
AglM and VNG1048G, Two Haloarchaeal UDP-Glucose Dehydrogenases, Show Different Salt-Related Behaviors


Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Growth Conditions
2.2. Plasmid Construction
2.3. Protein Purification
2.4. Size-Exclusion Chromatography
2.5. UDP-Glucose Dehydrogenase Activity Assay
2.6. Bioinformatics
2.7. Homology Modeling
3. Results
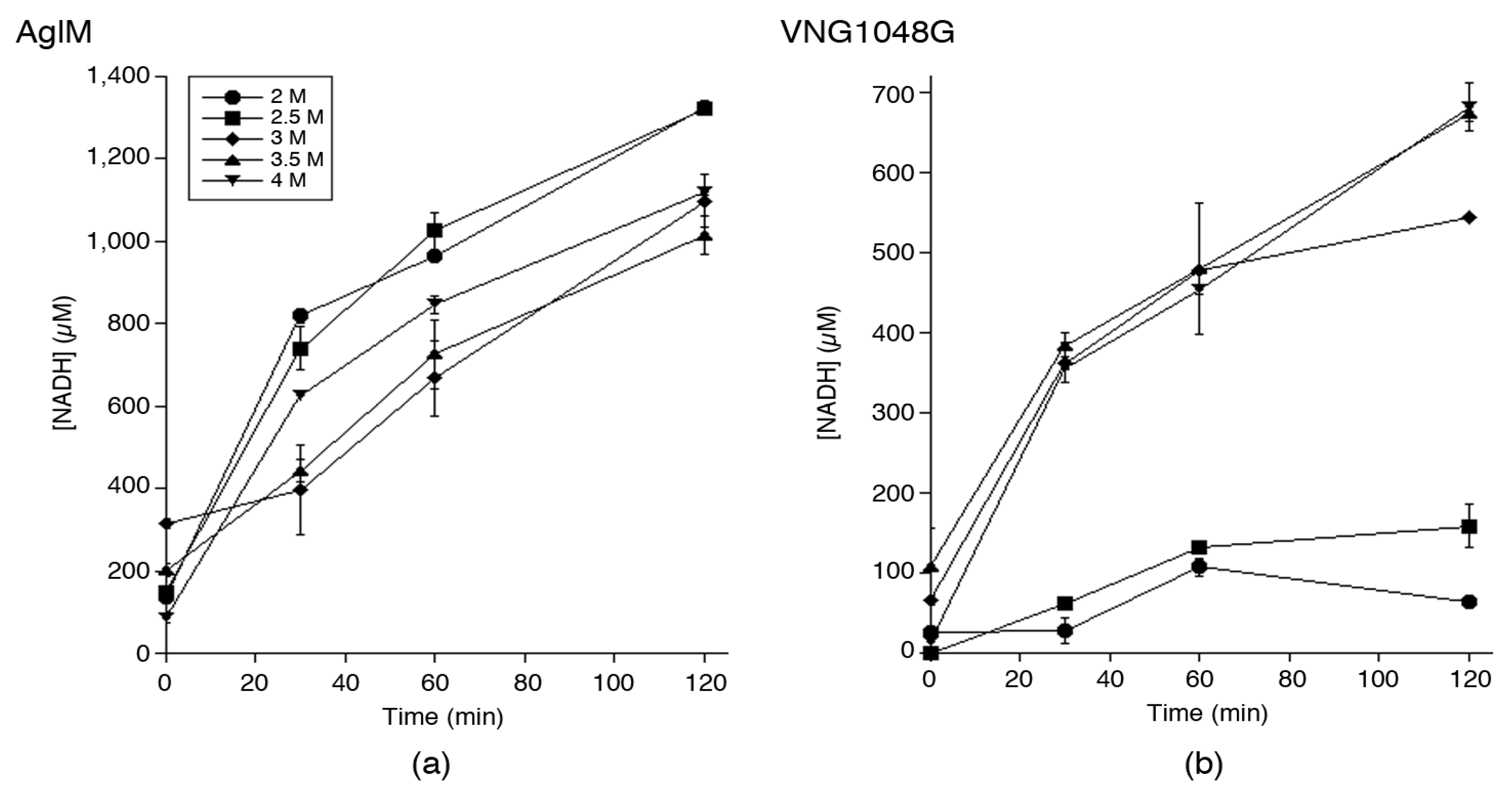
3.1. Hfx. volcanii AglM and Hbt. salinarum VNG1048G Activities Are Differently Affected by Decreasing Salinity
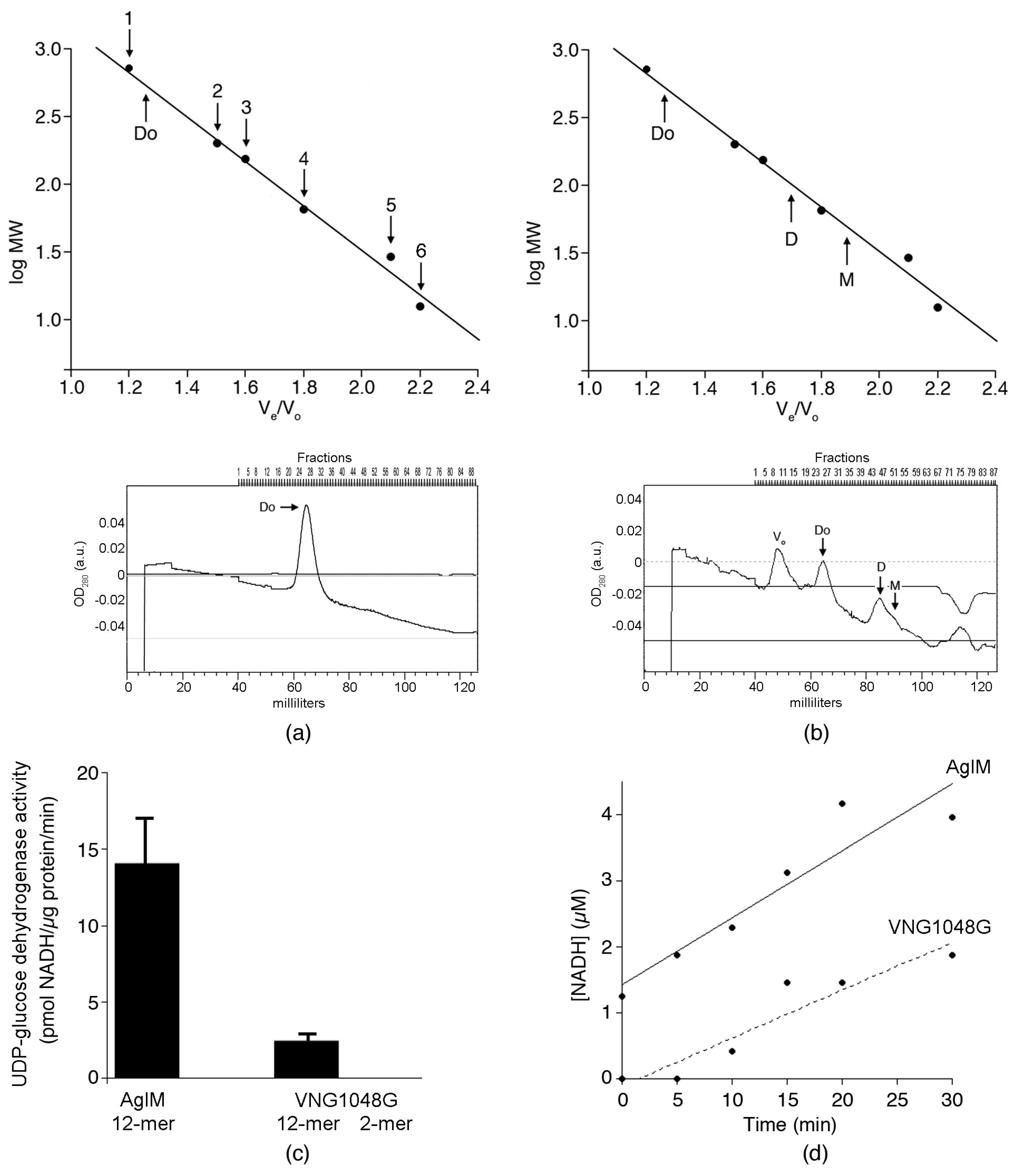
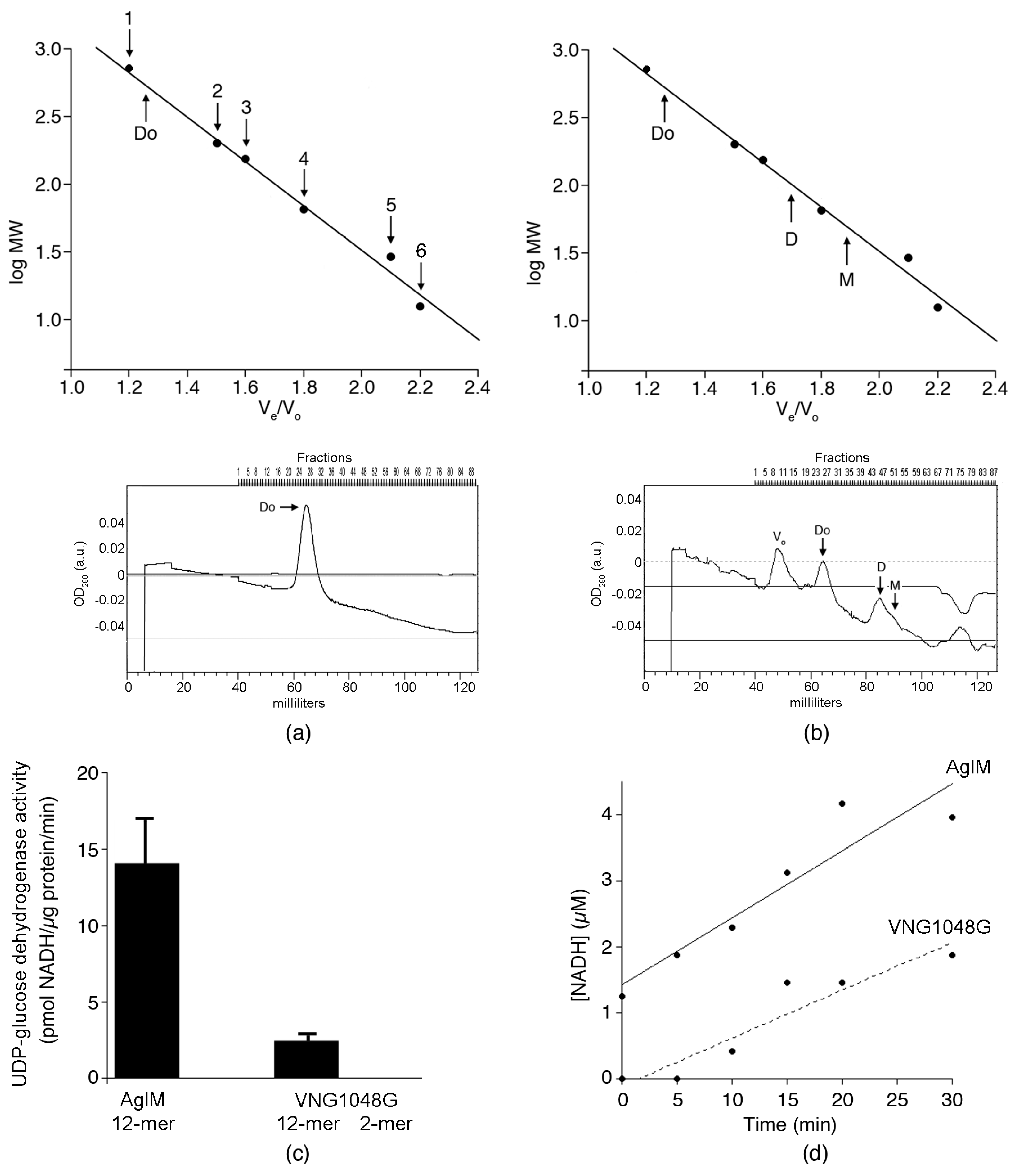
3.2. In 2 M NaCl, AglM, and VNG1048G Differ in Terms of Oligomeric Status
3.3. VNG1048G Contains More Acidic and Fewer Basic Residues than AglM
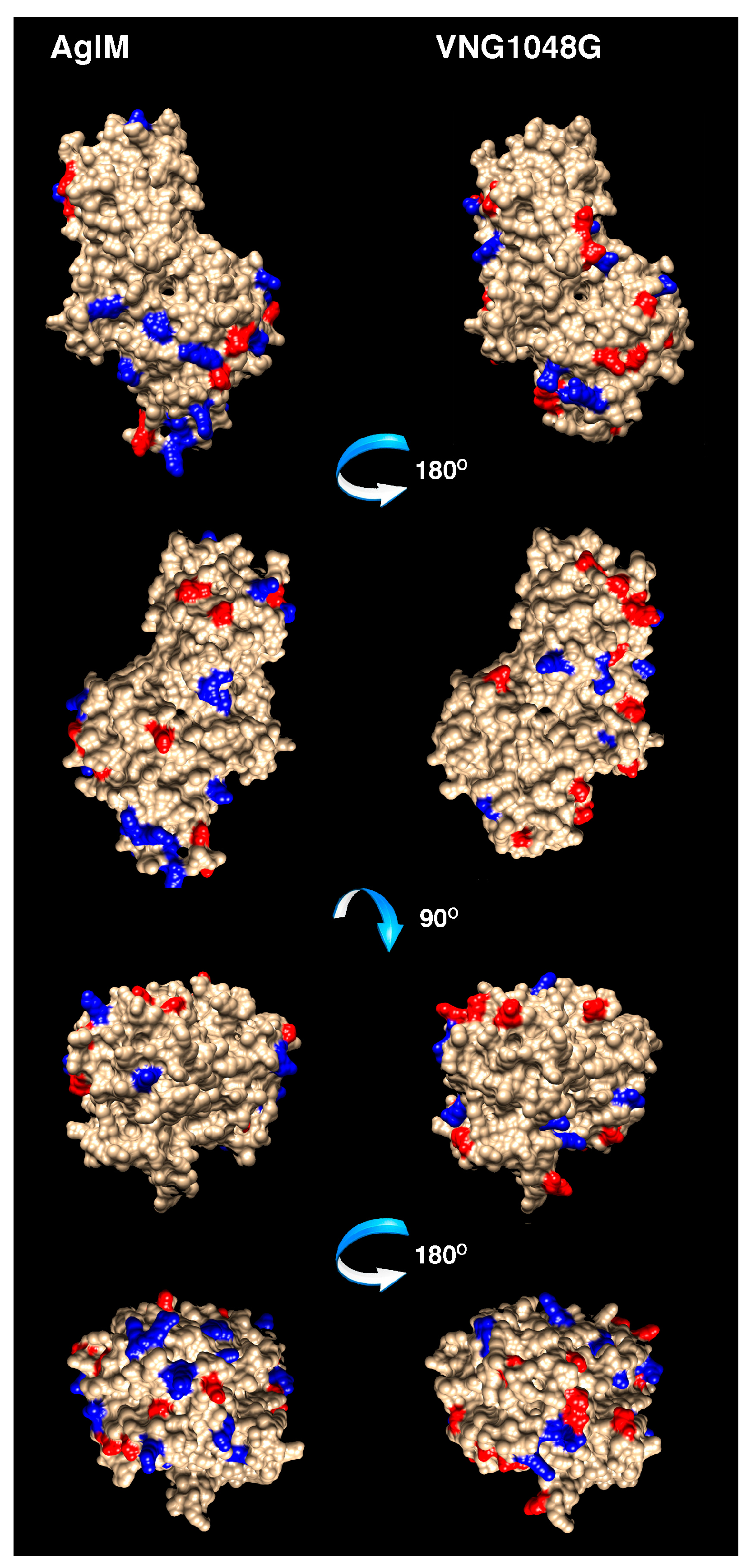
3.4. More Acidic and Fewer Basic Residues are Surface-Exposed in VNG1048G Than in AglM
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rothschild, L.J.; Mancinelli, R.L. Life in extreme environments. Nature 2001, 409, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Canganella, F.; Wiegel, J. Extremophiles: From abyssal to terrestrial ecosystems and possibly beyond. Naturwissenschaften 2011, 98, 253–279. [Google Scholar] [CrossRef] [PubMed]
- Lanyi, J.K. Salt-dependent properties of proteins from extremely halophilic bacteria. Bacteriol. Rev. 1974, 38, 272–290. [Google Scholar] [PubMed]
- Madern, D.; Ebel, C.; Zaccai, G. Halophilic adaptation of enzymes. Extremophiles 2000, 4, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, S.; Yoshimune, K.; Wakayama, M.; Moriguchi, M.; Nishikawa, K. Unique amino acid composition of proteins in halophilic bacteria. J. Mol. Biol. 2003, 327, 347–357. [Google Scholar] [CrossRef]
