Phosphate Activation via Reduced Oxidation State Phosphorus (P). Mild Routes to Condensed-P Energy Currency Molecules

Abstract
:
1. Introduction
2. Experimental Section
2.1. Materials and General Analytical Methods
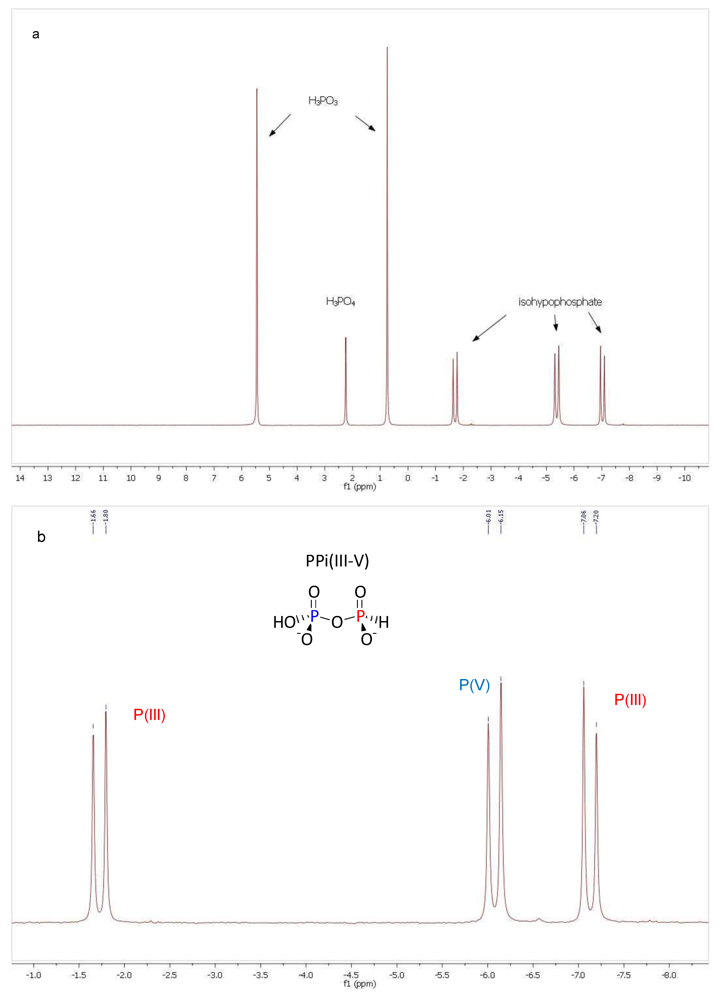
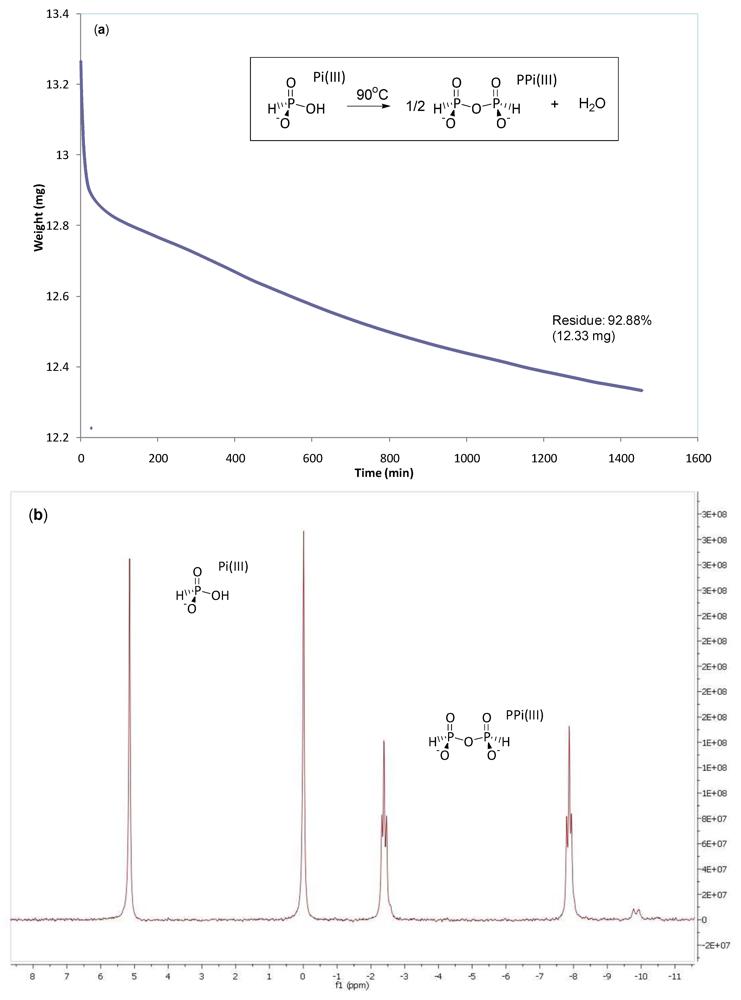
2.2. Production of Pyrophosphite PPi(III)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | 4 Days | 5 Days | 6 Days |
|---|---|---|---|
| 4.0 | 24(2) | 24(4) | 30(1) |
| 4.5 | 12(0) | 14(1) | 15(1) |
| 5.0 | 8(1) | 10(1) | 17(0) |
| 5.5 | 12(0) | 14(1) | 15(1) |
| 5.8 | 4(1) | 5(0) | 6(0) |
| 6.0 | 0 | 0 | 0 |
| Time (h) | pH 3 | pH 4 | |
| 24 | 61(4) | 39(3) | |
| 48 | 61(3) | 16(1) | |
| 72 | 62(4) | 20(10) | |
| 336 | 35(5) | 38(0) | |
2.3. Hveradalur Lake Geothermal Field Experiments: Site
2.4. Hveradalur Lake Geothermal Field Experiments: ICP-AES and ICP-MS-HPLC Analyses
- (i)
- Samples were pre-filtered using Whatman Number 1 filter papers.
- (ii)
- Samples were then diluted to 70 mL and re-filtered using Pall Corporation Acrodisc 32 mm 0.45 µm syringe filters to remove un-dissolved particulates.
- (iii)
- Nalgene bottles (30 mL capacity) were acid washed by filling with 50% HCl and leaving for 24 h. They were rinsed 3 times with tap water and 3 times with deionized water then left to air dry.
- (iv)
- Two portions of 35 mL of sample were placed in an acid-washed 30 mL Nalgene bottle.
- (v)
- Concentrated nitric acid (ca. 2 drops) was added to one of the portions and the lids screwed on tightly and the bottles labeled.
- (vi)
