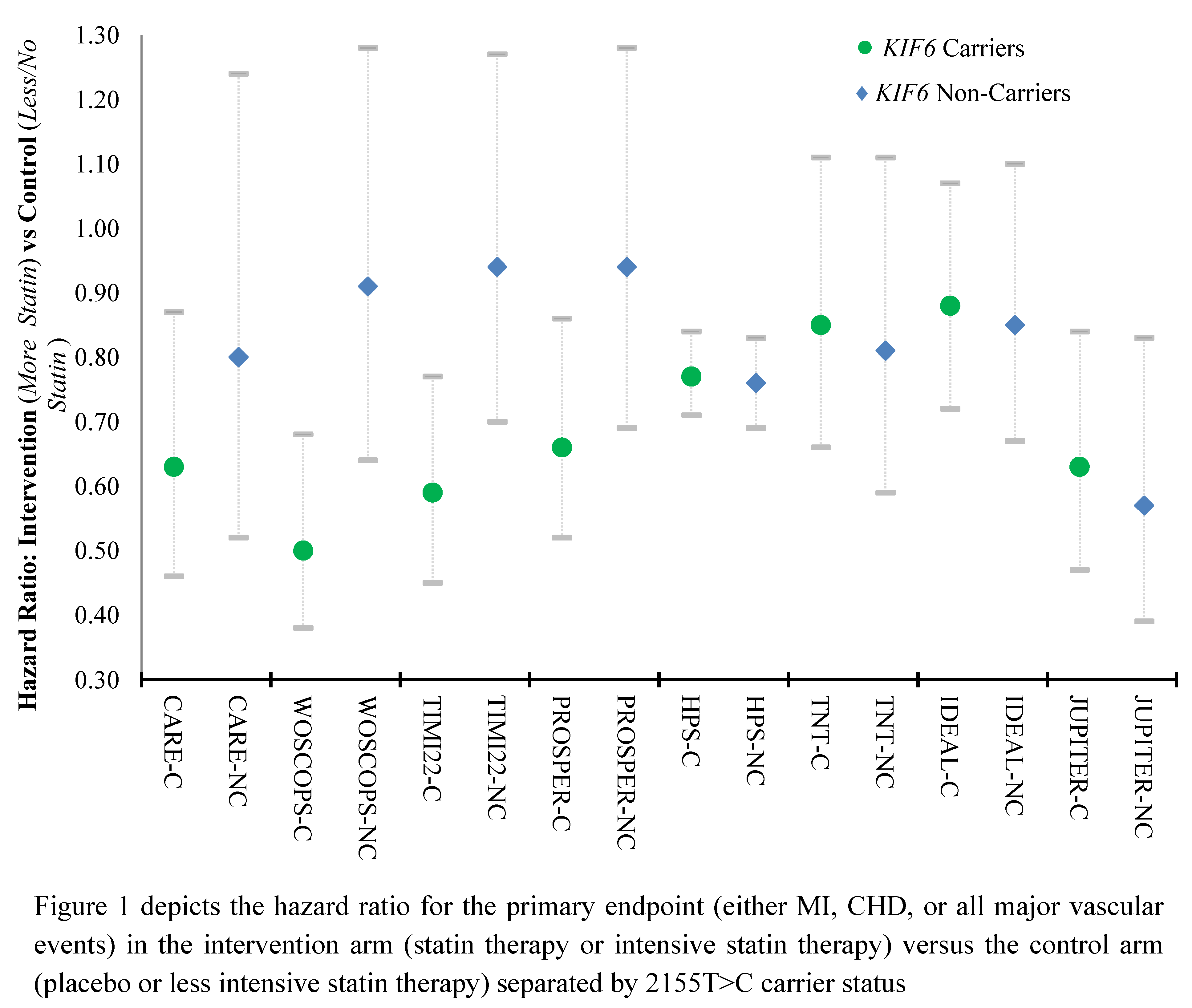
Solute carrier organic anion transporter family member 1B1 (
SLCO1B1) is a predictive marker of statin-related myopathy (SRM) which is a significant barrier to optimal adherence. Although there are many clinical factors that may predispose a patient to SRM, recent evidence suggests that SRM has a very strong genetic component. In fact, up to half of the SRM associated with simvastatin, one of the most commonly prescribed statins, may be attributable to a single genetic variant in
SLCO1B1 [
57]. Furthermore, recent evidence has demonstrated that patients carrying certain variants in
SLCO1B1 are twice as likely to show signs of intolerance to the first statin they are prescribed, which can lead to trial-and-error prescribing and unnecessary drug churn [
26]. These findings suggest that identification of patients with variant forms of
SLCO1B1 could mitigate SRM and subsequent low adherence.
