
Highly Conserved Elements and Chromosome Structure Evolution in Mitochondrial Genomes in Ciliates

 ,
, 
 ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Highly Conserved Elements in Mitochondrial Genome in Ciliates (Ciliophora)
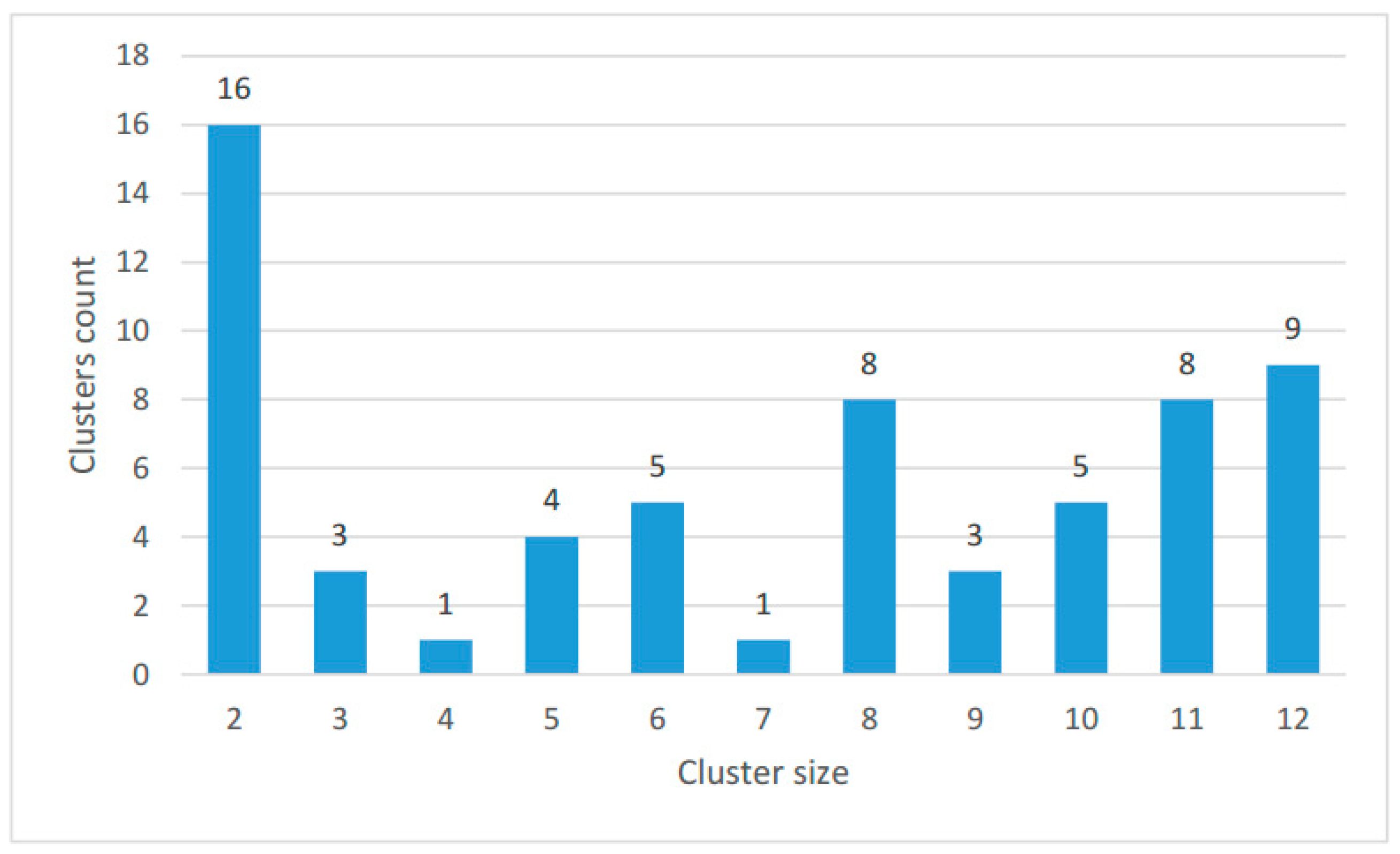
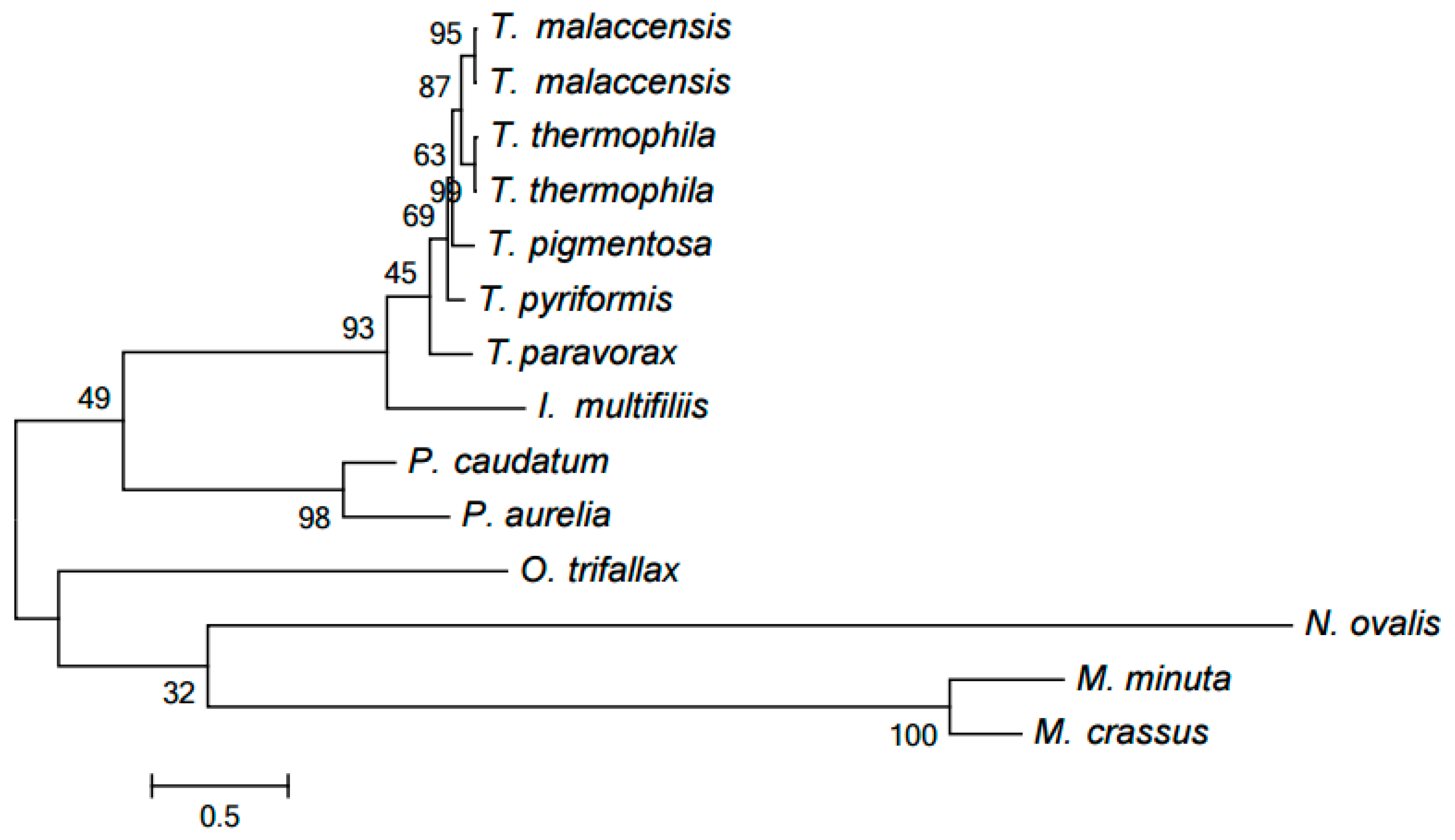
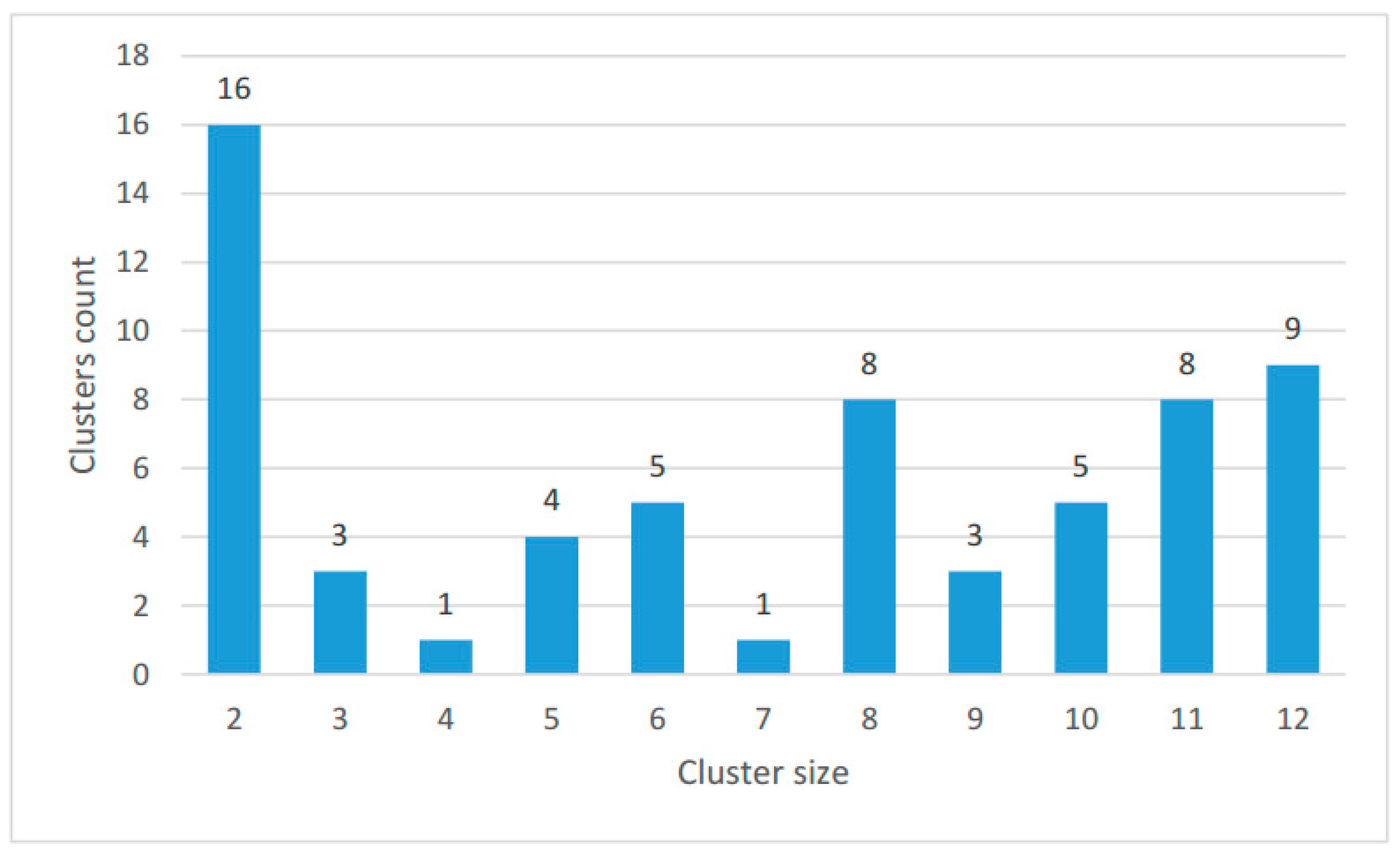
2.2. Clustering of Proteins Encoded in Mitochondria in Ciliates
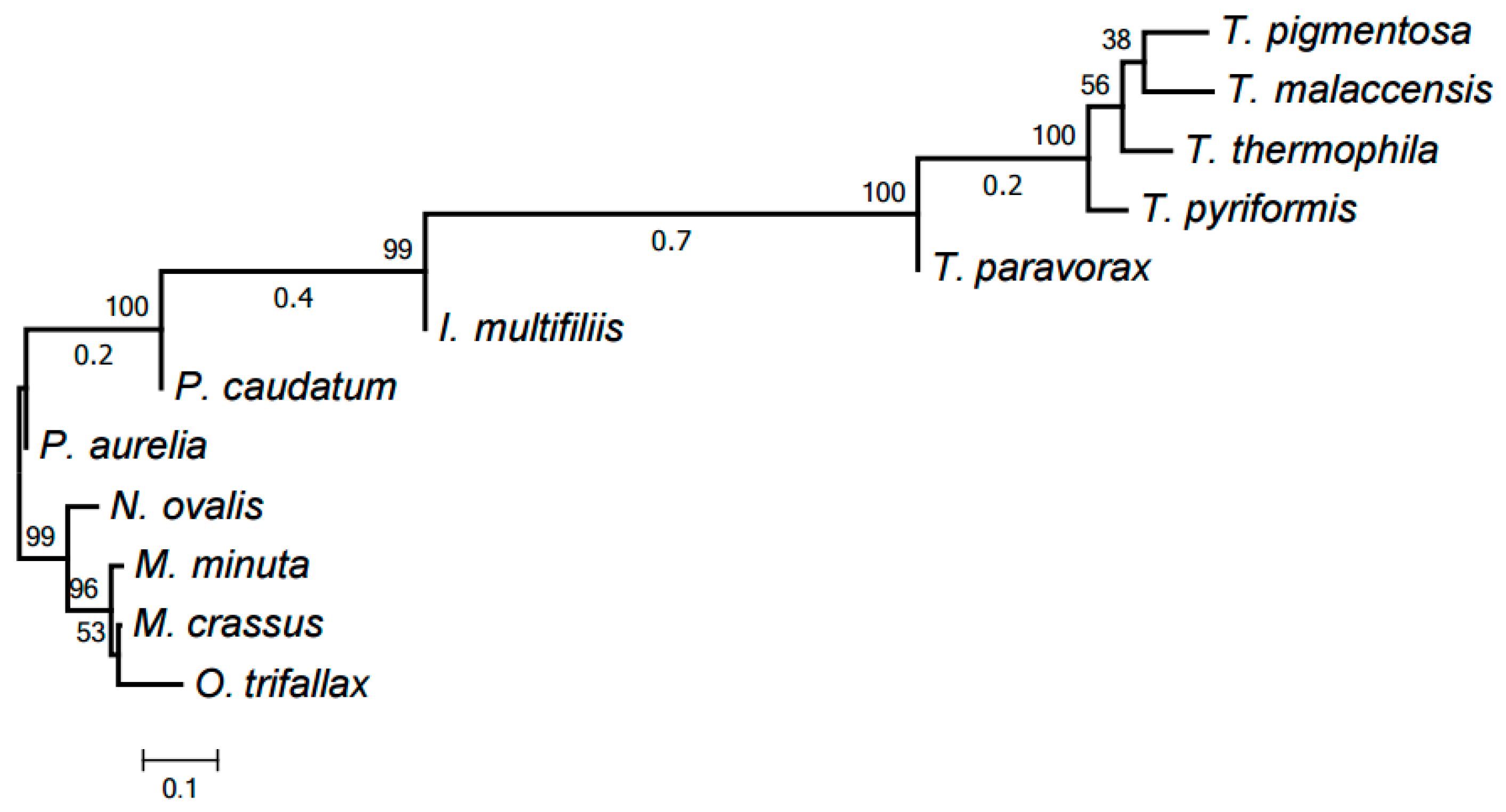
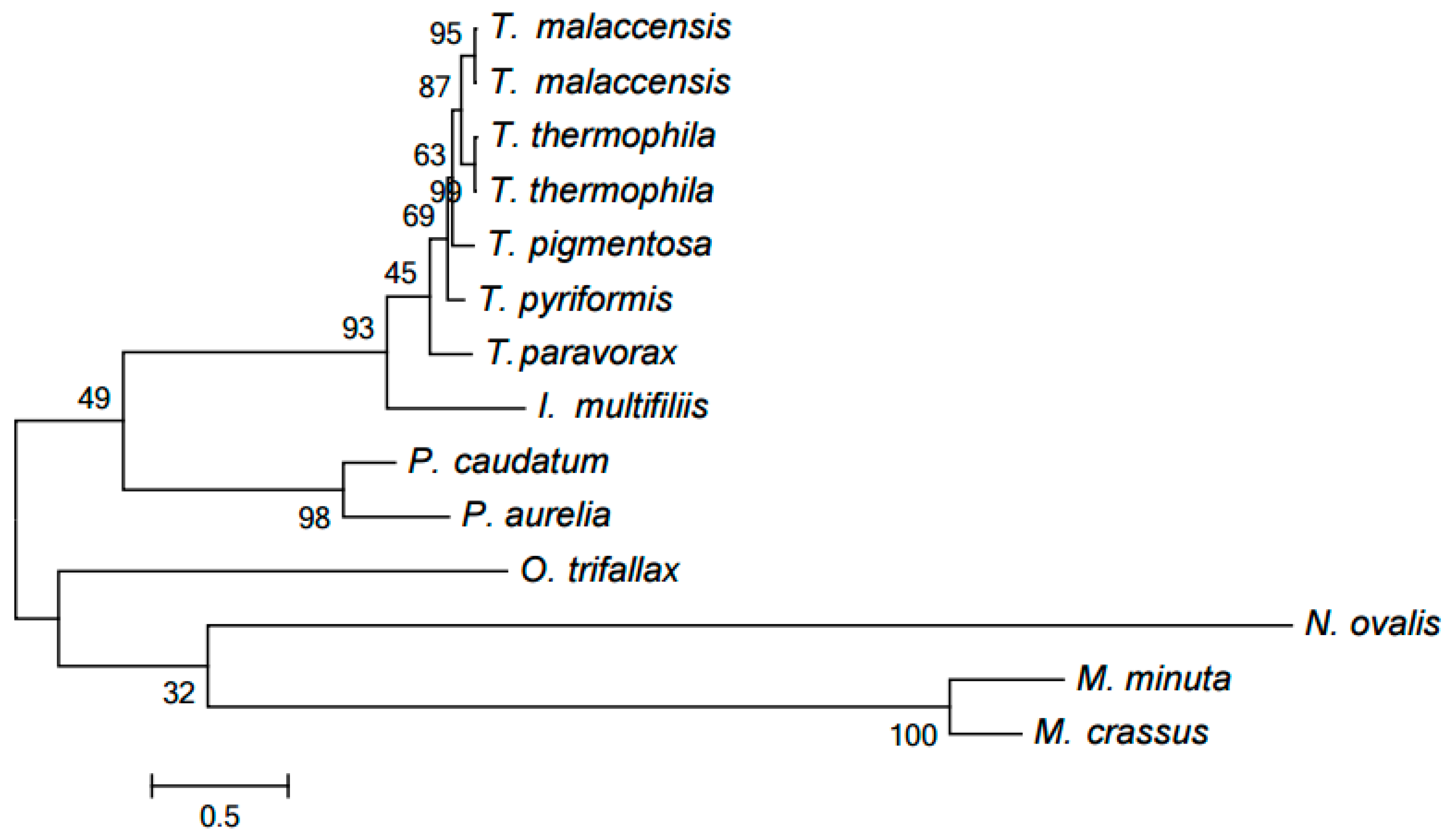
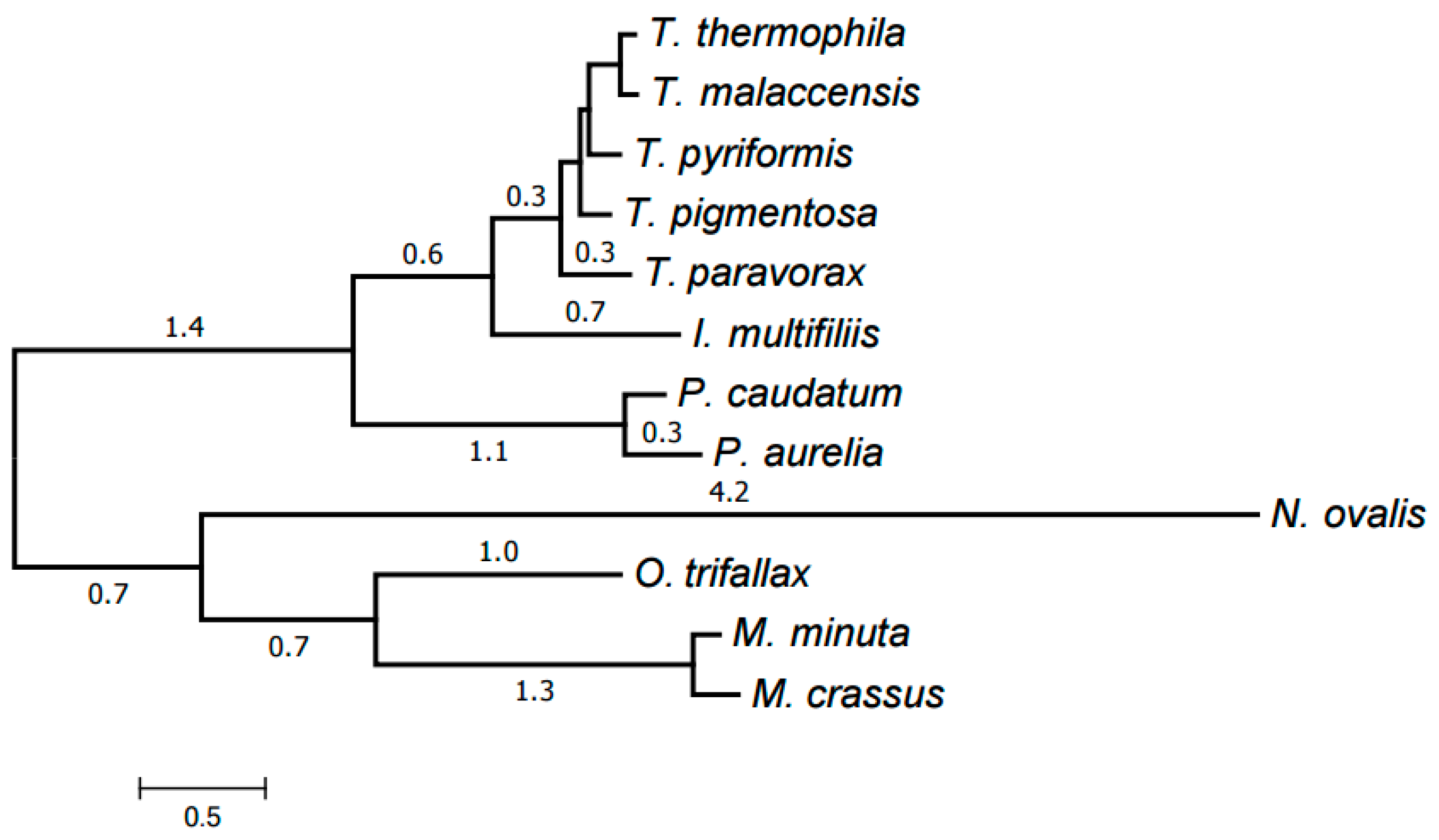
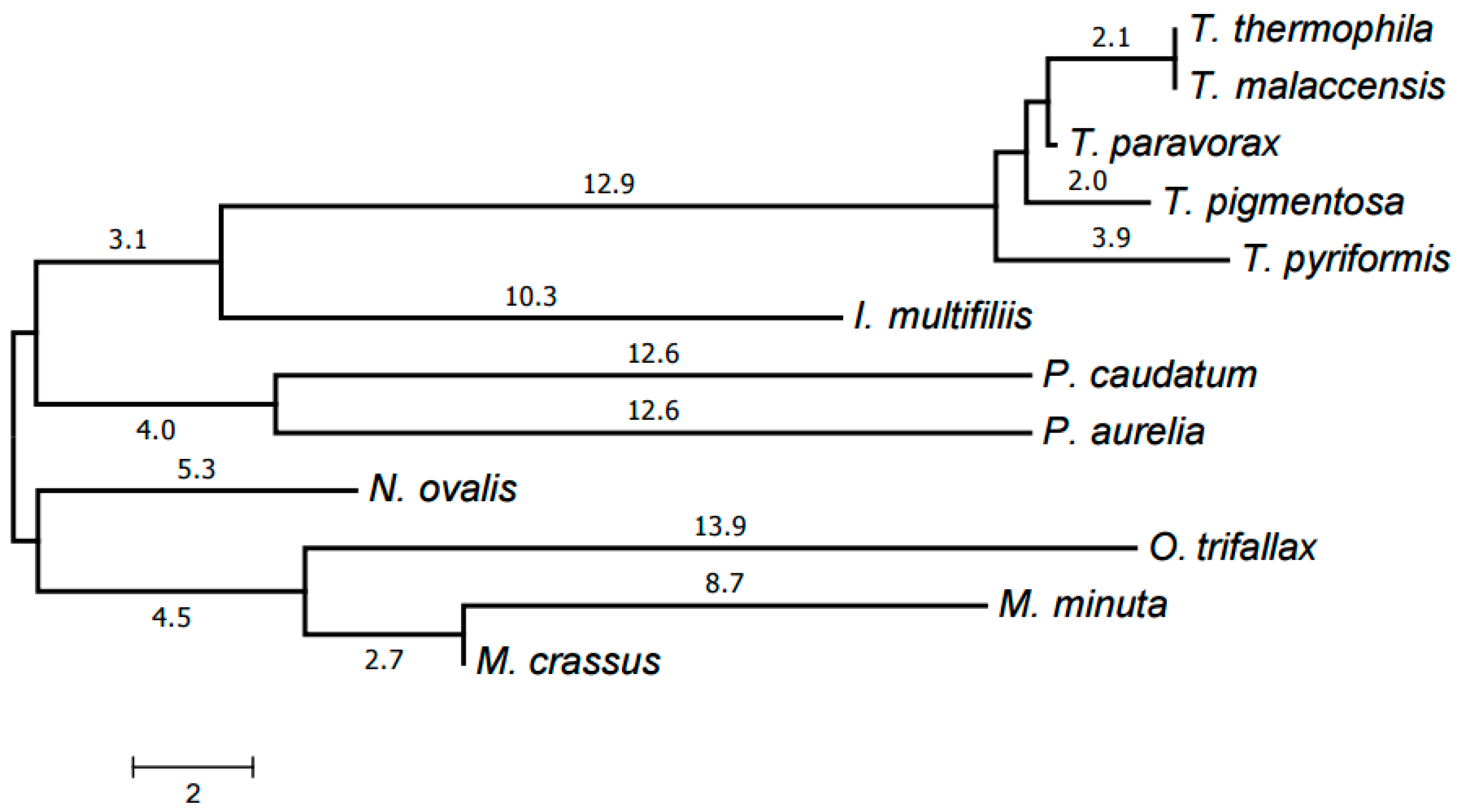
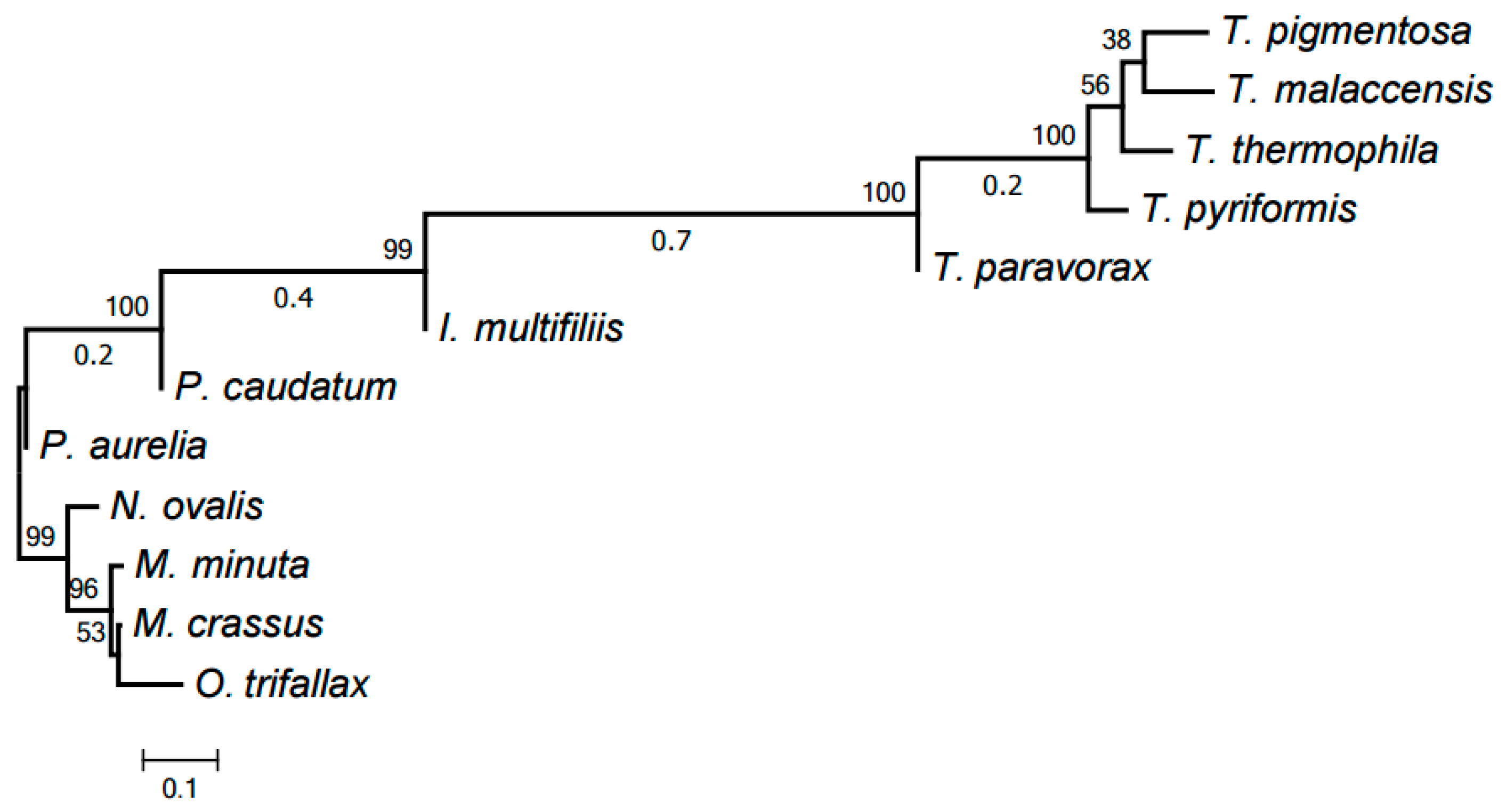
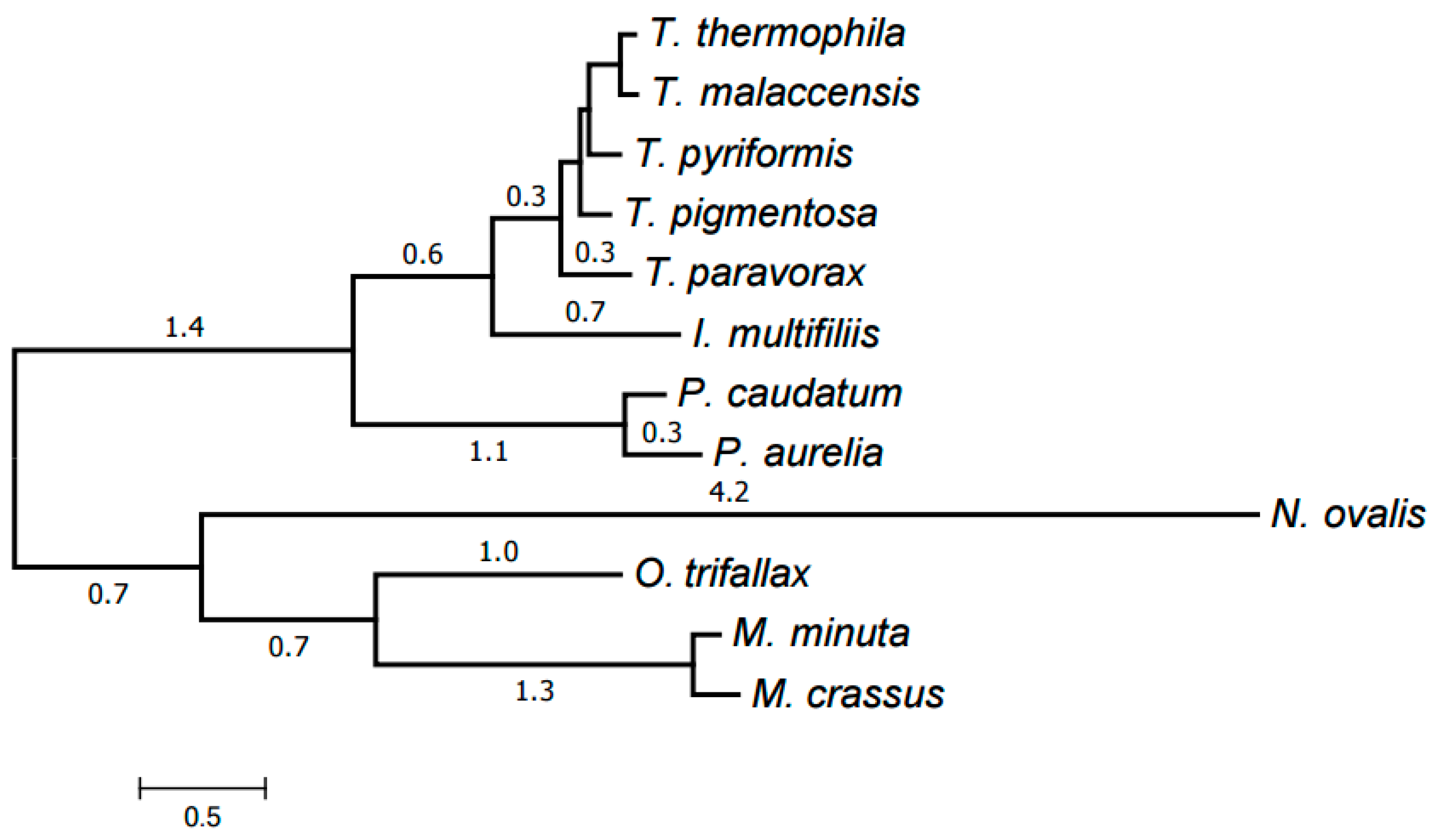
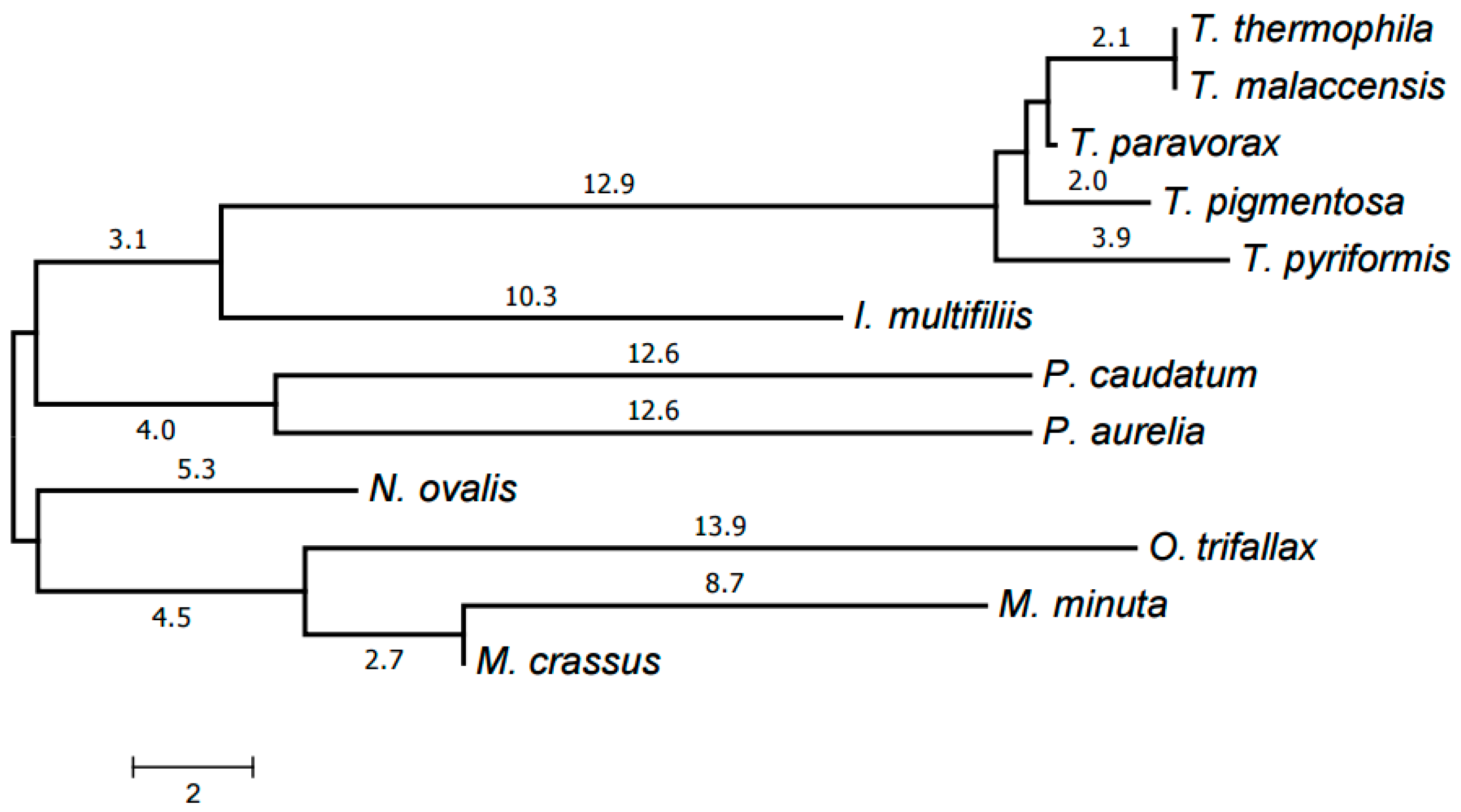
2.3. Evolution of Mitochondrial Chromosome Structure in Ciliates
2.4. Reconstruction of Mitochondrial Chromosome Structure in Ciliates
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Garmash, E.V. Mitochondrial respiration of the photosynthesizing cell. Russ. J. Plant Physiol. 2016, 63, 13–25. [Google Scholar] [CrossRef]
- Karnkowska, A.; Vacek, V.; Zubáčová, Z.; Treitli, S.C.; Petrželková, R.; Eme, L.; Novák, L.; Žárský, V.; Barlow, L.D.; Herman, E.K.; et al. A eukaryote without a mitochondrial organelle. Curr. Biol. 2016, 26, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Van Hoek, A.H.; van Alen, T.A.; Sprakel, V.S.; Hackstein, J.H.; Vogels, G.D. Evolution of anaerobic ciliates from the gastrointestinal tract: Phylogenetic analysis of the ribosomal repeat from Nyctotherus ovalis and its relatives. Mol. Biol. Evol. 1998, 15, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, R.M.; Ricard, G.; van Alen, T.A.; Duarte, I.; Dutilh, B.E.; Burgtorf, C.; Kuiper, J.W.; van der Staay, G.W.; Tielens, A.G.; Huynen, M.A.; et al. The organellar genome and metabolic potential of the hydrogen-producing mitochondrion of Nyctotherus ovalis. Mol. Biol. Evol. 2011, 28, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Kairo, A.; Fairlamb, A.H.; Gobright, E.; Nene, V. A 7.1 kb linear DNA molecule of Theileria parva has scrambled rDNA sequences and open reading frames for mitochondrially encoded proteins. EMBO J. 1994, 13, 898–905. [Google Scholar] [PubMed]
- Vanyatinsky, V.F.; Mirsoeva, L.M.; Poddubnaya, A.V. Bolezni Ryb [Fish Diseases]; Musselius, V.A., Ed.; Food Processing Industry: Moscow, Russia, 1979. (In Russian) [Google Scholar]
- De Graaf, R.M.; van Alen, T.A.; Dutilh, B.E.; Kuiper, J.W.P.; van Zoggel, H.J.A.A.; Huynh, M.B.; Görtz, H.-D.; Huynen, M.A.; Hackstein, J.H.P. The mitochondrial genomes of the ciliates Euplotes minuta and Euplotes crassus. BMC Genom. 2009, 10, 514. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.A. Ichthyophthirius multifiliis Fouquet and ichthyophthiriosis in freshwater teleosts. Adv. Parasitol. 2005, 59, 159–241. [Google Scholar] [PubMed]
- Preer, J.R., Jr.; Preer, L.B.; Jurand, A. Kappa and other endosymbionts in Paramecium aurelia. Bacteriol. Rev. 1974, 38, 113–163. [Google Scholar] [PubMed]
- Barth, D.; Berendonk, T.U. The mitochondrial genome sequence of the ciliate Paramecium caudatum reveals a shift in nucleotide composition and codon usage within the genus Paramecium. BMC Genom. 2011, 12, 272. [Google Scholar] [CrossRef] [PubMed]
