1. Introduction
The presence of organoarsenic in marine organisms was first reported almost 100 years ago in the classic studies of Jones [
1] and Chapman [
2], and today, it is well known that many organoarsenic species occur naturally at high concentrations in marine organisms [
3,
4]. Over 200 organoarsenic species have been described with arsenosugars (AsSug), the most dominant species in marine algae, whereas arsenobetaine (AB) represents the prominent species in most marine animals [
5]. The first lipid-soluble As (arsenolipid) species were identified later, in 1988 by Morita and Shibata [
6], but since then, marine organisms have proven to be a rich source of various types of arsenolipids (As-lipids). These include fatty acids [
7,
8], hydrocarbons [
8,
9], fatty alcohols [
10], phospholipids [
11,
12,
13,
14], di- and tri-acylglycerols [
14], and an unusual ether-bound phytyl 2-
O-methyl arsenosugar [
15].
Several studies have contributed to our understanding of the occurrence of arsenolipids [
16,
17,
18,
19,
20,
21,
22], but we still have only limited knowledge about the metabolism of these compounds and their behavior in natural systems. In this regard, two recent studies by Glabonjat et al. [
19,
22] have revealed the likely significance of phytyl 2-
O-methyl arsenosugars in the natural cycling of arsenic. In culture experiments, the unicellular alga
Dunaliella tertiolecta transformed arsenate primarily into arsenoribosides including phytyl 2-
O-methyl arsenosugar, and in a field study in the Great Salt Lake, an extreme hypersaline environment, this compound was a significant organoarsenical in sediments, its likely origin being planktonic
Dunaliella species that are dominant in the overlying water body.
Further advances in our understanding of arsenic cycling await studies that can directly compare results from field observations with controlled laboratory experiments. Guided by the observations of Glabonjat et al. and hypothesizing the significance of ether-bound isoprenoyl 2-O-methyl arsenosugars, we sought a halotolerant/halophilic phytoplanktonic organism that might be abundant in extreme environments and in addition would be amenable to culture experiments. Here, we report an investigation using high-performance liquid chromatography (HPLC)–mass spectrometry and X-ray spectroscopic methods to examine the arsenic species in cultured Picocystis strain ML as well as in collected samples representing a basic food web in Mono Lake, California, USA.
2. Materials and Methods
2.1. Research Site
Mono Lake is a closed basin soda lake located in central, eastern California on the edge of the Great Basin Desert. In contrast to the pH-neutral Great Salt Lake, Mono Lake is alkaline (pH = 9.8), moderately hypersaline (salinity ~80–90 g/L), and contains abundant dissolved inorganic arsenic (~200 µM) [
23] and inorganic phosphate (~1,000 µM) [
24]. When monomictic, the lake supports high levels (499–641 g/m
2) of annual primary production [
24,
25]. The most conspicuous and abundant member of the phytoplankton community is
Picocystis strain ML, a eukaryotic picoplankter (diameter ~3 µm). Strain ML is responsible for at least 50% of the lake’s primary productivity, is distributed throughout Mono Lake’s water column, is readily adapted to low light levels and the chemically harsh reducing conditions of the lake below the oxycline, and is grazed down by brine shrimp (
Artemia monica), the lake’s dominant zooplankter [
26]. When grown at low light levels, strain ML exhibits a trilobite morphology [
27] reminiscent of Disney’s famous Mickey Mouse [
25]. Transcriptomic investigations of the lake’s water column indicated that cryptic photosynthetic activity by strain ML was discernable in the depths beneath, where light could no longer be instrumentally detected [
28].