4.1. SLCO1B1 Physiology
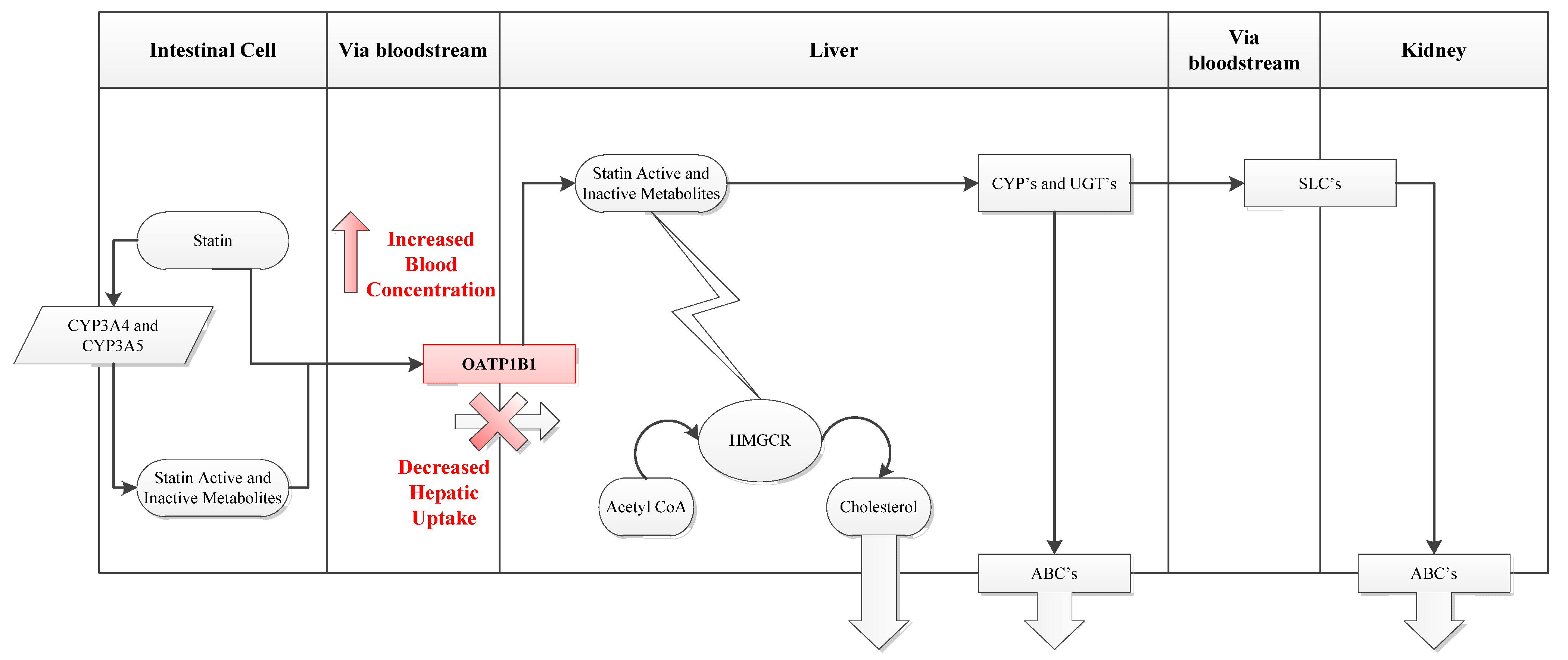
For statins to perform their function they must first reach the liver. The uptake of statins from portal blood into hepatocytes across the phospholipid bilayer occurs primarily through the organic anion-transporting polypeptide 1B1 (OATP1B1) influx transporter which is expressed on the basolateral membrane of human hepatocytes (
Figure 2). OATP1B1 transport appears to be rate limiting for hepatocyte uptake and hence distribution and metabolism of many statins. Consequently, modifications in this transporter have been significantly associated with the risk of SRM [
26,
58,
59]. OATP1B1 is encoded by the gene
SLCO1B1 whose
*5 allele (Val174Ala, 521T>C) has been shown to interfere with localization of the transporter to the plasma membrane, leading to decreased liver uptake and greater systemic statin concentrations and hence greater muscle statin exposure [
60].