- Brunk, C.F.; Lee, L.C.; Tran, A.B.; Li, J. Complete sequence of the mitochondrial genome of Tetrahymena thermophila and comparative methods for identifying highly divergent genes. Nucleic Acids Res. 2003, 31, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Burger, G.; Zhu, Y.; Littlejohn, T.G.; Greenwood, S.J.; Schnare, M.N.; Lang, B.F.; Gray, M.W. Complete sequence of the mitochondrial genome of Tetrahymena pyriformis and comparison with Paramecium aurelia mitochondrial DNA. J. Mol. Biol. 2000, 297, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.J. Mitochondrial genomes of the ciliates. Int. Rev. Cytol. 1992, 141, 1–64. [Google Scholar] [PubMed]
- Edqvist, J.; Burger, G.; Gray, M.W. Expression of mitochondrial protein-coding genes in Tetrahymena pyriformis. J. Mol. Biol. 2000, 297, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Swart, E.C.; Nowacki, M.; Shum, J.; Stiles, H.; Higgins, B.P.; Doak, T.G.; Schotanus, K.; Magrini, V.J.; Minx, P.; Mardis, E.R.; et al. The Oxytricha trifallax mitochondrial genome. Genome Biol. Evol. 2012, 4, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Moradian, M.M.; Beglaryan, D.; Skozylas, J.M.; Kerikorian, V. Complete mitochondrial genome sequence of three tetrahymena species reveals mutation hot spots and accelerated nonsynonymous substitutions in Ymf genes. PLoS ONE 2007, 2, E650. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, A.E.; Cummings, D.J. Replication of linear mitochondrial DNA from Paramecium: Sequence and structure of the initiation-end crosslink. Proc. Natl. Acad. Sci. USA 1981, 78, 7341–7345. [Google Scholar] [CrossRef] [PubMed]