- Tadeo, X.; López-Méndez, B.; Trigueros, T.; Laín, A.; Castaño, D.; Millet, O. Structural basis for the aminoacid composition of proteins from halophilic archea. PLoS Biol. 2009, 12, e1000257. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Bag, S.K.; Das, S.; Harvill, E.T.; Dutta, C. Molecular signature of hypersaline adaptation: Insights from genome and proteome composition of halophilic prokaryotes. Genome Biol. 2008, 9, R70. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.J.; Lewis, H.; Trejo, E.; Winston, V.; Evilia, C. Protein adaptations in archaeal extremophiles. Archaea 2013, 2013, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zaccai, G.; Cendrin, F.; Haik, Y.; Borochov, N.; Eisenberg, H. Stabilization of halophilic malate dehydrogenase. J. Mol. Biol. 1989, 208, 491–500. [Google Scholar] [CrossRef]
- Dym, O.; Mevarech, M.; Sussman, J.L. Structural features that stabilize halophilic malate dehydrogenase from an archaebacterium. Science 1995, 267, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Frolow, F.; Harel, M.; Sussman, J.L.; Mevarech, M.; Shoham, M. Insights into protein adaptation to a saturated salt environment from the crystal structure of a halophilic 2Fe-2S ferredoxin. Nat. Struct. Biol. 1996, 3, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Danson, M.J.; Hough, D.W. Structure, function and stability of enzymes from the Archaea. Trends Microbiol. 1998, 6, 307–314. [Google Scholar] [CrossRef]
- Mevarech, M.; Frolow, F.; Gloss, L.M. Halophilic enzymes: Proteins with a grain of salt. Biophys. Chem. 2000, 86, 155–164. [Google Scholar] [CrossRef]
- Britton, K.L.; Baker, P.J.; Fisher, M.; Ruzheinikov, S.; Gilmour, D.J.; Bonete, M.J.; Ferrer, J.; Pire, C.; Esclapez, J.; Rice, D.W. Analysis of protein solvent interactions in glucose dehydrogenase from the extreme halophile Haloferax mediterranei. Proc. Natl. Acad. Sci. USA 2006, 103, 4846–4851. [Google Scholar] [CrossRef] [PubMed]