Figure 2.
Statin Uptake Pathway. (a) SLCO1B1 encodes the OATP1B1 influx transporter. (b) OATP1B1 transport is particularly important for hepatic accessibility of statins. The transporter contributes to liver uptake of statins including first pass clearance from the portal circulation so that decreased transport results in increases systemic (including muscle) exposure to statin. (c) HMGCR = 3-hydroxy-3-methyl-glutaryl-CoA reductase, CYP = cytochrome P450 isoenzymes, UGT = UDP-glucuronyl transferase class of enzymes, SLC = solute carrier group of membrane transporters, ABC = ATP-binding cassette transporters.
Figure 2.
Statin Uptake Pathway. (a) SLCO1B1 encodes the OATP1B1 influx transporter. (b) OATP1B1 transport is particularly important for hepatic accessibility of statins. The transporter contributes to liver uptake of statins including first pass clearance from the portal circulation so that decreased transport results in increases systemic (including muscle) exposure to statin. (c) HMGCR = 3-hydroxy-3-methyl-glutaryl-CoA reductase, CYP = cytochrome P450 isoenzymes, UGT = UDP-glucuronyl transferase class of enzymes, SLC = solute carrier group of membrane transporters, ABC = ATP-binding cassette transporters.
The genotypic frequencies for the variants of
SLCO1B1 vary by ethnicity and some reduced function alleles are relatively common, such as the presence of
*5 in 8%–20% of Caucasians. The best-documented haplotypes thought to play an important role in modulating the risk of SRM are shown in
Table 3. Of note, the
*15 haplotype, with an allele frequency of 10% in Japanese, carries the same 521T>C substitution as
*5 in combination with the 388A>G SNP and represents another risk allele for myopathy in patients receiving statin therapy.
Table 3.
SLCO1B1 Haplotypes in Various Ethnic Groups. Adapted with permission from Oshiro
et al. [
60].
Table 3.
SLCO1B1 Haplotypes in Various Ethnic Groups. Adapted with permission from Oshiro et al. [60].
| Nucleotide Change (s) | rsID | Protein Variation (s) | Haplotype | Transporter Effect | OATP1B1 substrate serum conc. | Allele Frequency (%) a | Ref. |
|---|
| AA | J | As | C |
|---|
| None | N/A | N/A | *1A | Normal | Baseline | | | | | [60] |
| 388A>G | 2306283 | Asn130Asp | *1B | Increased | Decreased | 74–78 | 54 | 58–81 | 37–46 | [60] |
| 521T>C | 4149056 | Val174Ala | *5 | Reduced | Increased | 1–4 | 0.7 | 6–19 | 12–20 | [60] |
| 521T>C+ 388A>G | 4149056+ 2306283 | Val174Ala+ Asn130Asp | *15 | Reduced | Increased | | 10 | | | [60] |
4.3. Clinical Evidence
If toxicity is correlated with muscle exposure to drug levels, then logically, increased drug concentrations in the blood should be reflected in an altered side effect profile in patients with reduced transport. Several safety studies have evaluated how the rates of adverse drug reactions, most commonly myopathy, vary by genotype. To date, five studies have evaluated the risk of SRM as a function of genetic variation in
SLCO1B1 (
Table 5).
Table 5.
SLCO1B1 and Risk of Myopathy. S = Simvastatin, A = Atorvastatin, R = Rosuvastatin, P = Pravastatin, C = Cerivastatin, RR = relative risk, OR = Odds Ratio, ULN = Upper Limit of Normal.