- Rubanov, L.I.; Seliverstov, A.V.; Zverkov, O.A.; Lyubetsky, V.A. A method for identification of highly conserved elements and evolutionary analysis of superphylum Alveolata. BMC Bioinform. 2016, 17, 385. [Google Scholar] [CrossRef] [PubMed]
- Lyubetsky, V.A.; Gershgorin, R.A.; Seliverstov, A.V.; Gorbunov, K.Y. Algorithms for reconstruction of chromosomal structures. BMC Bioinform. 2016, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Kühn, K.; Bohne, A.-V.; Liere, K.; Weihe, A.; Börner, T. Arabidopsis phage-type RNA polymerases: Accurate in vitro transcription of organellar genes. Plant Cell 2007, 19, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, L.P. Origin of Group Identity: Viruses, Addiction and Cooperation; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Lyubetsky, V.A.; Seliverstov, A.V.; Zverkov, O.A. Transcription regulation of plastid genes involved in sulfate transport in Viridiplantae. BioMed Res. Int. 2013, 2013, 413450. [Google Scholar] [CrossRef] [PubMed]
- The Ciliophora Mitochondria-Encoded Protein Clusters. Available online: http://lab6.iitp.ru/mpc/cilio/ (accessed on 25 November 2016).
- Kim, S.; Kwack, K.B. A Fast Comparison Algorithm to Measure the Accuracy of Ortholog Clusters. Curr. Bioinf. 2016, 11, 324–329. [Google Scholar] [CrossRef]
- The ChromoGGL Programs. Available online: http://lab6.iitp.ru/en/chromoggl/ (accessed on 25 November 2016).
- Seliverstov, A.V. Monomials in quadratic forms. J. Appl. Ind. Math. 2013, 7, 431–434. [Google Scholar] [CrossRef]