- Due to the concentration of sulphur, the acid treated samples were further diluted to allow accurate measurement of the sulphur content of each sample.
- (vii)
- Two 1.5 mL aliquots were taken from each of the acid treated samples. Two drops of toluene was added as an antibacterial agent. The sub-samples were subsequently submitted for ICP analysis.
| Sample ζ | ICP-AES † (μgL−1) | ICP-MS ‡ (μgL−1) |
|---|---|---|
| Blank 1 | –0.04(1) | –0.02(0) |
| A1 (KHL–UCL3; 40; 3.6) | 0.53(2) | 0.39(3) |
| A2 (KHL–BPR; 79.5; 4.0) | 1.01(3) | 0.92(0) |
| A3 (KHL–MP1; 87.4; 1.6) | 21.98(3) | 17.17(5) |
| A4 (KHL–LP1; 93.5; 3.1) | 2.66(2) | 1.03(5) |
| A5 (KHL–UCL5; 89.2; 4.7) | 1.20(2) | 1.50(4) |
| A6 (KHL–LP3; 79.2; 2.5) | 0.62(0) | 0.47(1) |
| A7 (KHL–LP4; 87.8; 3.3) | 0.77(2) | 0.70(2) |
| A8 (KHL–MP3; 84.7; 2.7) | 2.28(6) | 2.00(3) |
2.5. Hveradalur Lake Geothermal Field Experiments: 31P-NMR Analyses
- (i)
- A 10 mL aliquot of the acidified sample (prepared as above) was taken in a 15 mL Falcon tube.
- (ii)
- Sample was treated with NaOH (1 M) to pH 12 and left for 1–2 min.
- (iii)
- Sample was gravity filtered to remove precipitate (hydrated ferrous oxides and hydroxides). These oxides were subsequently collected and shown to contain negligible amounts of phosphorus via EDX measurements.
- (iv)
- Sample was treated with HCl (1 M) to pH 4.
- (v)
- A 0.5 mL aliquot was taken and analyzed by 31P-NMR spectroscopy (500 MHz Bruker Avance, 320 scans, 300 K) using capillary D2O inserts.
- (vi)
- Sample was reduced to dryness and residues dried overnight in 50 °C oven.
- (vii)
- Residues were ground to fine powder in mortar and pestle.
