
The Effect of Limited Diffusion and Wet–Dry Cycling on Reversible Polymerization Reactions: Implications for Prebiotic Synthesis of Nucleic Acids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
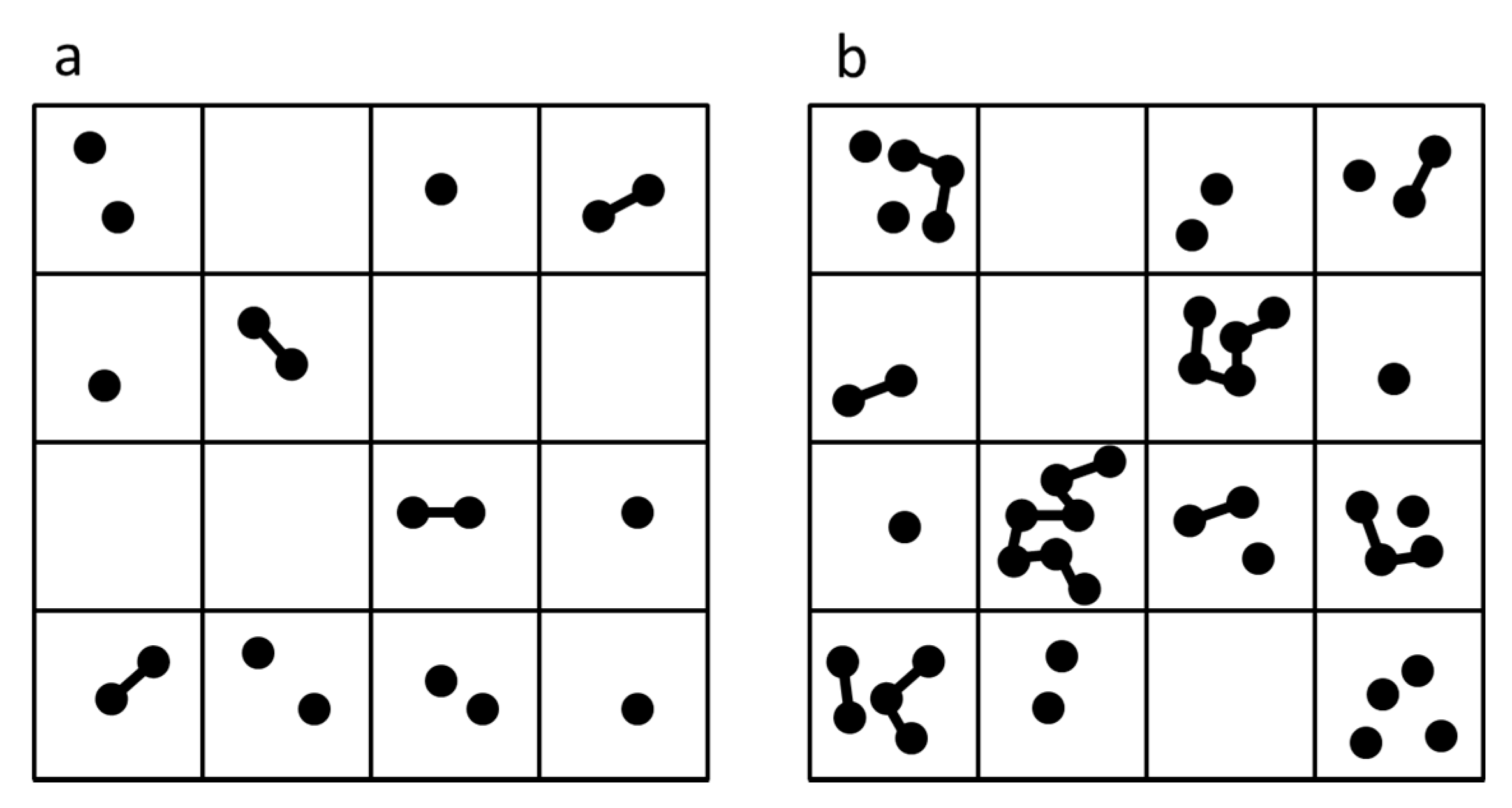
3.1. Dimer Formation Model
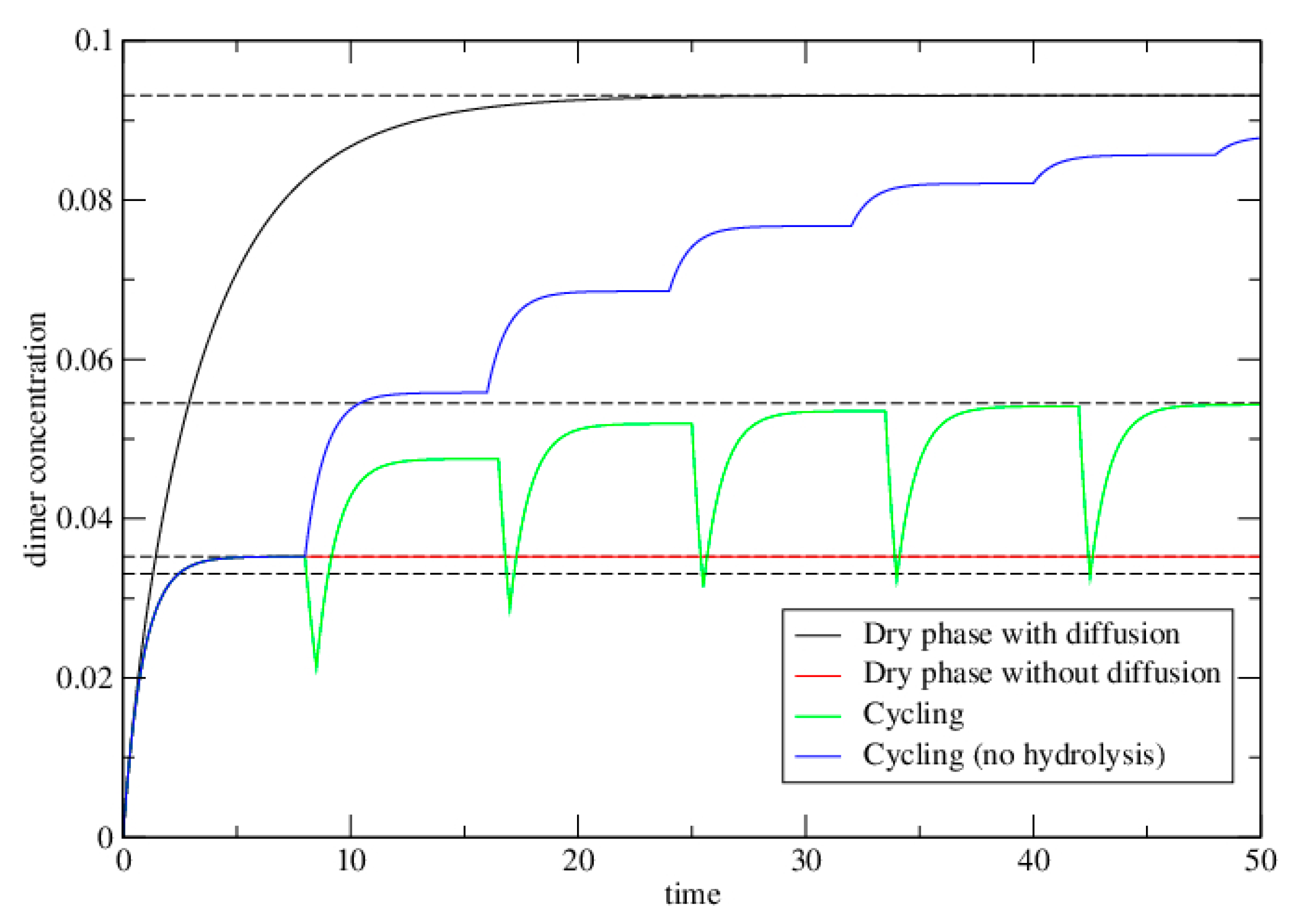
3.1.1. Case 1—Dry Phase with Diffusion Permitted
3.1.2. Case 2—Dry Phase without Diffusion
3.1.3. Case 3—Alternating Dry and Wet Cycles
3.2. Polymerization Model
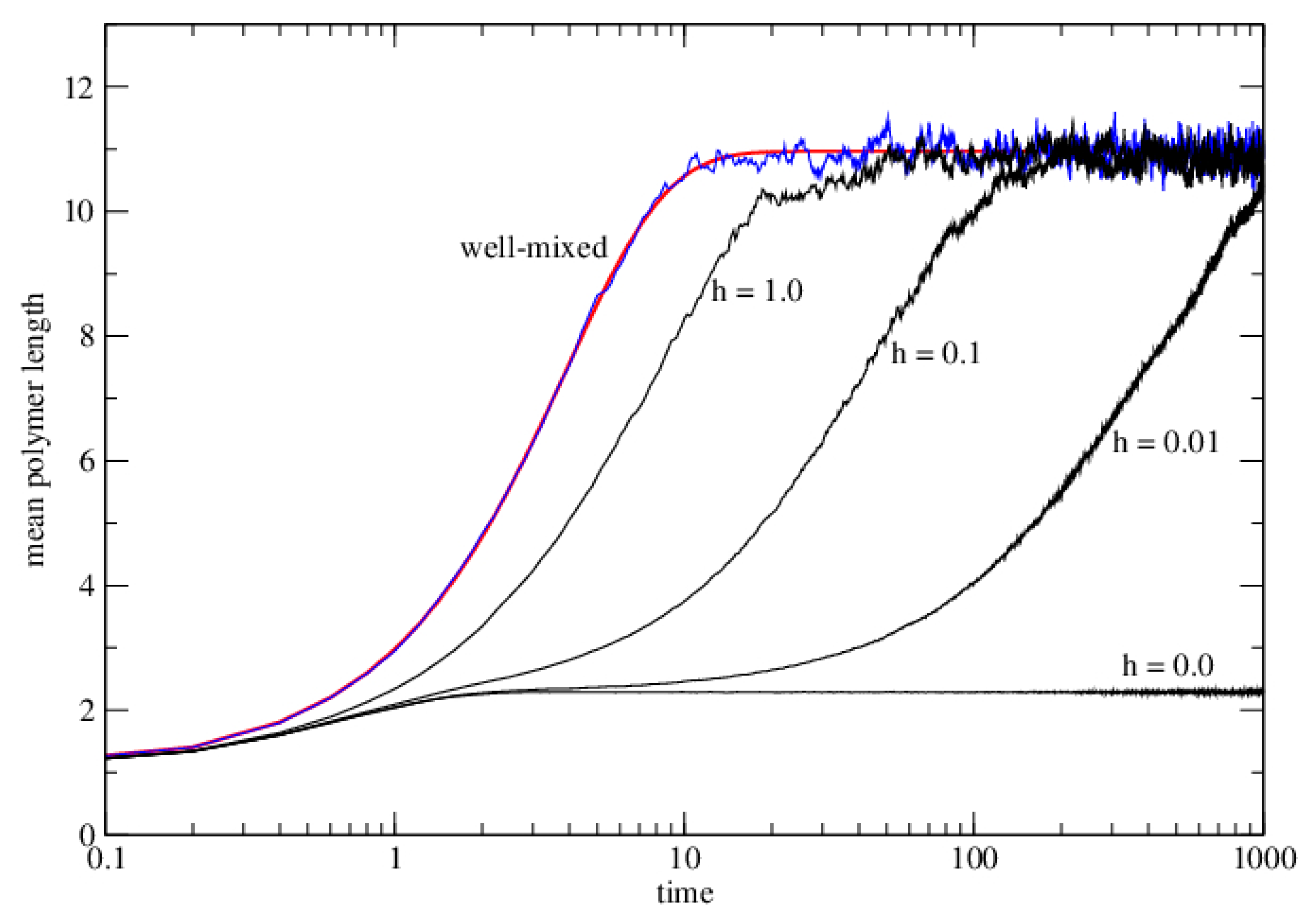
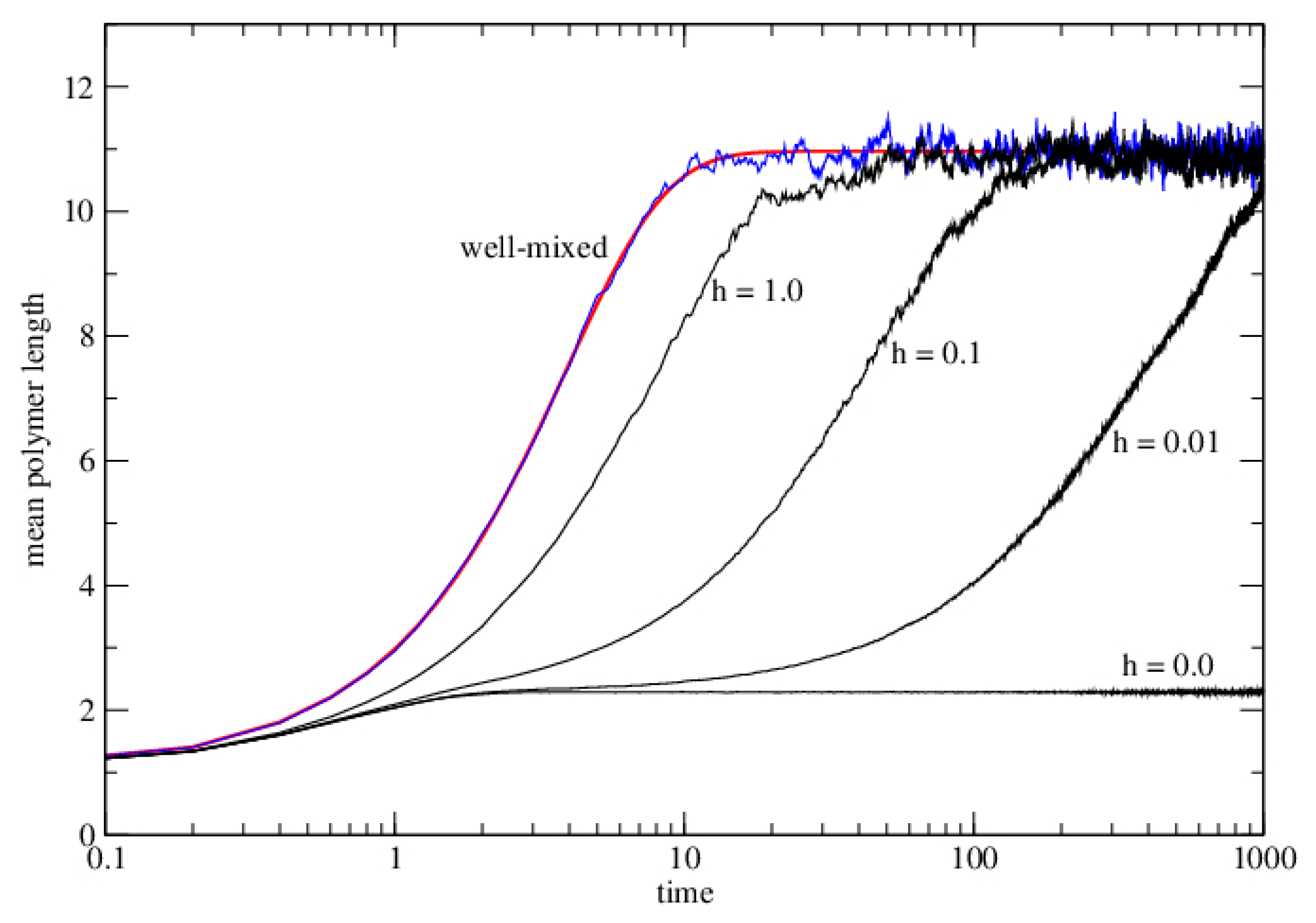
3.2.1. Well-Mixed Case
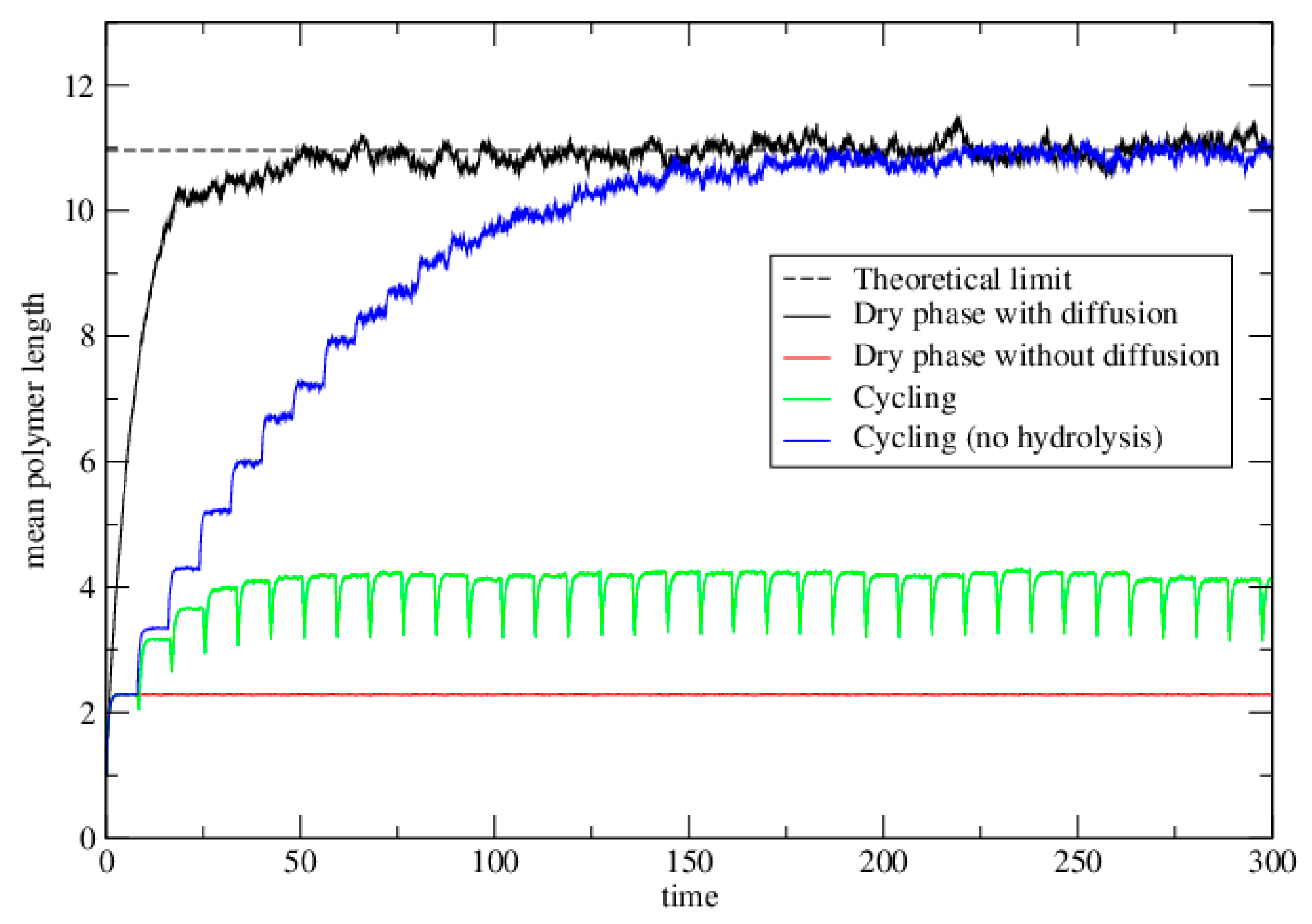
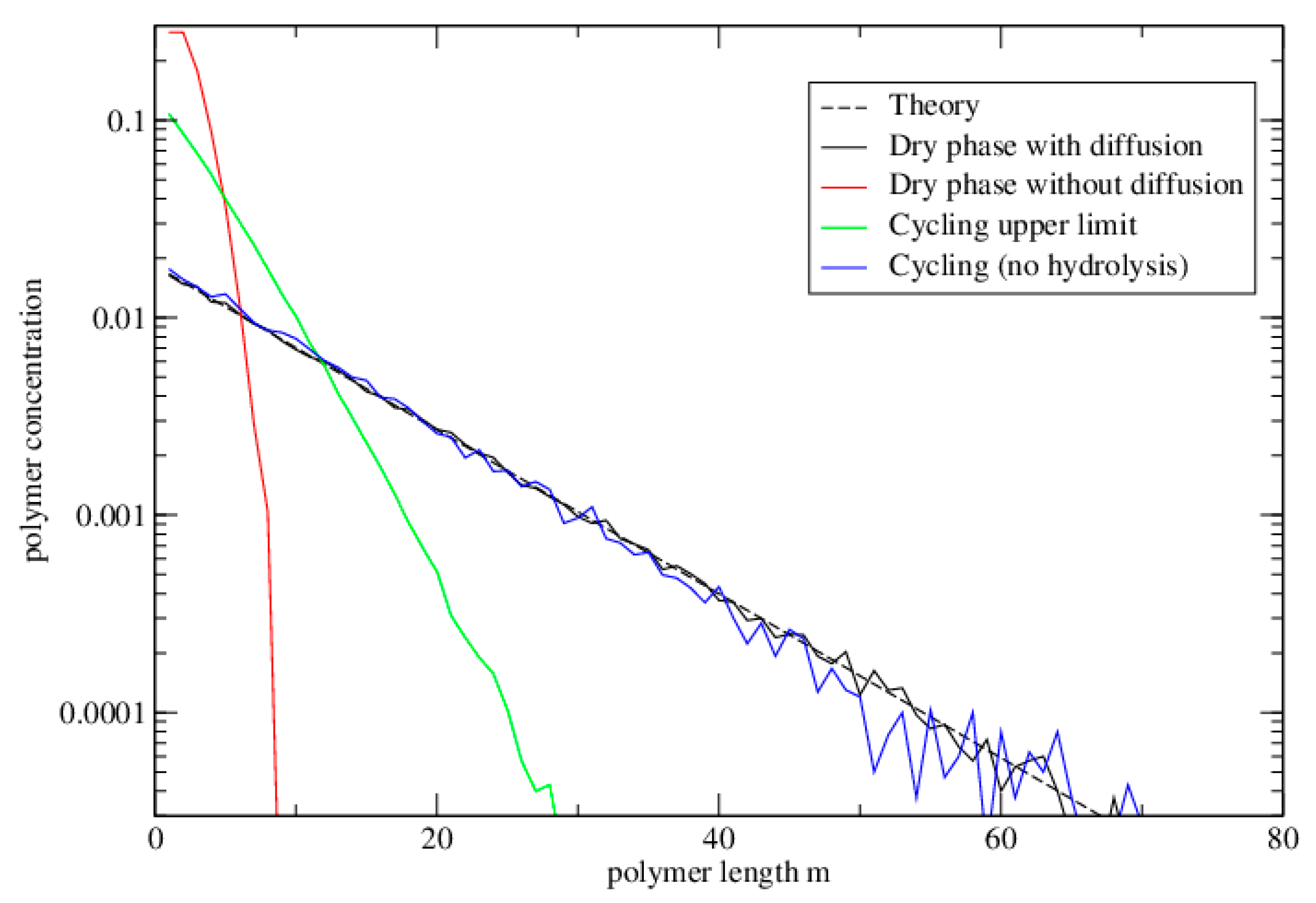
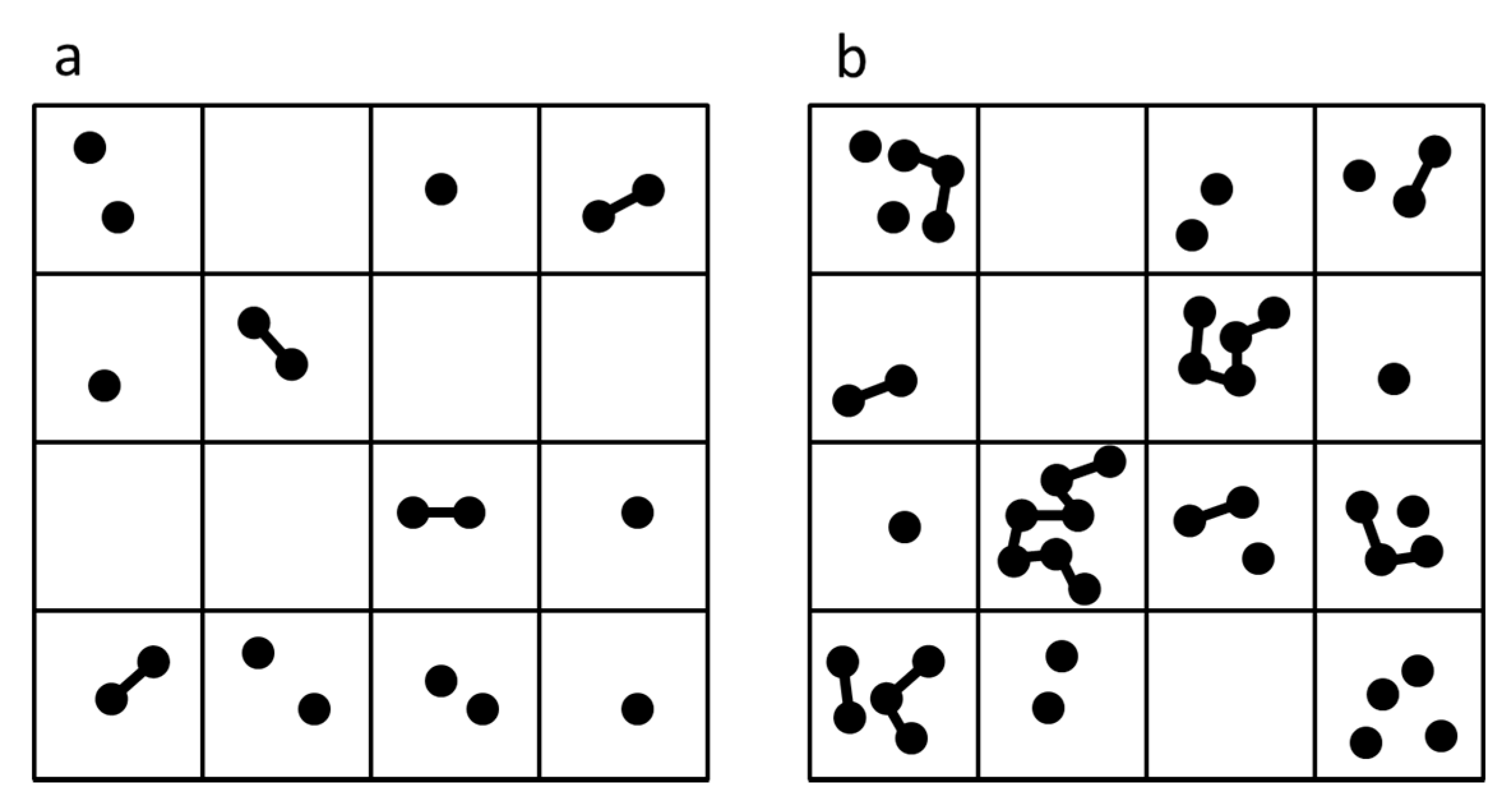
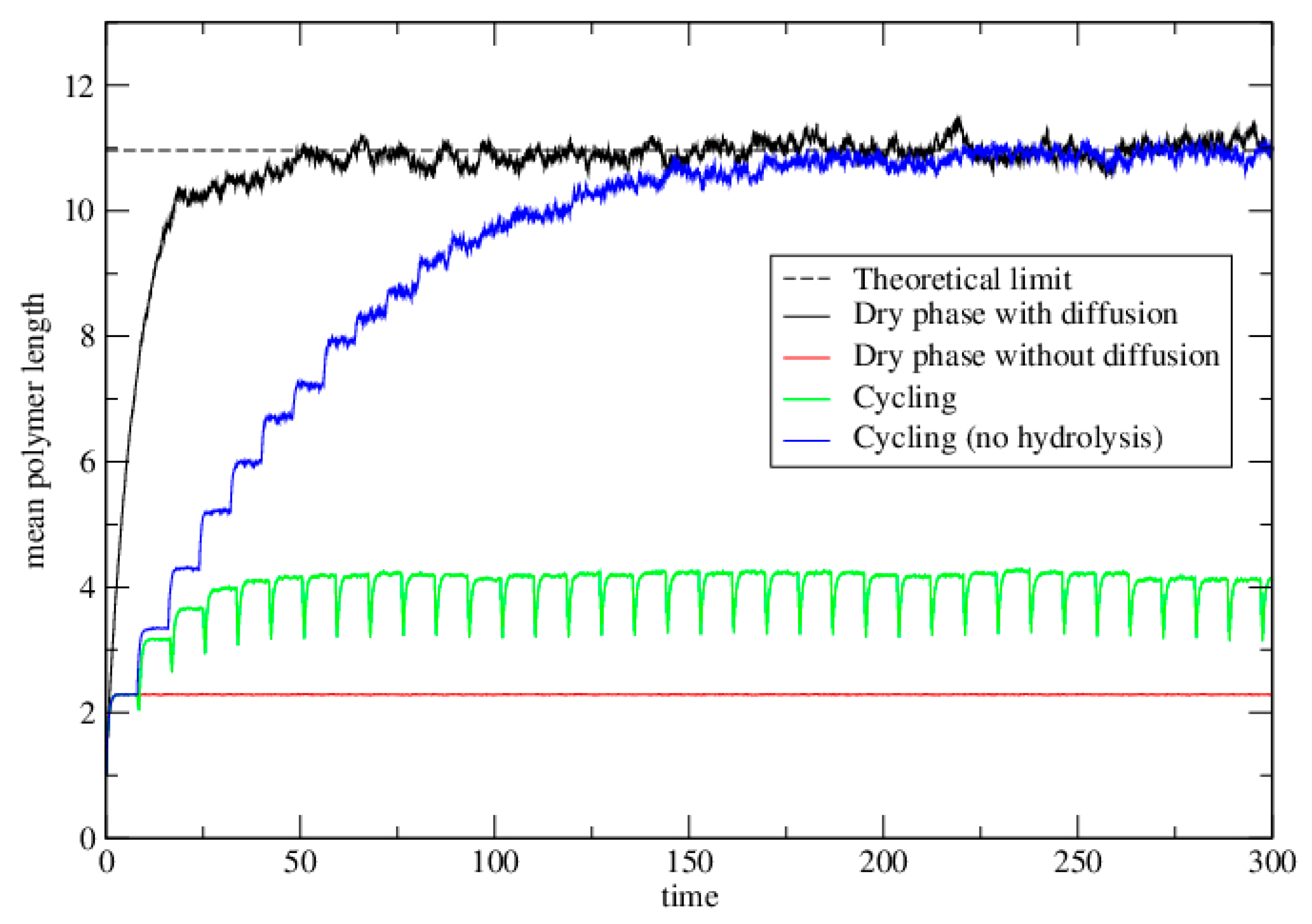
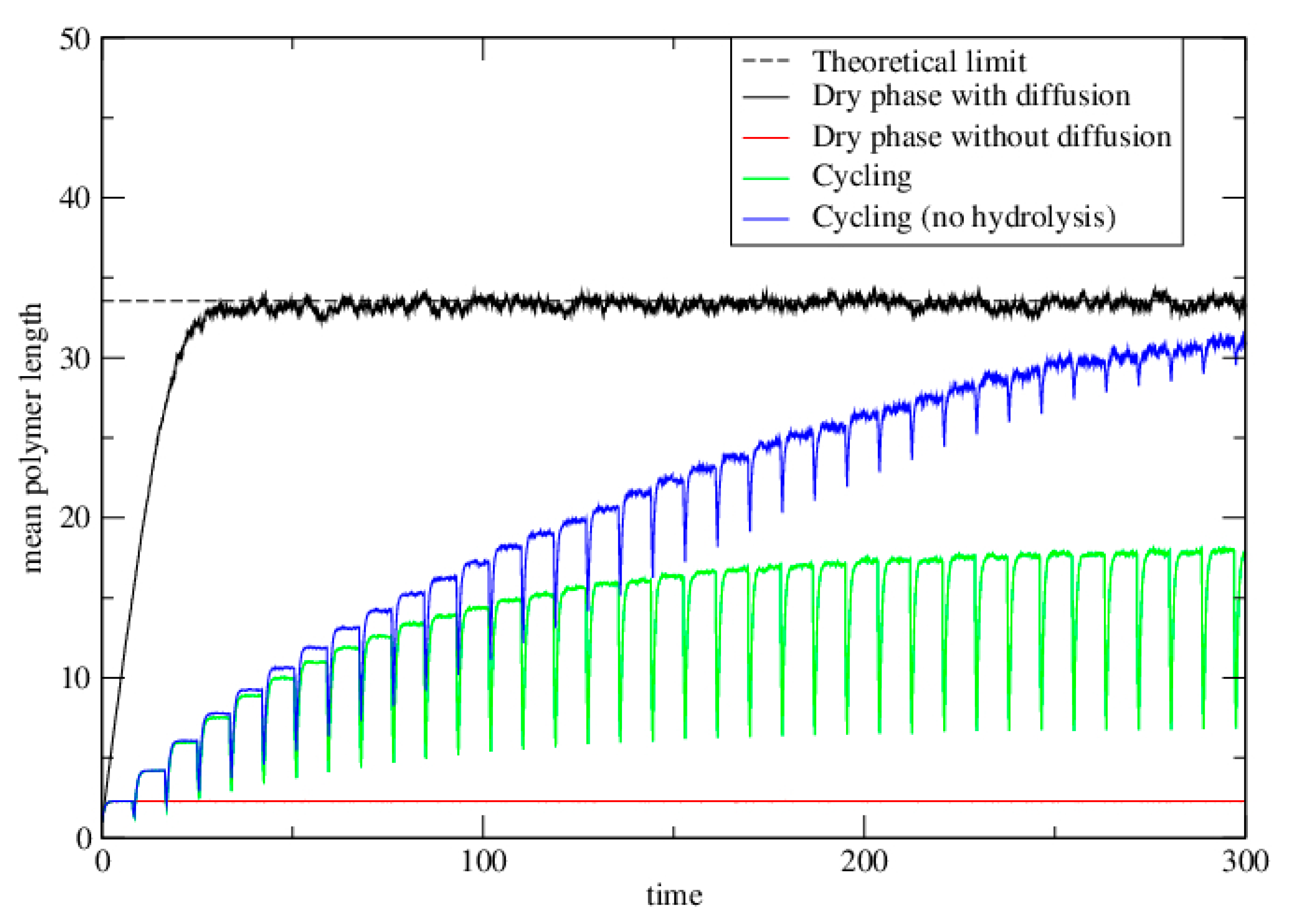
3.2.2. Lattice Model for Polymerization
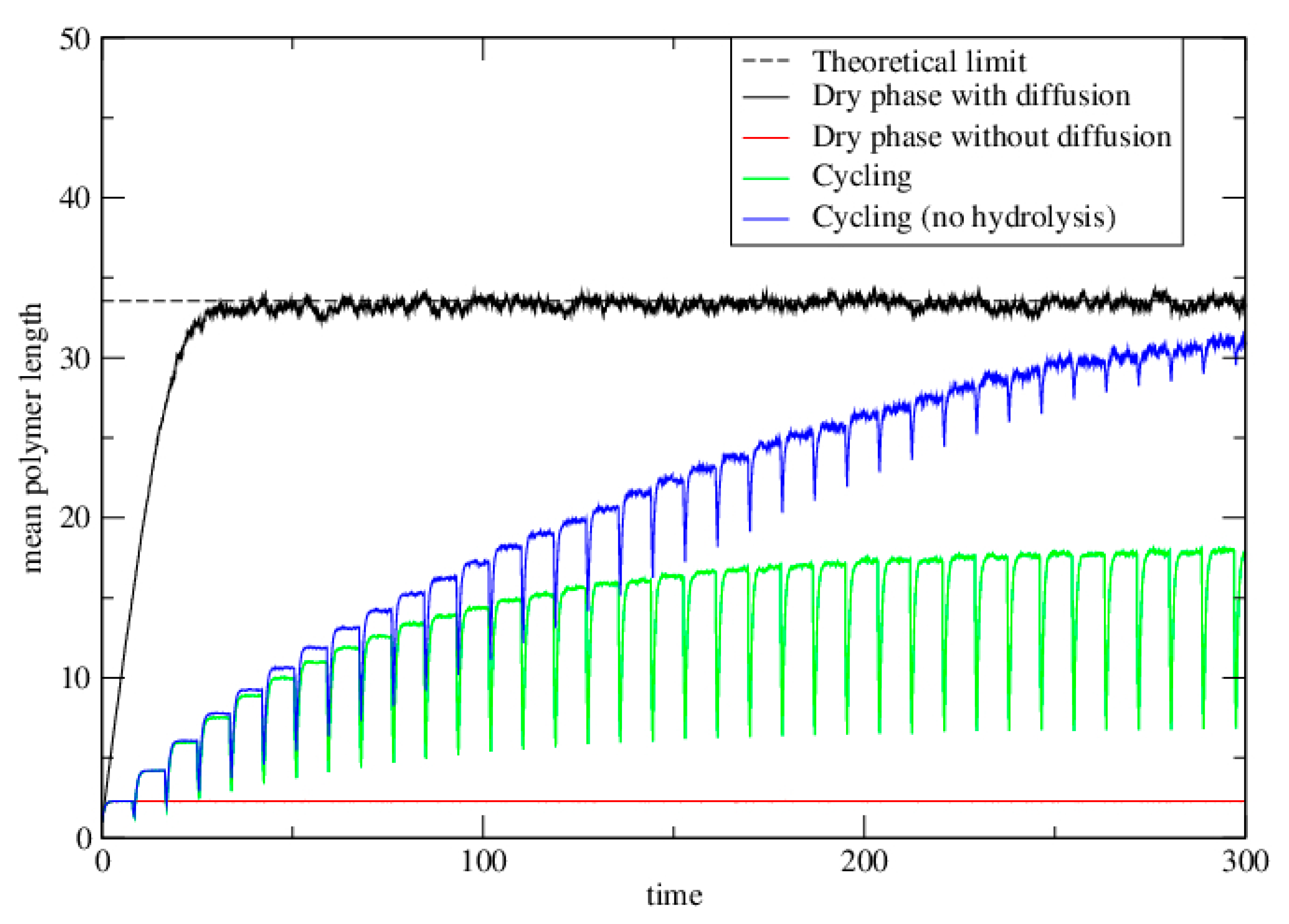
3.2.3. Lattice Model for Polymerization with Wet–Dry Cycling
3.3. Polymerization Model with Monomer Influx
4. Discussion
Acknowledgments
Conflicts of Interest
References