- Gorbunov, K.Y.; Gershgorin, R.A.; Lyubetsky, V.A. Rearrangement and inference of chromosome structures. Mol. Biol. 2015, 49, 327–338. [Google Scholar] [CrossRef]
- Gorbunov, K.Y.; Lyubetsky, V.A. A linear algorithm of the shortest transformation of graphs under different operation costs. Inf. Process. 2016, 16, 223–236. (In Russian) [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Lyubetsky, V.A.; Seliverstov, A.V.; Zverkov, O.A. Elaboration of the homologous plastid-encoded protein families that separate paralogs in Magnoliophytes. Math. Biol. Bioinform. 2013, 8, 225–233. (In Russian) [Google Scholar] [CrossRef]
- Zverkov, O.A.; Seliverstov, A.V.; Lyubetsky, V.A. Plastid-encoded protein families specific for narrow taxonomic groups of algae and protozoa. Mol. Biol. 2012, 46, 717–726. [Google Scholar] [CrossRef]
- Zverkov, O.A.; Seliverstov, A.V.; Lyubetsky, V.A. A Database of plastid protein families from red algae and Apicomplexa and expression regulation of the moeB gene. BioMed Res. Int. 2015, 2015, 510598. [Google Scholar] [CrossRef] [PubMed]
- Zverkov, O.A.; Seliverstov, A.V.; Lyubetsky, V.A. Regulation of expression and evolution of genes in plastids of rhodophytic branch. Life 2016, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2014, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Philippe, H. Computing Bayes factors using thermodynamic integration. Syst. Biol. 2006, 55, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Brinkmann, H.; Philippe, H. Suppression of long-branch attraction artefacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 2007, 7 (Suppl. 1), S4. [Google Scholar] [CrossRef] [PubMed]