- Siglioccolo, A.; Paiardini, A.; Piscitelli, M.; Pascarella, S. Structural adaptation of extreme halophilic proteins through decrease of conserved hydrophobic contact surface. BMC Struct. Biol. 2011, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Bohm, G.; Jaenicke, R. A structure-based model for the halophilic adaptation of dihydrofolate reductase from Halobacterium volcanii. Protein Eng. 1994, 7, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Elcock, A.H.; McCammon, J.A. Electrostatic contributions to the stability of halophilic proteins. J. Mol. Biol. 1998, 280, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Deole, R.; Challacombe, J.; Raiford, D.W.; Hoff, W.D. An extremely halophilic proteobacterium combines a highly acidic proteome with a low cytoplasmic potassium content. J. Biol. Chem. 2013, 288, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G.; Merlino, A. Molecular bases of protein halotolerance. Biochim. Biophys. Acta 2014, 1844, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Ortega, G.; Diercks, T.; Millet, O. Halophilic protein adapation results from syntergistic residue-ion interactions in the folded and unfolded states. Chem. Biol. 2015, 22, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Jarrell, K.F.; Ding, Y.; Meyer, B.H.; Albers, S.V.; Kaminski, L.; Eichler, J. N-linked glycosylation in Archaea: A structural, functional and genetic analysis. Microbiol. Mol. Biol. Rev. 2014, 78, 304–341. [Google Scholar] [CrossRef] [PubMed]
- Eichler, J. Extreme sweetness: Protein glycosylation in Archaea. Nat. Rev. Microbiol. 2013, 11, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, L.; Guan, Z.; Yurist-Doutsch, S.; Eichler, J. Two distinct N-glycosylation pathways process the Haloferax volcanii S-layer glycoprotein upon changes in environmental salinity. mBio 2013, 4, e00716–e00713. [Google Scholar] [CrossRef] [PubMed]