- Joyce, G.F. The antiquity of RNA-based evolution. Nature 2002, 418, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G.; Lehman, N. The RNA World: Molecular cooperation at the origins of life. Nat. Rev. Genet. 2015, 16, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.J.; von Kiedrowski, G.; Lehman, N. Systems chemistry on ribozyme self-construction: Evidence for anabolic autocatalysis in a recombination network. Angew. Chem. Int. Ed. 2008, 47, 8424–8428. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, T.A.; Joyce, G.F. Self-sustained replication of an RNA enzyme. Science 2009, 323, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Attwater, J.; Wochner, A.; Holliger, P. In-ice evolution of RNA polymerase activity. Nat. Chem. 2013, 5, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P.B.; Steitz, T.A. The structural basis of ribosome activity in peptide bond synthesis. Science 2000, 289, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Higgs, P.G. Origin of Self-replicating Biopolymers: Autocatalytic Feedback can Jump-start the RNA World. J. Mol. Evol. 2009, 69, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Higgs, P.G. Comparison of the roles of nucleotide synthesis, polymerization and recombination in the origin of autocatalytic sets of RNAs. Astrobiology 2011, 11, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Higgs, P.G.; Wu, M. The importance of stochastic transitions for the origin of life. Orig. Life. Evol. Biopsph. 