- Rota-Stabelli, O.; Yang, Z.; Telford, M.J. MtZoa: A general mitochondrial amino acid substitutions model for animal evolutionary studies. Mol. Phylogenet. Evol. 2009, 52, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Seliverstov, A.V.; Lysenko, E.A.; Lyubetsky, V.A. Rapid evolution of promoters for the plastome gene ndhF in flowering plants. Russ. J. Plant Physiol. 2009, 56, 838–845. [Google Scholar] [CrossRef]
- Lyubetsky, V.A.; Rubanov, L.I.; Seliverstov, A.V. Lack of conservation of bacterial type promoters in plastids of Streptophyta. Biol. Direct. 2010, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.Y.; Eddy, S.R.; Floden, E.W.; Gardner, P.P.; Jones, T.A.; Tate, J.; et al. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015, 43, D130–D137. [Google Scholar] [CrossRef] [PubMed]
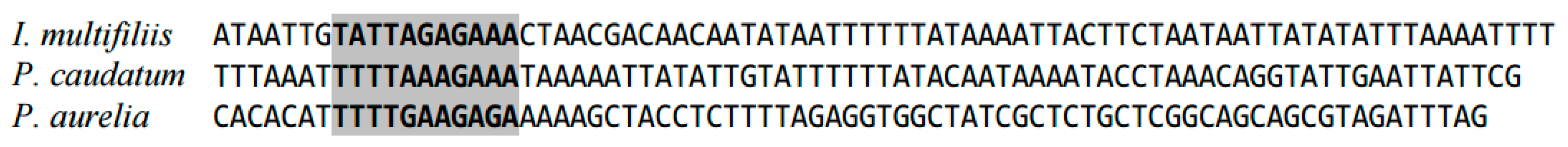

{kind=link}
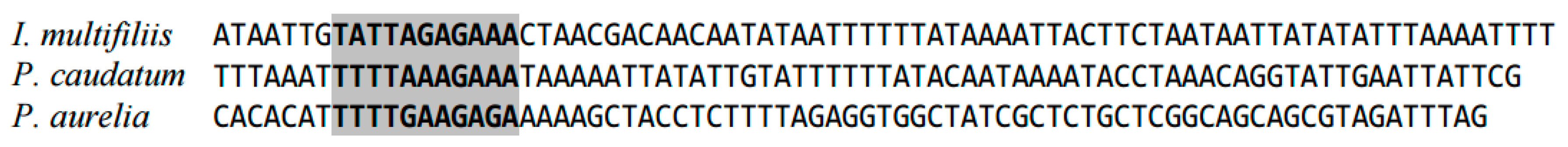{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | 1st Position | Sequence Fragments |
|---|---|---|
| HCE 287 | ||
| T. malaccensis | 2984 | AATTTAAATACTTGCATTAAGACTAATCGTGG |
| T. pigmentosa | 2988 | AATTTAAATACTTGCATTAAGACTAATCGTGG |
| T. pyriformis | 2988 | AATTTAAAAGCTTGCATTAATACTAATCTTGG |
| T. thermophila | 2943 | AATTTAAACACTTGCATTAAAACTAATCTTGG |
| HCE 299 | ||
| T. malaccensis | 10523 | GACACACCATATGAATTTAAATCATTAATAATTCAA |
| T. pigmentosa | 10558 | GATAAACCATATGAATTTAAATTATTACTAATTAAA |
| T. pyriformis | 10589 | GATAGACCATAAGAATTTAAGTCATTATTTATTCAA |