- Lechner, J.; Wieland, F. Structure and biosynthesis of prokaryotic glycoproteins. Annu. Rev. Biochem. 1989, 58, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Kandiba, L.; Eichler, J. Deciphering a pathway of Halobacterium salinarum N-glycosylation. MicrobiologyOpen 2015, 4, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Yurist-Doutsch, S.; Magidovich, H.; Ventura, V.V.; Hitchen, P.G.; Dell, A.; Eichler, J. N-glycosylation in Archaea: On the coordinated actions of Haloferax volcanii AglF and AglM. Mol. Microbiol. 2010, 75, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Mullakhanbhai, M.F.; Larsen, H. Halobacterium volcanii spec. nov., a Dead Sea halobacterium with a moderate salt requirement. Arch. Microbiol. 1975, 104, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Stoeckenius, W.; Rowen, R. A morphological study of Halobacterium halobium and its lysis in media of low salt concentration. J. Cell Biol. 1967, 34, 365–393. [Google Scholar] [CrossRef] [PubMed]
- Mevarech, M.; Werczberger, R. Genetic transfer in Halobacterium volcanii. J. Bacteriol. 1985, 162, 461–462. [Google Scholar] [PubMed]
- Plavner, N.; Eichler, J. Defining the topology of the N-glycosylation pathway in the halophilic archaeon Haloferax volcanii. J. Bacteriol. 2008, 190, 8045–8052. [Google Scholar] [CrossRef] [PubMed]
- Reuter, C.J.; Maupin-Furlow, J.A. Analysis of proteasome-dependent proteolysis in Haloferax volcanii cells, using short-lived green fluorescent proteins. Appl. Environ. Microbiol. 2004, 70, 7530–7538. [Google Scholar] [CrossRef] [PubMed]
- Irihimovitch, V.; Eichler, J. Post-translational secretion of fusion proteins in the halophilic archaea Haloferax volcanii. J. Biol. Chem. 2003, 278, 12881–12887. [Google Scholar] [CrossRef] [PubMed]