2012, 42, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Higgs, P.G. The origin of life is a spatially localized stochastic transition. Biol. Direct 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.A.; Huynh, C.; Higgs, P.G. The origin and spread of a cooperative replicase in a prebiotic chemical system. J. Theor. Biol. 2015, 364, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Cafferty, B.J.; Hud, N.V. Abiotic synthesis of RNA in water: A common goal of prebiotic chemistry and bottom-up synthetic biology. Curr. Opin. Chem. Biol. 2014, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Ferris, J.P. One-step, regioselective synthesis of up to 50-mers of RNA oligomers by montmorillonite catalysis. J. Am. Chem. Soc. 2006, 128, 8914–8919. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.C.; Aldersley, M.F.; Delano, J.W.; Ferris, J.P. Mechanism of montmorillonite catalysis in the formation of RNA oligomers. J. Am. Chem. Soc. 2009, 131, 13369–13374. [Google Scholar] [CrossRef] [PubMed]
- Burcar, B.T.; Cassidy, L.M.; Moriarty, E.M.; Joshi, P.C.; Coari, K.M.; McGown, L.B. Potential pitfalls in MALDI-TOF MS analysis of abiotically synthesized RNA oligonucleotides. Orig. Life Evol. Biosph. 2013, 43, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Burcar, B.T.; Jawed, M.; Shah, H.; McGown, L.B. In situ imidazole activation of ribonucleotides for abiotic RNA oligomerization reactions. Orig. Life Evol. Biosph. 2015, 45, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Monnard, P.A.; Kanavarioti, A.; Deamer, D.W. Eutectic phase polymerization of activated ribonucleotide mixtures yields quasi-equimolar incorporation of purine and pyrimidine nucleobases. J. Am. Chem. Soc. 2003, 125, 13734–13740. [Google Scholar] [CrossRef] [PubMed]
- Verlander, M.S.; Lohrmann, R.; Orgel, L.E. Catalysis for the self-polymerization of adenosine cyclic 2′,3′-phosphate. J. Mol. Evol. 1973, 2, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, S.; Vlassov, A.; Benner, S.; Coombs, A.; Olasagasti, F.; Deamer, D. Lipid-assisted synthesis of RNA-like polymers from mononucleotides. Orig. Life Evol. Biosph. 2008, 38, 57–74. [Google Scholar] [CrossRef] [PubMed]