Table 5.
SLCO1B1 and Risk of Myopathy. S = Simvastatin, A = Atorvastatin, R = Rosuvastatin, P = Pravastatin, C = Cerivastatin, RR = relative risk, OR = Odds Ratio, ULN = Upper Limit of Normal.
| Study | Drug | n | Allele(s) | Clinical Endpoint | Outcome |
|---|
| SEARCH [59] | S 80 mg | 175 | *5 | Definite or incipient myopathy | OR = 4.7 per copy (p = 3 × 10−28) |
| HPS [59] | S 40 mg | 1,664 | *5 | Definite or incipient myopathy | OR = 2.6 per copy (p = 0.004) |
| STRENGTH [58] | S20→80 mg | 452 | *5 | Composite adverse event (CAE) defined as discontinuation for any side effect, myalgia, or CK>3x ULN | S: OR = 1.7 per copy (p = 0.03) |
| P10→40 mg |
| A10→80 mg |
| GO-DARTS [26] | All Statins, all doses | 4,141 | *1B, *5, *15 | Intolerance as defined by an increase in CK (1xULN>CK<3xULN) or ALT and aberrant prescription patterns | OR = 2.05, (p = 0.043) |
| Marciante et al., 2011 [70] | C | 917 | *5 | Rhabdomyolysis | OR = 1.89, (p = 0.002) |
The myopathy risk associated with
SLCO1B1 was first reported in a study reported by the Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group [
59]. The authors studied two cohorts of clinically severe cases and controls from large trials involving approximately 12,000 and 20,000 participants who were treated with 80 mg and 40 mg of simvastatin per day, respectively. The investigators observed a significant association between SRM and a single marker in the
SLCO1B1 gene (rs4363657,
p = 3 × 10
−28). This association was then confirmed in a second cohort which included patients who were randomly assigned to 40 mg of simvastatin per day (see
Table 6).
Table 6.
Myopathy Risk in SEARCH Stratified by SLCO1B1 Genotype [
59].
Table 6.
Myopathy Risk in SEARCH Stratified by SLCO1B1 Genotype [59].
| Genotype a | Population Frequency | Cumulative Percentage with Myopathy |
|---|
| Year 1 | Year 5 |
|---|
| Wild Type | 73% | 0.34% | 0.63% |
| Heterozygote | 24.9% | 1.38% | 2.83% |
| Homozygote | 2.1% | 15.25% | 18.55% |
The results from the retrospective genetic association study in SEARCH and HPS were subsequently validated in the prospective randomized STRENGTH (Statin Response Examined by Genetic Haplotype Markers) study [
58]. In STRENGTH, subjects (n = 509) were randomized to ascending doses of atorvastatin 10→80 mg, simvastatin 20→80 mg, or pravastatin 10→40 mg. A composite adverse event (CAE) was defined as discontinuation for any side effect, myalgia, or CK>3 times upper limit of normal (ULN) during follow-up. Of the five candidate genes evaluated, including
CYP2D6,
CYP2C8,
CYP2C9,
CYP3A4, and
SLCO1B1, only
SLCO1B1*5 was associated with CAE (37%
vs. 25% in carriers and wild type patients respectively,
p = 0.03) and more significantly for those with CAE exclusive of significant CK elevation (
p ≤ 0.03). Furthermore, a gene-dosage effect was observed (percent with CAE in those with 0, 1, or 2 of the variant (*5) alleles: 19%, 27%, and 50%,
p = 0.01 for the trend). Importantly, only allele carriers receiving ascending doses of simvastatin showed significantly heightened risk of CAE compared to patients who carried no alleles (16%
vs. 34%,
p = 0.01). This is in contrast to patients receiving atorvastatin and pravastatin who showed non-significant changes in CAE risk based on allele carriage (19%
vs. 27%,
p = 0.3 and 22%
vs. 22%,
p = 0.97 for atorvastatin and pravastatin respectively).Since carriers of 521T>C mutations experienced higher rates of myalgia, a significant obstacle to optimal adherence, these same patients should have prescribing patterns reflective of intolerance such as switching to a different statin at a lower or equivalent dose, reducing the dose of the same statin, or discontinuation of statin therapy. This hypothesis was the aim of the GO-DARTS (Genetics of Diabetes Audit and Research) study which examined whether
SLCO1B1 variants were associated with general statin intolerance in a large population of patients with type 2 diabetes receiving statins as part of routine clinical care. This observational incident cohort analysis used information from 4,196 genotyped patients in the GO-DARTS database, which is part of an ongoing research initiative in the Tayside, Scotland (population 400,000) community to track the treatment and health outcomes of individuals with diabetes [
26]. Information captured in this database included detailed clinical information for individuals with diabetes from 1990 to the present including all pharmacy records, lab test results, and other clinical data related to diabetes care. This study particularly focused on mild manifestations of myopathy, and patients with CK > 3×ULN
iv [
71] were excluded from analysis. For purposes of this study, intolerance was defined as a composite measure of abnormal lab values, alanine transaminase (ALT) and CK, and relevant adjustments to the prescription of each patient.