2.2. Cell Cultures and Growth of Picocystis Strain ML
Picocystis strain ML was originally isolated from Mono Lake enrichments by classical dilution/streaking methods at Bigelow Labs National Center for Marine Algae and Microbiota and, when purified, transferred to the USGS in Menlo Park, CA. The strain was cultured in Bigelow L1 seawater-based medium [
29] in 500 mL conical flasks for several years after its isolation. Illumination was attained by placing flasks atop a lab bench under general fluorescent lighting (8 h per day) at room temperature (~20 °C). Strain ML tolerates a wide range of salinities, being able to grow from essentially freshwater up to 260 g/L [
26]. For the experiments, growth was on one of two phosphate regimens: either kept at the L1 recipe concentration (37 µM) or augmented with 1000 µM KH
2PO
4. Experimental variables for both phosphate conditions were the incubation of cells in medium without any arsenic addition or augmented with sodium arsenate (0.8 mM) or sodium arsenite (0.4 mM). Arsenic oxyanion concentrations were determined by HPLC in subsamples taken over the course of the 50–60 days incubations [
30], and growth was followed on subsamples by optical density (A
680). Cells were harvested by centrifugation, washed with sterile L1 medium that lacked arsenic oxyanions, and re-centrifuged. Cell pellets were stored frozen until analyzed by X-ray spectroscopy at the Stanford Linear Accelerator or express-shipped (frozen on dry ice) to the University of Graz, Austria, for the extraction and analysis of arsenic compounds.
2.3. Transmission Electron Microscopy of Picocystis Strain ML
Cells were initially fixed in their culture medium with glutaraldehyde (2.5% final concentration), then post fixed (1% osmium tetroxide), dehydrated, and embedded in Spurr’s as described previously [
31]. Ultrathin sections were observed on a JEOL 1210 TEM (JEOL, Peabody MA) at 80 kV equipped with an ORCA HR digital camera (Hamamatsu, Bridgewater NJ).
2.4. Collection of Field Samples from Mono Lake
Environmental samples were collected in October 2018 at station 6 (lat 37.95, long 119.03 on Mono Lake, CA. A zooplankton net tow (80 µm mesh) was conducted at 0.5 m depth to collect brine shrimp. Discrete water samples were collected at 12, 17, and 20 m depth using a 4-L Niskin closing bottle. Sampling was preceded by a CTD cast (SeaBird SBE 19) to determine water column physical characteristics (e.g., salinity, temperature), light penetration (PAR), and dissolved oxygen concentration. Water was screened (10 µm Nitex) to exclude brine shrimp while filling 2-L Nalgene sample bottles. Water samples were vacuum filtered within several hours of collection using 0.8 µm nylon filters (47 mm diameter). The so obtained plankton samples were frozen and shipped together with the filters. Sediment cores were collected from station 6 using a bottom grab that upon retrieval was sub-cored in duplicate (0–10 cm depth). All samples were frozen on site with dry ice and stored at −80 °C and then shipped frozen to University of Graz for further analysis of arsenic species as outlined below.
2.5. Arsenic Species Extractions from Lab-grown Picocystis Cells
Extractions of arsenic species were performed in sequence, starting with lipid-soluble arsenicals, followed by aqueous arsenic phases, and finally by a trifluoroacetate (TFA)-based extraction of the “recalcitrant” phase residual after the first two extractions. The non-extractable arsenic remaining in the pellets was solubilized by microwave-assisted acid digestion. Duplicate samples of cells were combined, freeze-dried (0.05 mbar), and the fine powder homogenized with a spatula. For arsenolipids, about 10 mg (weighed to a precision of 0.01 mg) of each sample was transferred into an Eppendorf vial (1.5 mL; polypropylene) in duplicate and extracted with pyridine (>98%; 500 µL) in an ultrasonic bath (15 min at 30 °C) followed by mixing on a rotatory cross (45 min at 20 °C). The suspension was centrifuged (10,000 × g, 10 min., 10 °C) and the liquid phase (ca 450 µL) transferred to a new Eppendorf vial before the remaining pellet was re-extracted with pyridine as described above. The two extracts were combined and lyophilized (10 mbar, 30 °C, 10 h). The dry lipid-extracts were stored (−20 °C) and re-dissolved in pyridine (300 µL) just prior to measurement by reversed phase-high performance liquid chromatography (RP-HPLC) coupled to an inductively coupled plasma mass spectrometer (ICPMS) and simultaneously to an electro spray ionization mass spectrometer (ESMS). The remaining extracted pellets were dried and stored (−20 °C) until the subsequent aqueous extraction was performed.