- DeGuzman, V.; Vercoutere, W.; Shenasa, H.; Deamer, D. Generation of oligonucleotides under hydrothermal conditions by non-enzymatic polymerization. J. Mol. Evol. 2014, 78, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, L.; Maurel, M.C.; Deamer, D. Salt-promoted synthesis of RNA-like molecules in simulated hydrothermal conditions. J. Mol. Evol. 2015, 80, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Toppozini, L.; Dies, H.; Deamer, D.W.; Rheinstadter, M.C. Adenosine monophosphase forms ordered arrays in multilamellar lipid matrices: Insights into assembly of nucleic acid for primitie life. PLoS ONE 2013, 8, e62810. [Google Scholar] [CrossRef] [PubMed]
- Himbert, S.; Chapman, M.; Deamer, D.W.; Rheinstadter, M.C. Organization of nucleotides in different environments and the formation of pre-polymers. Scientific Reports 2016. submitted. [Google Scholar]
- Forsythe, J.G.; Yu, S.S.; Mamajanov, I.; Grover, M.A.; Krishnamurthy, R.; Fernandez, F.M.; Hud, N.V. Ester-mediated amide bond formation driven by wet-dry cycles: A possible path to polypeptides on the prebiotic Earth. Angew. Chem. Ind. Ed. 2015, 54, 9871–9875. [Google Scholar] [CrossRef] [PubMed]