This study confirmed the association of the *5 allele with statin intolerance (OR = 2.05, 95% CI: 1.02–4.09, p = 0.04), and further showed that *5 allele carriers have a doubled risk for intolerance to their originally prescribed statin. These results were observed in a population where moderate and severe cases of myopathy were excluded, therefore representing better the sub-pathological end of the spectrum of statin related muscle effects likely to be the more significant driver of correlated non-adherence in terms of numbers. This study suggests that the muscle toxicity associated with SLCO1B1 is represented in prescribing patterns suggestive of intolerance, and may ultimately prove to be useful as a prospective intervention.
Although the majority of evidence for
SLCO1B1-related SRM has been around simvastatin, cerivastatin, a drug that was recalled due to its risk of rhabdomyolysis [
4], has also recently been shown to be effected by this locus. In an analysis by Marciante
et al., a candidate gene study (examining
CYP2C8,
UGT1A1,
UGT1A3, and
SLCO1B1) and a GWAS study were performed on 185 cerivastatin-induced rhabdomyolysis cases matched to statin-using controls from Cardiovascular Health Study (n = 374) and Vascular Health Study (n = 358) [
70], A subsequent in vitro functional analysis for 521T>C was also performed in stable HEK293 cells. Permutation test results showed an association between cerivastatin-induced rhabdomyolysis and the
*5 allele (OR = 1.89,
p = 0.002). In functional studies, this variant reduced transport by 40% compared with the reference transporter (
p < 0.001). This study extends the results of simvastatin-centered trials to cerivastatin and functional studies provide a potential causal association.
4.4. SLCO1B1 as an Adherence Intervention
Clinical evidence shows a strong association between carriage of alleles of
SLCO1B1 and both mild myalgia and clinically severe myopathy [
58,
59]. Furthermore,
SLCO1B1 induced muscle toxicity has also been associated with lower levels of drug tolerance [
26]. Since there is a gradient of effect for variations in this transporter across the statin class [
72], it may be possible to personalize statin treatment for the patient’s effectiveness goals as well as their predisposition to myopathy according to
SLCO1B1 genotype. In fact various groups, including the Clinical Pharmacogenomics Implementation Consortium, have drafted specific treatment recommendations that can provide clinicians with a practical starting point for how to implement a patient’s 521T>C status into their treatment [
72,
73].
Although the evidence from the AKROBATS trial may not fully support the utility of genomic testing in itself in improving patient adherence [
27], the concept is compelling and the use of
SLCO1B1 may go one step further by not only affecting a patient’s sense of self-efficacy [
32] but also reducing the probability of myopathy, an independent barrier to optimal adherence. Most importantly, decisions based on this intervention could lead to less atherosclerosis and cardiovascular events. Currently there is no direct clinical evidence that the personalized prescribing of statins using a patient’s
SLCO1B1*5 status will improve their medication adherence; however, previous analyses suggest that this is a logical conclusion and will be an important hypothesis to evaluate in future analyses.


{kind=link}
{kind=link}