- (viii)
- Residues were dry heated to ca. 90 °C for 72 h on a sand bath under flowing N2 (ca. 1 bubble per second).
- (ix)
- Residues were dissolved in deionized water (ca. 0.5 mL) and adjusted to pH 7.2 using aqueous Na2CO3 solution (1 M).
- (x)
- Sample was analyzed by 31P-NMR spectroscopy (500 MHz Bruker Avance, 2048 scans, 300 K) using capillary D2O inserts.
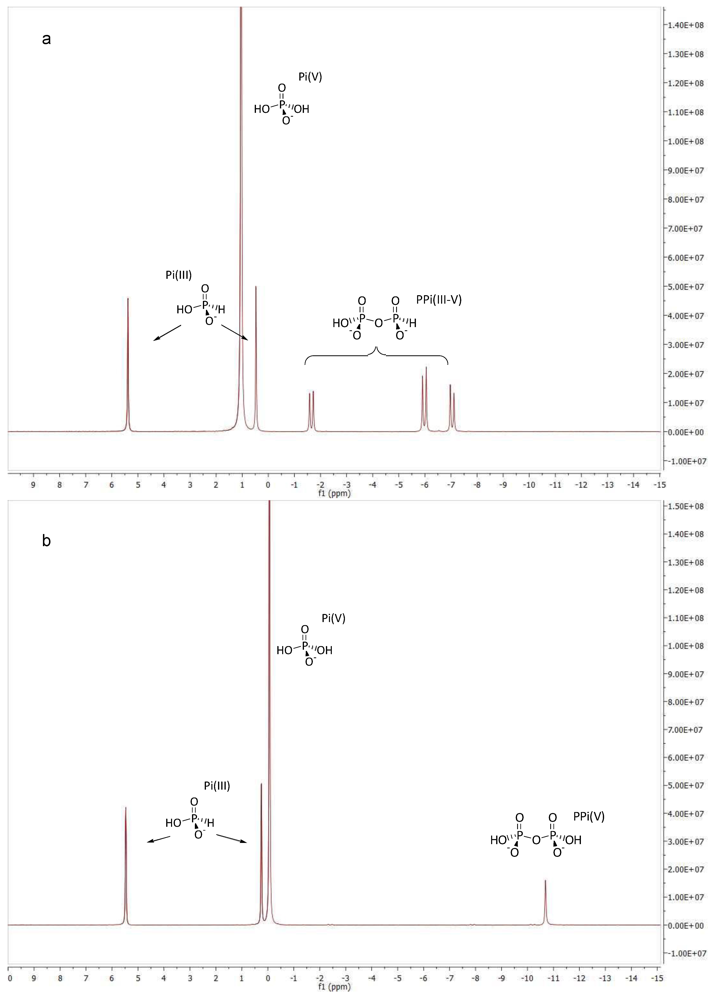
2.6. Conversion of PPi(III) to PPi(III–V)
2.7. Conversion of PPi(III–V) to PPi(V)
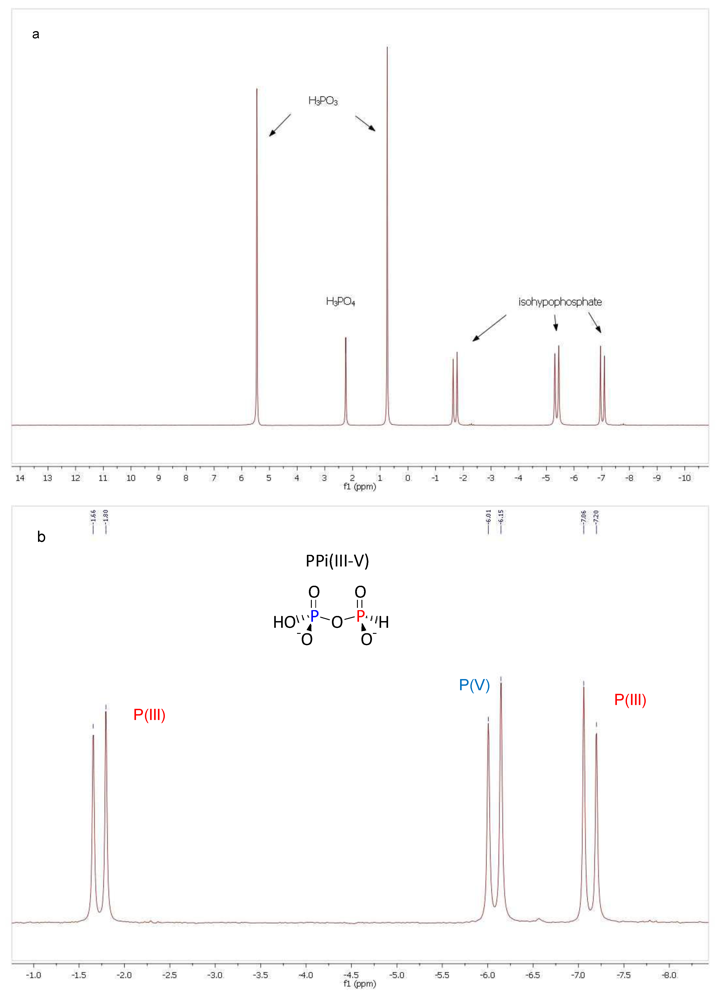

3. Results and Discussion
3.1. Pyrophosphite Formation and Geological Provenance
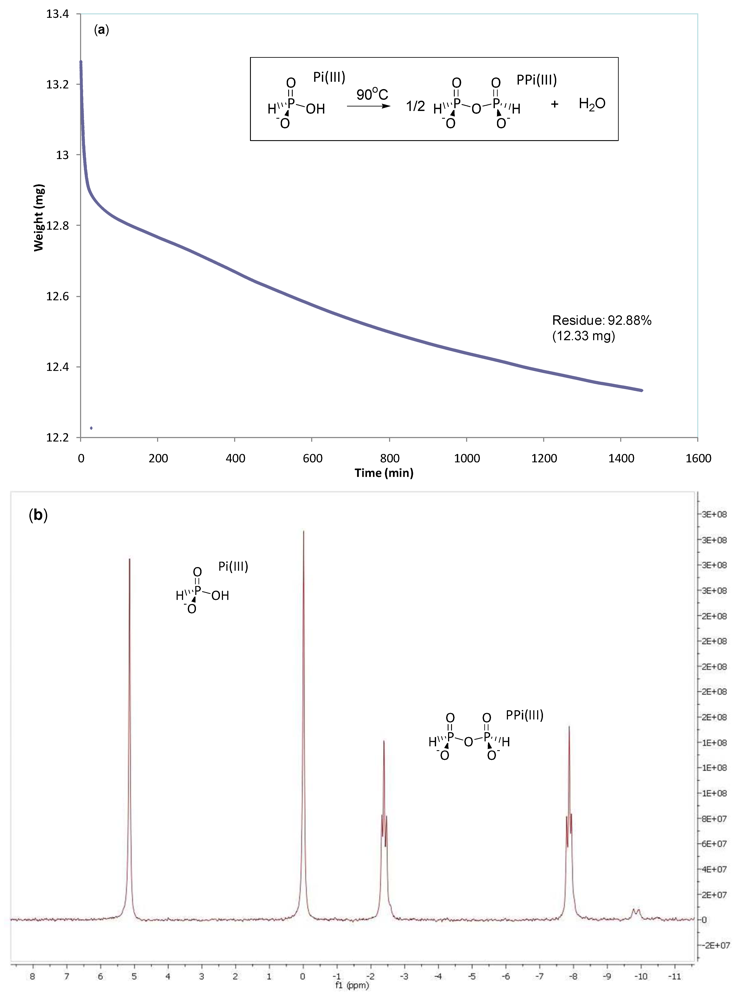
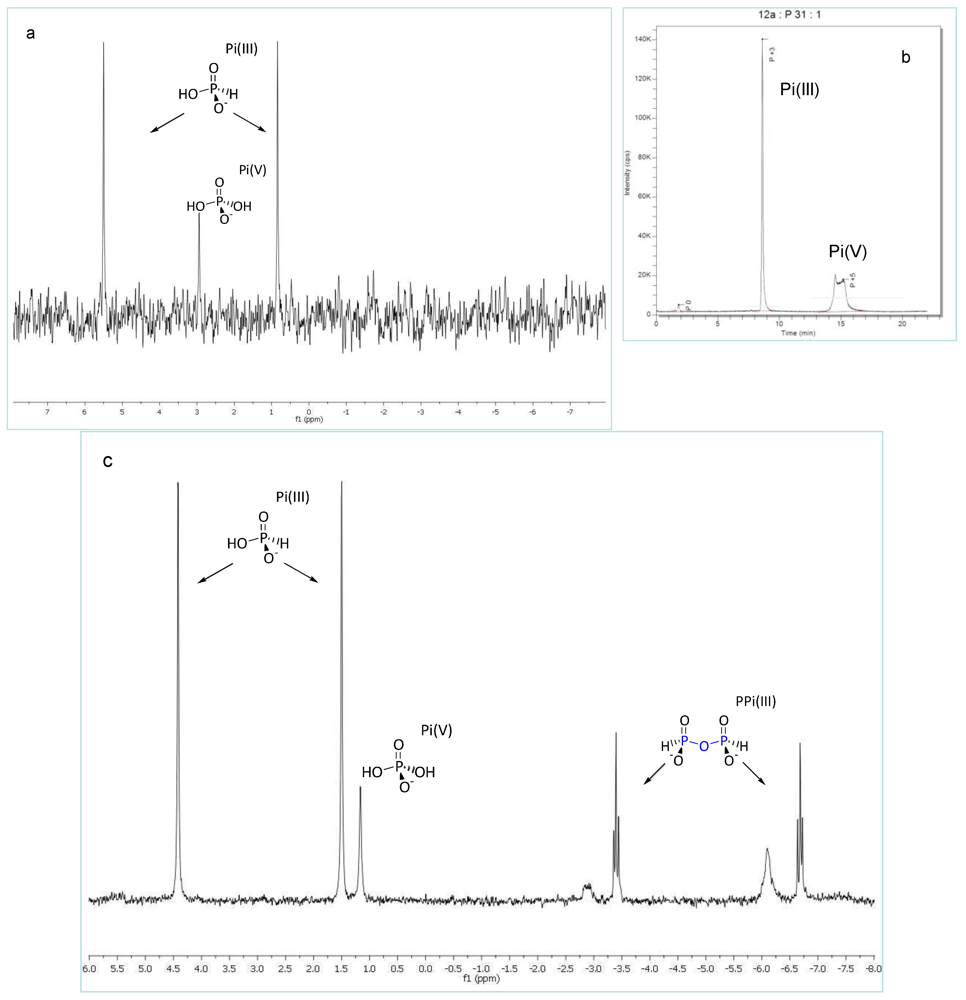
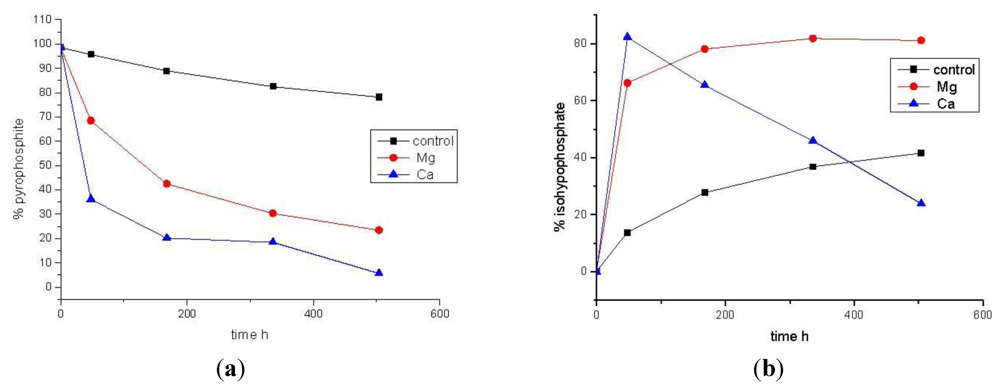
3.2. Phosphonylation of Pi(V) with PPi(III) in Aqueous Solution. Formation of PPi(III–V) and PPi(V)
| Time (h) | Control [% total P] | MgCl2 [% total P] | CaCl2 [% total P] | |||
|---|---|---|---|---|---|---|
| PPi(III) | PPi(III–V) | PPi(III) | PPi(III–V) | PPi(III) | PPi(III–V) | |
| 0 | 98.6 | 0 | 98.6 | 0 | 98.6 | 0 |
| 48 | 95.7 | 13.8 | 68.5 | 66.2 | 36.2 | 82.3 |
| 168 | 89.0 | 27.7 | 42.5 | 78.1 | 20.2 | 65.4 |