The post lipid-extraction dried pellets were re-weighed prior to extraction with aqueous buffer. The extractions were performed in the same way as described above except that 20 mM ammonium bicarbonate was employed as the solvent (pH 8.5 adjusted with ammonia; 500 µL). The extracts were dried, stored, and re-dissolved in water (500 µL) just before anion-exchange and cation-exchange HPLC-ICPMS measurements. In order to recover any possible polar water-soluble arsenicals (aqueous As) that were entrained with the arsenolipid extraction, we also performed a liquid/liquid partitioning of the lipid-extract with dichloromethane (DCM)/aqueous buffer, which was also analyzed by HPLC-ICPMS. To do so, a subsample of the re-dissolved pyridine-extract (150 µL out of 300 µL total) was transferred to a new vial, evaporated to dryness (10 mbar, 30 °C, 5 h), re-dissolved in DCM (500 µL), and extracted with aqueous buffer (20 mM ammonium bicarbonate, pH 8.5; 500 µL) on a vortex mixer (1 min, 20 °C). After centrifugation (10,000 × g, 10 min., 10 °C), the water-phase was transferred to a new vial and the procedure repeated with another portion of aq. buffer (500 µL) added to the remaining DCM. The two aqueous phases were combined, evaporated to dryness, stored (−20 °C), and re-dissolved in water (500 µL) just before ion exchange HPLC-ICPMS. Once again, after this analysis the extracted residual pellets were re-dried and stored (−20 °C) until subsequent final extraction with TFA to access the “recalcitrant” arsenic phase (see below).
To access any recalcitrant aqueous As species that resisted the first two extractions, we performed a harsher procedure under acidic aqueous conditions. The pellets were treated with aqueous TFA (1%, v/v; 500 µL) using the same procedure described above (two sequential extractions), and after extraction, the samples were combined, evaporated to dryness, and re-dissolved in water (1 mL) just prior to ion exchange HPLC-ICPMS analysis. The remaining pellets were also lyophilized and stored (−20 °C) until acid digestion.
The final remaining pellets were weighed before microwave assisted digestion with nitric acid (≥65%, 2 × sub-boiled; 2 mL; in closed quartz vessels) and internal standard (aqueous 100 µg/L Ge, In, Te; 1 mL) in an UltraClaveIV microwave digestion system (40 bar argon start pressure; 250 °C for 30 min). The digests were transferred to polypropylene tubes (15 mL) with water and diluted to a final volume of 10 mL before being analyzed by ICPMS. This procedure solubilized the entire pellet, allowing access to the previously non-extractable arsenic remaining in the sample after the three previous extraction steps.
2.6. Extraction of Arsenic Species from Collected Field Samples
Upon arrival in Graz, samples were freeze-dried (0.05 mbar). Sediment cores were subdivided into four subsamples in Graz: 0–25, 25–50, 50–75, and 75–100 mm depths. Sediment and Artemia samples were homogenized to a fine powder with an agate mortar. About 50 mg of Artemia, whole filters containing phytoplankton from each depth (~5 mg dry biomass per filter), and ~200 mg of sediment subsamples (all weighed to a precision of 0.01 mg) were transferred to Eppendorf vials (1.5 mL) in duplicate.
Extractions of arsenic species from collected Mono Lake samples were performed as described above for strain ML but with the following modifications: (i) Lipids were extracted with a mixture of CHCl3/EtOH (2 + 1, v/v; 500 µL) containing 1% NH3 (v/w); organic extracts of two sequential extractions were combined, dried, and the resultant residue stored (−20 °C) before being re-dissolved in MeOH (500 µL) directly before measurement by RP-HPLC-ICPMS/ESMS. Extracted pellets and sediments were also dried and stored until aqueous extraction. (ii) The re-weighed post lipid-extracted dry pellets and sediments were extracted employing an aqueous ammonia solution (1% NH3, v/w; 500 µL), and the combined extracts of two sequential extractions were dried, stored, and re-dissolved in water (1 mL) just before ion exchange HPLC-ICPMS measurements. Extracted pellets and sediments were dried and stored until TFA-extraction. (iii) Liquid/liquid partitioning of the lipid-extracts was performed on subsamples of the re-dissolved alkaline CHCl3/EtOH-extract (250 µL out of 500 µL total), which were dried, re-dissolved in CHCl3 (500 µL), and extracted with water (500 µL) on a vortex mixer followed by centrifugation. The aqueous layers of two sequential partitions were combined, dried, stored, and re-dissolved in water (1 mL) before ion exchange HPLC-ICPMS. (iv) The remaining pellets and sediments were acid-digested as described for cultured Picocystis cells.