- Kandiba, L.; Aitio, O.; Helin, J.; Guan, Z.; Permi, P.; Bamford, D.H.; Eichler, J.; Roine, E. Diversity in prokaryotic glycosylation: An archaeal-derived N-linked glycan contains legionaminic acid. Mol. Microbiol. 2012, 84, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.E.; Mosimann, S.C.; van de Rijn, I.; Tanner, M.E.; Strynadka, N.C. The first structure of UDP-glucose dehydrogenase reveals the catalytic residues necessary for the two-fold oxidation. Biochemistry 2000, 39, 7012–7023. [Google Scholar] [CrossRef] [PubMed]
- Egger, S.; Chaikuad, A.; Kavanagh, K.L.; Oppermann, U.; Nidetzky, B. Structure and mechanism of human UDP-glucose 6-dehydrogenase. J. Biol. Chem. 2011, 286, 23877–23887. [Google Scholar] [CrossRef] [PubMed]
- Egger, S.; Chaikuad, A.; Kavanagh, K.L.; Oppermann, U.; Nidetzky, B. UDP-glucose dehydrogenase: Structure and function of a potential drug target. Biochem. Soc. Trans. 2010, 38, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, H.; Kawai, T.; Yoneda, K.; Ohshima, T. Structure of a UDP-glucose dehydrogenase from the hyperthermophilic archaeon Pyrobaculum islandicum. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Sommer, B.J.; Barycki, J.J.; Simpson, M.A. Characterization of human UDP-glucose dehydrogenase. CYS-276 is required for the second of two successive oxidations. J. Biol. Chem. 2004, 279, 23590–23596. [Google Scholar] [CrossRef] [PubMed]
- Soppa, J. From genomes to function: Haloarchaea as model organisms. Microbiology 2006, 152, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Leigh, J.A.; Albers, S.V.; Atomi, H.; Allers, T. Model organisms for genetics in the domain Archaea: Methanogens, halophiles, Thermococcales and Sulfolobales. FEMS Microbiol. Rev. 2011, 35, 577–608. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.; Popescu, A.O.; Borges, P.; Mil-Homens, D.; Moreira, L.M.; Sa-Correia, I.; Fialho, A.M.; Frazao, C. Structure of Burkholderia cepacia UDP-glucose dehydrogenase (UGD) BceC and role of Tyr10 in final hydrolysis of UGD thioester intermediate. J. Bacteriol. 2011, 193, 3978–3987. [Google Scholar] [CrossRef] [PubMed]
- Satomura, T.; Kusumi, K.; Ohshima, T.; Sakuraba, H. Identification and characterization of UDP-glucose dehydrogenase from the hyperthermophilic archaon, Pyrobaculum islandicum. Biosci. Biotechnol. Biochem. 2011, 75, 2049–2051. [Google Scholar] [CrossRef] [PubMed]
- Gainey, P.A.; Pestell, T.C.; Phelps, C.F. A study of the subunit structure and the thiol reactivity of bovine liver uridine diphosphate glucose dehydrogenase. Biochem. J. 1972, 129, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Franzen, J.S.; Ishman, R.; Feingold, D.S. Half-of-the-sites reactivity of bovine liver uridine diphosphoglucose dehydrogenase toward iodoacetate and iodoacetamide. Biochemistry 1976, 15, 5665–5671. [Google Scholar] [CrossRef] [PubMed]
- Franzen, J.S.; Ashcom, J.; Marchetti, P.; Cardamone, J.J., Jr.; Feingold, D.S. Induced versus pre-existing asymmetry models for the half-of-the-sites reactivity effect in bovine liver uridine diphosphoglucose dehydrogenase. Biochim. Biophys. Acta 1980, 614, 242–255. [Google Scholar] [CrossRef]
- Bonneté, F.; Ebel, C.; Zaccai, G.; Eisenberg, H. Biophysical study of halophilic malate dehydrogenase in solution: Revised subunit structure and solvent interactions of native and recombinant enzyme. J. Chem. Soc. Faraday Trans. 1993, 89, 2659–2666. [Google Scholar] [CrossRef]