| 336 | 82.5 | 36.8 | 30.3 | 81.8 | 18.4 | 45.9 |
| 504 | 78.2 | 41.6 | 23.4 | 81.1 | 5.6 | 23.9 |
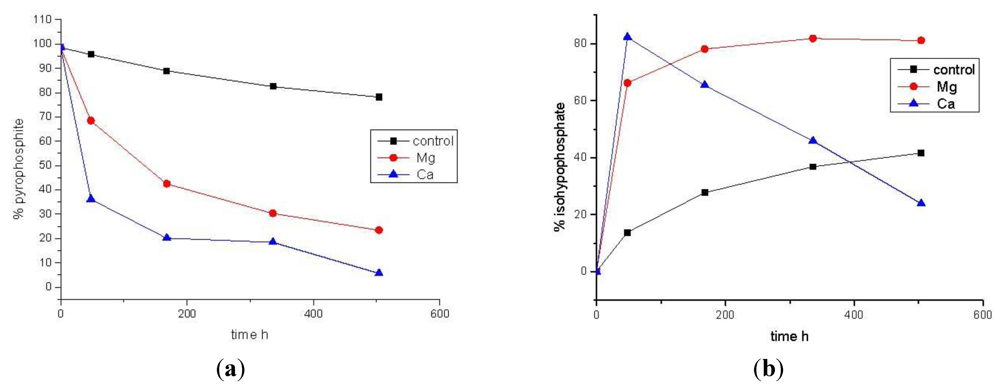
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Harold, F.M. The Vital Force: A Study of Bioenergetics; W.H. Freeman: New York, NY, USA, 1986. [Google Scholar]
- Dimroth, P.; Kaim, G.; Matthey, U. Crucial role of the membrane potential for ATP synthesis by F(1)F(0) synthases. J. Exp. Biol. 2000, 203, 51–59. [Google Scholar]
- Bochud-Allemann, N.; Schneider, A. Mitochondrial substrate level phosphorylation is essential for growth of procyclic Trypanosoma brucei. J. Biol. Chem. 2002, 277, 32849–32854. [Google Scholar] [CrossRef]
- Baltscheffsky, M.; Schultz, A.; Baltscheffsky, H. H+-proton-pumping inorganic pyrophosphatase: A tightly membrane-bound family. FEBS Lett. 1999, 452, 121–127. [Google Scholar] [CrossRef]
- Serrano, A.; Perez-Castineira, J.R.; Baltscheffsky, H.; Baltscheffsky, M. Proton-pumping inorganic pyrophosphatases in some archaea and other extremophilic prokaryotes. J. Bioenerg. Biomembr. 2004, 36, 127–133. [Google Scholar] [CrossRef]
- Serrano, A.; Perez-Castineira, J.R.; Baltscheffsky, M.; Baltscheffsky, H. H+-PPases: Yesterday, today and tomorrow. IUBMB Life 2007, 59, 76–83. [Google Scholar] [CrossRef]