| T. thermophila | 10500 | GATAGACCATATGAATTTAAATCATTATTAATTCAA |
| HCE 290 | ||
| T. malaccensis | 4810 | ATAAAATAAGTTCTAAAAATGTGTATTAATTCCTTAAACATTTA |
| T. paravorax | 5270 | ATAAAATAAGTTCTTAATATATGTATAAATTCTTTAAACATTTA |
| T. pigmentosa | 4811 | ATAAAATATGTTCTAAAAATATGTATTAATTCTTTAAACATTTA |
| T. pyriformis | 4839 | ATAAAATAAGTTCTAAAAATATGTATCAATTCTTTAAACATTTA |
| HCE 234 | ||
| T. malaccensis | 4788 | TTTTTTTAAATATCTAAAAGTAATAAAATAAGTTCTAAA |
| T. paravorax | 5248 | TTTTTTTAAATATCTAAATGTTATAAAATAAGTTCTTAA |
| T. pigmentosa | 4789 | TTTTTTAAAATATCTAAAAGTTATAAAATATGTTCTAAA |
| T. pyriformis | 4817 | TTTTTTGATATATCTAAAAGTGATAAAATAAGTTCTAAA |
| T. thermophila | 4756 | TTTTTTTAAATATCTAAAAGTAATAAAATAAGTTCTAAA |
| HCE 138 | ||
| I. multifiliis | 1364 | TTTAGGTGCAGCTAT |
| I. multifiliis | 47702 | TATAGCTGCACCTAAAAAAAAAAAA |
| T. malaccensis | 27009 | AATAGCCGCACCTAAAAGAAAAAAATCTA |
| T. paravorax | 26884 | AATAGCTGCTCCAAAAAGAAAAAAATCAA |
| T. pigmentosa | 26364 | AATAGCCGCACCTAAAAGAAAAAAATCCA |
| T. pyriformis | 26770 | AATGGCCGCACCTAAAAGAAAAAAATCAA |
| T. thermophila | 27061 | AATAGCCGCACCTAAAAGAAAAAAATCTA |
| HCE 315 | ||
| T. malaccensis | 26891 | ATAACGTATTTACAATAAAAAAATAAT |
| T. pigmentosa | 26211 | TCAACGTATTTACAATAAAATAATAAA |
| T. pyriformis | 26678 | TTAACGAATTTACAATAAAAAAATAAA |
| T. thermophila | 26921 | TTAACGTATCTACAATAAAAAAATAAA |
| Locus | Species | Proteins | Clusters | Singletons |
|---|---|---|---|---|
| NC_015981.1 | Ichthyophthirius multifiliis | 41 | 39 | 2 |
| GQ903131.1 | Moneuplotes crassus | 29 | 25 | 4 |
| GQ903130.1 | Moneuplotes minuta | 36 | 30 | 6 |