- Yamamura, A.; Ichimura, T.; Kamekura, M.; Mizuki, T.; Usami, R.; Makino, T.; Ohtsuka, J.; Miyazono, K.; Okai, M.; Nagata, K.; et al. Molecular mechanism of distinct salt-dependent enzyme activity of two halophilic nucleoside diphosphate kinases. Biophys. J. 2009, 96, 4692–4700. [Google Scholar] [CrossRef] [PubMed]
- Timpson, L.M.; Liliensiek, A.K.; Alsafadi, D.; Cassidy, J.; Sharkey, M.A.; Liddell, S.; Allers, T.; Paradisi, F. A comparison of two novel alcohol dehydrogenase enzymes (ADH1 and ADH2) from the extreme halophile Haloferax volcanii. Appl. Microbiol. Biotechnol. 2013, 97, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Madern, D.; Pfister, C.; Zaccai, G. Mutation at a single acidic amino acid enhances the halophilic behaviour of malate dehydrogenase from Haloarcula marismortui in physiological salts. Eur. J. Biochem. 1995, 230, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Russell, R.J.; Hough, D.W.; Danson, M.J. Site-directed mutagenesis and halophilicity of dihydrolipoamide dehydrogenase from the halophilic archaeon, Haloferax volcanii. Eur. J. Biochem. 1997, 248, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Irimia, A.; Ebel, C.; Madern, D.; Richard, S.B.; Cosenza, L.W.; Zaccai, G.; Vellieux, F.M. The oligomeric states of Haloarcula marismortui malate dehydrogenase are modulated by solvent components as shown by crystallographic and biochemical studies. J. Mol. Biol. 2003, 326, 859–873. [Google Scholar] [CrossRef]
- Esclapez, J.; Pire, C.; Bautista, V.; Martínez-Espinosa, R.M.; Ferrer, J.; Bonete, M.J. Analysis of acidic surface of Haloferax mediterranei glucose dehydrogenase by site-directed mutagenesis. FEBS Lett. 2007, 581, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Christian, J.H.; Waltho, J.A. Solute concentrations within cells of halophilic and non-halophilic bacteria. Biochim. Biophys. Acta 1962, 65, 506–508. [Google Scholar] [CrossRef]