- Ernester, L. Molecular Mechanism in Bioenergetics; Elsevier: Amsterdam, The Netherlands, 1992. [Google Scholar]
- Lipscomb, W.N.; Strater, N. Recent advances in zinc enzymology. Chem. Rev. 1996, 96, 2375–2433. [Google Scholar] [CrossRef]
- Pross, A. Toward a general theory of evolution: Extending Darwinian theory to inanimate matter. J. Syst. Chem. 2011, 2, 1–14. [Google Scholar] [CrossRef]
- Keefe, A.D.; Miller, S.L. Are polyphosphates or phosphate esters prebiotic reagents? J. Mol. Evol. 1995, 41, 693–702. [Google Scholar]
- Eschenmoser, A. Etiology of potentially primordial biomolecular structures: From VitaminB12 to the nucleic acids and an inquiry into the chemistry of life’s origin: A retrospective. Angew. Chem. Int. Ed. 2011, 50, 12412–12472. [Google Scholar] [CrossRef]
- Babich, L.; Hartog, A.F.; van der Horst, M.A.; Wever, R. Continuous-flow reactor-based enzymatic synthesis of phosphorylated compounds on a large scale. Chem. Eur. J. 2012, 18, 6604–6609. [Google Scholar] [CrossRef]
- Holm, N.G.; Baltscheffsky, H. Links between hydrothermal environments, pyrophosphate, Na+ and early evolution. Orig. Life Evol. Biosph. 2011, 41, 483–493. [Google Scholar] [CrossRef]