We decided to apply two different extraction procedures for arsenolipids: extraction using pyridine for
Picocystis strain ML or a mixture of CHCl
3/EtOH under alkaline conditions, which was necessary to release arsenolipids from silica-containing matrices [
11,
32], e.g., natural Mono Lake plankton collected on nylon filters that contained a SiO
2-support core. We also used the same two procedures with CRM 7405-a (Hijiki) and obtained no significant differences (p > 0.05 in one-way ANOVA), although we observed a tendency towards slightly lower (ca 5–10%) arsenolipid recoveries when we employed the pyridine extraction method. Nevertheless, extraction with pure pyridine provides several advantages compared to alkaline CHCl
3/EtOH: first, it obviates the need for alcohols during extraction and reduces the risk of chemical reaction; second, it avoids chlorinated solvents which often cause interferences during ICPMS determinations because of
35Cl
40Ar polyatomic interference on
75As; and third, the pyridine extraction method extracts less water-soluble arsenicals. For these reasons, we decided to use the pyridine extraction method when possible.
2.7. Chemical Analysis of Extracted Arsenicals
2.7.1. Determination of Arsenolipids
Separation of As species was performed on an ACE Super-Hexyl-Phenyl column (4.6 × 250 mm; 5 µm particles) under gradient elution conditions. This method was adapted from Finke et al. [
33]; mobile phase A was water containing 25 mM acetic acid adjusted to pH 9.2 with NH
3, and B was MeOH containing 25 mM acetic acid and 0.5% NH
3 (v/v). We used the following gradient: 0–25 min, 40–100% B; 25–35 min, 100% B; 35–35.5 min, 100–40% B; and 35.5–42 min, 40% B. The flow rate was 1.0 mL/min, column temperature was 40 °C, and injection volume was 50 µL. The outflow of the HPLC-system was split (passive splitter) whereby 90% was directed to the ESMS (Agilent 6460) and the remaining 10% transported to the ICPMS (Agilent 7500ce or 7900). Between the splitter and the ICPMS a T-piece was inserted whereby we introduced our internal standard sheath solution (water incl. 0.1 vol% formic acid and 20 µg/L In, Ge, Te; 0.9 mL/min). Carbon compensation [
33] was achieved by pumping water/acetone (95 + 5, v/v) directly into the spray chamber with an external rotary pump (0.5 mL/min). The flow to the ESMS subsequently entered the instrument without further alteration. The ESMS was operated in positive SCAN mode (m/z 100–1100) with an applied ionization voltage of 5 kV; 2 kV nozzle voltage; fragmentor voltages were 135 or 200 V; nitrogen gas temperature was 350 °C with a flow of 12 L/min; and the nebulizer pressure was set to 25 psi.
Offline, the extracts were also analyzed by HPLC-high resolution-ESMS/MS using a Q-Exactive Quadrupole Orbitrap mass analyzer equipped with a HESI-II source. The HPLC conditions were adjusted to optimize ionization efficiency, which is considerably higher in positive electrospray ionization mode, when acidic mobile phases are used compared to the alkaline buffers previously applied for HPLC-ICPMS. The applied method was adapted from Glabonjat et al. [
22]. The column was a Shodex Asahipak ODP-50 (4.0 × 125 mm; 5 µm particles), and elution was performed using mobile phase A (water containing 0.1 vol% formic acid) and mobile phase B (MeOH containing 0.1 vol% formic acid) with the following gradient: 0–15 min, 60–100% B; 15–23 min, 100% B; 23–23.1 min, 100–60% B; and 23.1–30 min, 60% B. The flow was set to 0.5 mL/min, column held at 40 °C, and injection volume was 10 µL. The instrument was operated in positive ionization mode with 4.2 kV capillary voltage; data dependent MS/MS mode was chosen in m/z window of 120–1200; fragmentation of precursor ions was achieved with N
2 gas using collision energies of 20, 30, and 40 NCE; and the resolution was 70,000 (FWHM).