- Ginzburg, M.; Sachs, L.; Ginzburg, B.Z. Ion metabolism in a Halobacterium. I. Influence of age of culture on intracellular concentrations. J. Gen. Physiol. 1970, 55, 187–207. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Description | Sequence 1 |
|---|---|---|
| VNG1048G NdeI | Forward primer introducing an NdeI site at the start of VNG1048G for cloning into plasmids pWL-CBD and pET24b | ccccatatgGACGTGAGCATCGTTGGGAGTGGG |
| VNG1048G StuI rev | Reverse primer introducing a StuI site at the end of VNG1048G for cloning into plasmid pWL-CBD | gggaggcctCTACCAGGTGAGCCCGTCGTAGGTC |
| VNG1048G XhoI rev | Reverse primer introducing a XhoI site at the end of VNG1048G for cloning into plasmid pET24b | gggctcgagCCAGGTGAGCCCGTCGTAGGTCAGG |
| aglM NdeI | Forward primer introducing an NdeI site at the start of aglM for cloning into plasmids pWL-CBD and pET24b | gggcatatgGAACTCAGTATCATCGGGAG |
| aglM KpnI rev | Reverse primer introducing a KpnI site at the end of aglM for cloning into plasmid pWL-CBD | cccggtaccTCAGACGAGCGACTCGTAGTCG |
| aglM XhoI rev | Reverse primer introducing a XhoI site at the end of aglM for cloning into plasmid pET24b | cccggtaccGACGAGCGACTCGTAGTCGAG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandiba, L.; Eichler, J. AglM and VNG1048G, Two Haloarchaeal UDP-Glucose Dehydrogenases, Show Different Salt-Related Behaviors. Life 2016, 6, 31. https://doi.org/10.3390/life6030031
Kandiba L, Eichler J. AglM and VNG1048G, Two Haloarchaeal UDP-Glucose Dehydrogenases, Show Different Salt-Related Behaviors. Life. 2016; 6(3):31. https://doi.org/10.3390/life6030031
Chicago/Turabian StyleKandiba, Lina, and Jerry Eichler. 2016. "AglM and VNG1048G, Two Haloarchaeal UDP-Glucose Dehydrogenases, Show Different Salt-Related Behaviors" Life 6, no. 3: 31. https://doi.org/10.3390/life6030031