- Hill, A.; Orgel, L.E. Trimetaphosphate-induced addition of aspartic acid to oligo(glutamic acid)s. Helv. Chim. Acta 2002, 85, 4244–4254. [Google Scholar] [CrossRef]
- Yamagata, Y.; Watanabe, H.; Saitoh, M.; Namba, T. Volcanic production of polyphosphates and its relevance to prebiotic evolution. Nature 1991, 352, 516–519. [Google Scholar] [CrossRef]
- Pasek, M.A.; Kee, T.P. On the Origin of Phosphorylated Biomolecules. In Origins of Life: The Primal Self Organization; Egel, R., Lankenau, D.-H., Mulkidjanian, A.-Y., Eds.; Springer Verlag: Berlin, Germany, 2011; pp. 57–83. [Google Scholar]
- Hoskuldsson, A.; Sparks, R.S.J.; Carroll, M.R. Constraints on the dynamics of subglacial basalt eruptions from geologicaland geochemical observations at Kverkfjöll, NE-Iceland. Bull. Volcanol. 2006, 68, 689–701. [Google Scholar] [CrossRef]
- Ólafsson, M.; Torfason, H.; Grönvold, K. Surface Exploration and Monitoring of Geothermal Activity in the Kverkfjöll Geothermal Area, Central Iceland. In Proceedings of World Geothermal Congress, Beppu-Morioka, Japan, 28 May–10 June 2000; pp. 1539–1545.