- Mamajanov, I.; MacDonald, P.J.; Ying, J.; Duncanson, D.M.; Dowdy, G.R.; Walker, C.A.; Engelhart, A.E.; Fernandez, F.M.; Grover, M.A.; Hud, N.V.; et al. Ester formation and hydrolysis during wet-dry cycles: Generation of far-from-equilibrium polymers in a model prebiotic reaction. Macromolecules 2014, 47, 1334–1343. [Google Scholar] [CrossRef]
- Damer, B.; Deamer, D. Coupled phases and combinatorial selection in fluctuating hydrothermal pools: A scenario to guide experimental approaches to the origin of cellular life. Life 2015, 5, 872–887. [Google Scholar] [CrossRef] [PubMed]
- Szabo, P.; Scheuring, I.; Czaran, T.; Szathmary, E. In Silico Simulations Reveal that Replicators with Limited Dispersal Evolve Towards Higher Efficiency and Fidelity. Nature 2002, 420, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, N.; Hogeweg, P. Multilevel Selection in Models of Prebiotic Evolution II: A Direct Comparison of Compartmentalization and Spatial Self-Organization. PLoS Comput. Biol. 2009, 5, e1000542. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.I.; Grover, M.A.; Hud, N.V. Universal Sequence Replication, Reversible Polymerization and Early Functional Biopolymers: A Model for the Initiation of Prebiotic Sequence Evolution. PLoS ONE 2012, 7, e34166. [Google Scholar] [CrossRef] [PubMed]