2.7.2. Determination of Water-soluble Arsenicals
Cation-exchange HPLC-separations were performed according to Xiong et al. [
34] on an IonoSpher 5C column (3.0 × 200 mm; 5 µm particles) under isocratic elution conditions using aqueous 10 mM pyridine (adjusted with formic acid to pH 2.6) as mobile phase. The flow was set to 1.0 mL/min; column temperature was 30 °C; and injection volumes were 5 or 20 µL.
Anion-exchange HPLC-separations of
Picocystis strain ML extracts were performed after Glabonjat et al. [
19] using a PRP-X100 column (4.6 × 150 mm; 5 µm particles) under isocratic conditions with 5 mM malonic acid (adjusted to pH 5.8 with NH
3) at a flow of 1.0 mL/min; temperature was 30 °C (injection volumes were 20 µL). Field-collected samples were chromatographed on a Thermo AS14A Dionex IonPac column (3.0 × 150 mm, 5 µm particles) under isocratic elution conditions adapted from Narukawa et al. [
35] using aqueous 10 mM formic acid (adjusted to pH 4.0 with NH
3) as mobile phase; flow was 0.7 mL/min; temperature was 30 °C (injection volumes were 5 µL). The outflow of the HPLC-system ran directly into the ICPMS (Agilent 7500ce or 7900). The ICPMS operated in no-gas mode either without optional gas, or with addition of 10 vol% optional gas (Ar/CO
2, 99 + 1, v/v) for As-signal enhancement.
2.7.3. Determination of Non-extractable As in Digested Pellets
The digested pellets were introduced into the ICPMS (Agilent 7500ce or 7900) through an ASX-500 auto sampler. The ICPMS was operated in the collision mode (5 mL/min He) either without optional gas or with addition of 10 vol% optional gas (Ar/CO2, 99 + 1, v/v).
2.7.4. Quality Control of Extracted As Determinations
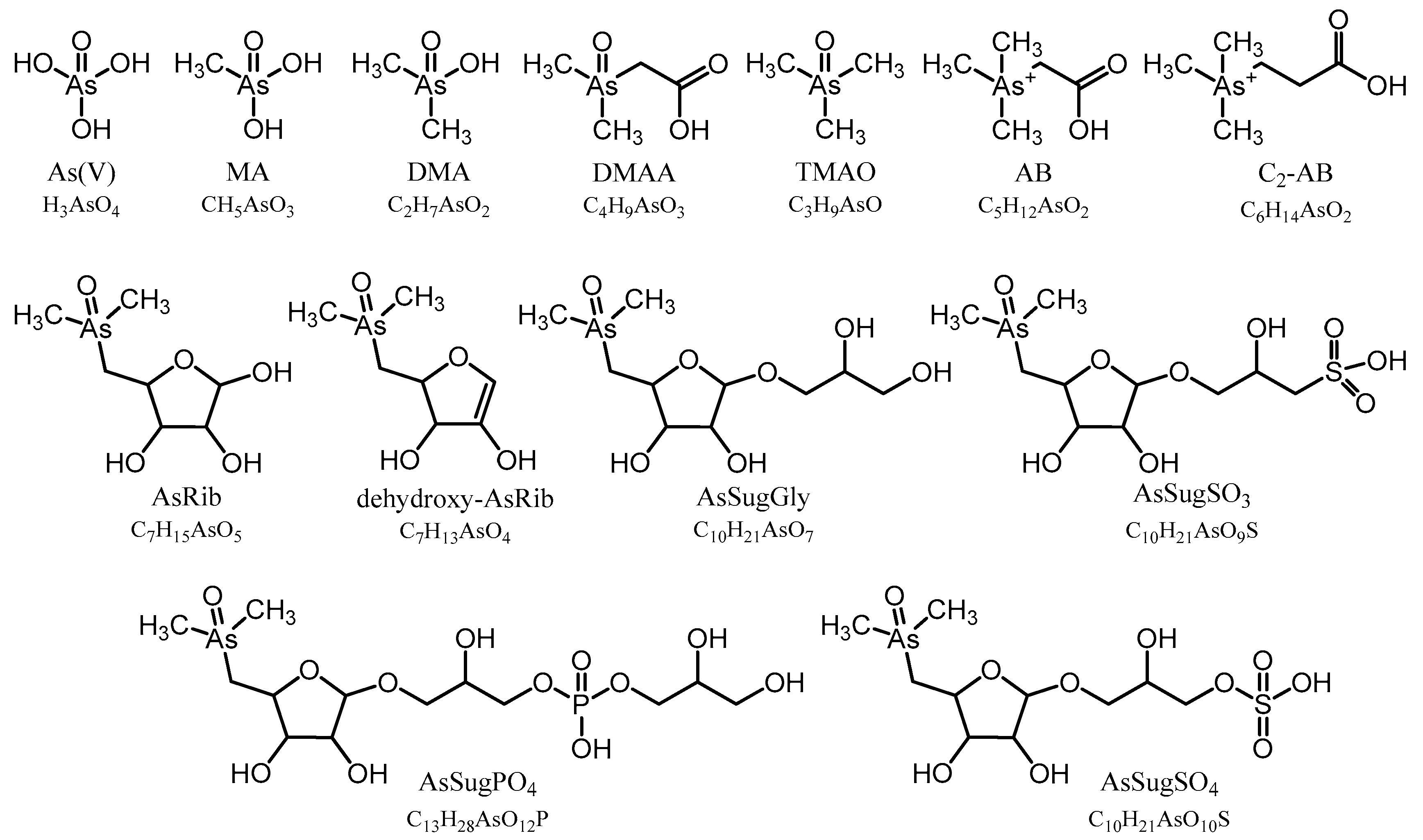