- Cousins, C.R.; Crawford, I.A.; Carrivick, J.; Gunn, M.; Harris, J.; Kee, T.; Karlsson, M.; Carmody, L.; Cockell, C.; Herschy, B.; et al. Glaciovolcanic hydrothermal environments in Iceland and implications for their detection on Mars. J. Volcanol. Geotherm. Res. 2013, 256, 61–77. [Google Scholar] [CrossRef]
- Blaser, B.; Worms, I.H.Z. Umanhydrisierungsreaktionen von P-O-P-Sauren,über die P-O-P-P-Saure. Anorg. Allgem. Chem. 1953, 301, 18–22. [Google Scholar] [CrossRef]
- Madhurambal, G.; Subha, R.; Mojumdar, S.C. Crystallization and thermal characterization of calcium hydrogen phosphate dihydrate crystals. J. Therm. Anal. Calorim. 2009, 96, 73–76. [Google Scholar] [CrossRef]
- Mesmer, R.E.; Carroll, R.L. The kinetics and mechanism of the hydrolysis of pyrophosphate. J. Am. Chem. Soc. 1966, 88, 1381–1387. [Google Scholar] [CrossRef]
- Mulkidjanian, A.Y.; Bychkov, A.Y.; Dibrova, D.V.; Galperin, M.Y.; Koonin, E.V. Origin of first cells at terrestrial, anoxic geothermal fields. Proc. Natl. Acad. Sci. USA 2012, 109, E821–E830. [Google Scholar]
- Pech, H.; Henry, A.; Khachikian, C.S.; Salmassi, T.M.; Hanrahan, G.; Foster, K.L. Detection of geothermal phosphite using high performanceliquid chromatography. Environ. Sci. Technol. 2009, 43, 7671–7675. [Google Scholar]
- Pasek, M.A.; Lauretta, D.S. Aqueous corrosion of phosphide minerals from iron meteorites: A highly reactive source of prebiotic phosphorus on the surface of the early earth. Astrobiology 2005, 5, 515–535. [Google Scholar]
- Bryant, D.E.; Kee, T.P. Direct evidence for the availability of reactive, water soluble phosphorus on the early Earth. H-Phosphinic acid from the Nantan meteorite. Chem. Commun. 2006, 22, 2344–2346. [Google Scholar]
- Pasek, M.A.; Dworkin, J.P.; Lauretta, D.S. A radical pathway for organic phosphorylation during schreibersite corrosion with implications for the origin of life. Geochim. Cosmochim. Acta 2007, 71, 1721–1736. [Google Scholar] [CrossRef]
- Pasek, M.A.; Lauretta, D.S. Extraterrestrial flux of potentially prebiotic C, N, and P. Orig. Life Evol. Biosph. 2008, 38, 5–21. [Google Scholar] [CrossRef]
- Bryant, D.E.; Greenfield, D.; Walshaw, R.D.; Evans, S.M.; Nimmo, A.E.; Smith, C.; Wang, L.; Pasek, M.A.; Kee, T.P. Electrochemical studies of iron meteorites: Phosphorus redox chemistry on the early Earth. Int. J. Astrobiol. 2009, 8, 27–36. [Google Scholar] [CrossRef]
- Benedix, G.K.; McCoy, T.J.; Kiel, K.; Love, S.G. A petrologic study of the IAB iron meteorites: Constraints on the formation of the IAB-Winonaite parent body. Meteorit. Planet. Sci. 2000, 35, 1127–1141. [Google Scholar] [CrossRef]
- Pasek, M.A.; Block, K. Lightning-induced reduction of phosphorus oxidation state. Nat. Geosci. 2009, 2, 553–556. [Google Scholar] [CrossRef]
- Chao, E.C.T.; Dwornik, E.J.; Littler, J. New data on the nickel-iron spherules from Southeast Asian tektites and their implications. Geochim. Cosmochim. Acta 1964, 28, 971–974. [Google Scholar] [CrossRef]
- Klöck, W.; Palme, H.; Tobsehall, H.J. Trace elements in natural metallic iron from Disko Island, Greenland. Contrib. Mineral. Petrol. 1986, 93, 273–282. [Google Scholar]