- Leu, K.; Kervio, E.; Obermayer, B.; Turk-MacLeod, R.M.; Yuan, C.; Luevano, J.M., Jr.; Chen, E.; Gerland, U.; Richert, C.; Chen, I.A. Cascade of reduced speed and accuracy after errors in enzyme-free copying of nucleic acid sequences. J. Am. Chem. Soc. 2013, 135, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Derr, J.; Manapat, M.L.; Rajamani, S.; Leu, K.; Xulvi-Brunet, R.; Joseph, I.; Nowak, M.A.; Chen, I.A. Prebiotically plausible mechanisms increase compositional diversity of nucleic acid sequences. Nucl. Acids Res. 2012, 40, 4711–4722. [Google Scholar] [CrossRef] [PubMed]
- Olasagasti, F.; Kim, H.J.; Pourmand, M.; Deamer, D.W. Non-enzymatic transfer of sequence information under plausible prebiotic conditions. Biochimie 2011, 93, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Mungi, C.V.; Singh, S.K.; Chugh, J.; Rajamani, S. Synthesis of barbituric acid containing nucleotides and their implications for the origin of primitive informational polymers. Phys. Chem. Chem. Phys. 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Breaker, R.R. Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2’ hydroxyl group. J. Am. Chem. Soc. 1999, 121, 5364–5372. [Google Scholar] [CrossRef]
- Emilsson, G.M.; Nakamura, S.; Roth, A.; Breaker, R.R. Ribozyme speed limits. RNA 2003, 9, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Soukup, G.A.; Breaker, R.R. Relationship between internucleotide linkage geometry and the stability of RNA. RNA 1999, 5, 1308–1325. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higgs, P.G. The Effect of Limited Diffusion and Wet–Dry Cycling on Reversible Polymerization Reactions: Implications for Prebiotic Synthesis of Nucleic Acids. Life 2016, 6, 24. https://doi.org/10.3390/life6020024
Higgs PG. The Effect of Limited Diffusion and Wet–Dry Cycling on Reversible Polymerization Reactions: Implications for Prebiotic Synthesis of Nucleic Acids. Life. 2016; 6(2):24. https://doi.org/10.3390/life6020024
Chicago/Turabian StyleHiggs, Paul G. 2016. "The Effect of Limited Diffusion and Wet–Dry Cycling on Reversible Polymerization Reactions: Implications for Prebiotic Synthesis of Nucleic Acids" Life 6, no. 2: 24. https://doi.org/10.3390/life6020024