| GU057832.1 | Nyctotherus ovalis | 35 | 13 | 22 |
| JN383843.1 | Oxytricha trifallax | 99 | 31 | 68 |
| NC_001324.1 | Paramecium aurelia | 46 | 41 | 5 |
| NC_014262.1 | Paramecium caudatum | 42 | 41 | 1 |
| NC_008337.1 | Tetrahymena malaccensis | 45 | 44 | 0 |
| NC_008338.1 | Tetrahymena paravorax | 44 | 43 | 1 |
| NC_008339.1 | Tetrahymena pigmentosa | 44 | 44 | 0 |
| NC_000862.1 | Tetrahymena pyriformis | 44 | 44 | 0 |
| NC_003029.1 | Tetrahymena thermophila | 45 | 44 | 0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gershgorin, R.A.; Gorbunov, K.Y.; Zverkov, O.A.; Rubanov, L.I.; Seliverstov, A.V.; Lyubetsky, V.A. Highly Conserved Elements and Chromosome Structure Evolution in Mitochondrial Genomes in Ciliates. Life 2017, 7, 9. https://doi.org/10.3390/life7010009
Gershgorin RA, Gorbunov KY, Zverkov OA, Rubanov LI, Seliverstov AV, Lyubetsky VA. Highly Conserved Elements and Chromosome Structure Evolution in Mitochondrial Genomes in Ciliates. Life. 2017; 7(1):9. https://doi.org/10.3390/life7010009
Chicago/Turabian StyleGershgorin, Roman A., Konstantin Yu. Gorbunov, Oleg A. Zverkov, Lev I. Rubanov, Alexandr V. Seliverstov, and Vassily A. Lyubetsky. 2017. "Highly Conserved Elements and Chromosome Structure Evolution in Mitochondrial Genomes in Ciliates" Life 7, no. 1: 9. https://doi.org/10.3390/life7010009
APA StyleGershgorin, R. A., Gorbunov, K. Y., Zverkov, O. A., Rubanov, L. I., Seliverstov, A. V., & Lyubetsky, V. A. (2017). Highly Conserved Elements and Chromosome Structure Evolution in Mitochondrial Genomes in Ciliates. Life, 7(1), 9. https://doi.org/10.3390/life7010009