- Bryant, D.E.; Greenfield, D.; Walshaw, R.D.; Johnson, B.R.G.; Herschy, B.; Smith, C.; Pasek, M.A.; Telford, R.; Scowen, I.; Munshi, T.; et al. Hydrothermal modification of the Sikhote-Alin iron meteorite under low pH geothermal environments. A plausibly prebiotic route to activated phosphorus on the early Earth. Geochim. Cosmochim. Acta 2013, 109, 90–112. [Google Scholar] [CrossRef]
- Miller, S.L.; Parris, M. Synthesis of pyrophosphate under primitive earth conditions. Nature 1964, 204, 1248–1250. [Google Scholar] [CrossRef]
- Steinman, G.; Kenyon, D.H.; Calvin, M. Dehydration condensation in aqueous solution. Nature 1965, 206, 707–708. [Google Scholar] [CrossRef]
- Beck, A.; Orgel, L.E. The formation of condensed phosphate in aqueous solution. Proc. Natl. Acad. Sci. USA 1965, 54, 664–667. [Google Scholar] [CrossRef]
- Weber, A.L. Formation of pyrophosphate, tripolyphosphate, and phosphorylimidazole with the thioester, N,S-diacetylcysteamine, as the condensing agent. J. Mol. Evol. 1981, 18, 24–29. [Google Scholar] [CrossRef]
- Weber, A.L. Formation of pyrophosphate on hydroxyapatite with thioesters as condensing agents. Biosystems 1982, 15, 183–189. [Google Scholar] [CrossRef]
- Hermes-Lima, M.; Vieyra, A. Pyrophosphate formation from phospho(enol)pyruvate adsorbed onto precipitated orthosphosphate: A model for prebiotic catalysis of transphosphorylations. Orig. Life Evol. Biosph. 1989, 19, 143–152. [Google Scholar] [CrossRef]
- Hermes-Lima, M. Model for prebiotic pyrophosphate formation: Condensation of precipitated orthophosphate at low temperature in the absence of condensing or phosphorylating agents. J. Mol. Evol. 1990, 31, 353–358. [Google Scholar] [CrossRef]
- Keefe, A.D.; Miller, S.L. Potentially prebiotic syntheses of condensed phosphates. Orig. Life Evol. Biosph. 1996, 26, 15–25. [Google Scholar] [CrossRef]
- De Zwart, I.I.; Meade, S.J.; Pratt, A.J. Biomimetic phosphoryl transfer catalysed by iron(II)-mineral precipitates. Geochim. Cosmochim. Acta 2004, 68, 4093–4098. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kee, T.P.; Bryant, D.E.; Herschy, B.; Marriott, K.E.R.; Cosgrove, N.E.; Pasek, M.A.; Atlas, Z.D.; Cousins, C.R. Phosphate Activation via Reduced Oxidation State Phosphorus (P). Mild Routes to Condensed-P Energy Currency Molecules. Life 2013, 3, 386-402. https://doi.org/10.3390/life3030386
Kee TP, Bryant DE, Herschy B, Marriott KER, Cosgrove NE, Pasek MA, Atlas ZD, Cousins CR. Phosphate Activation via Reduced Oxidation State Phosphorus (P). Mild Routes to Condensed-P Energy Currency Molecules. Life. 2013; 3(3):386-402. https://doi.org/10.3390/life3030386
Chicago/Turabian StyleKee, Terence P., David E. Bryant, Barry Herschy, Katie E. R. Marriott, Nichola E. Cosgrove, Matthew A. Pasek, Zachary D. Atlas, and Claire R. Cousins. 2013. "Phosphate Activation via Reduced Oxidation State Phosphorus (P). Mild Routes to Condensed-P Energy Currency Molecules" Life 3, no. 3: 386-402. https://doi.org/10.3390/life3030386