To monitor the analytical performance and stability of our methods, we used the previously characterized certified reference material (CRM) NMIJ 7405-a (Hijiki) obtained from the Metrology Institute of Japan (Tsukuba, Ibaraki, Japan) under each of the previously described conditions in triplicate. Individual As species (water- and lipid-soluble) were previously determined and are compared in
Table S1. We also analyzed an in-house reference material (
Dunaliella tertiolecta), produced during a previous study in our laboratory in Graz [
15], to further validate the applied methods for those As species absent in CRM-Hijiki.
To identify individual arsenic species and to monitor the analytical performance and stability of the applied methods, we used retention time matching between standard compounds and environmental samples based on HPLC-ICPMS and HPLC-ESMS for those compounds, where standards were available: As(V), MA, DMA, TMAO, AB, C
2-AB, AsRib, AsSugGly, AsSugSO
3, AsSugSO
4, AsSugPO
4, AsHC332, and AsHC360. When As standard compounds were not available, we used a combination of the previously determined CRM 7405-a (Hijiki) as a reference for AsPL720, AsPL958, AsPL986, AsPL1014 [
32]; and the in-house cultured
Dunaliella tertiolecta as reference material for AsIsop546, AsPL718, AsPL720, AsPL958, AsPL978, AsPL980, and AsPL982 [
15]. We also obtained accurate masses (Δm ≤ 2 ppm) of all identified arsenic species by means of HR-ESMS (
Table S2), except for As(V), which did not ionize under the given conditions. In the case of the two newly discovered isoprenoyl 2-
O-methyl arsenosugars, AsIsop408 and AsIsop422, we also performed fragmentation experiments based on high resolution ESMS and compared the obtained MS
2 spectra with fragmentation patterns of already known organic arsenicals (
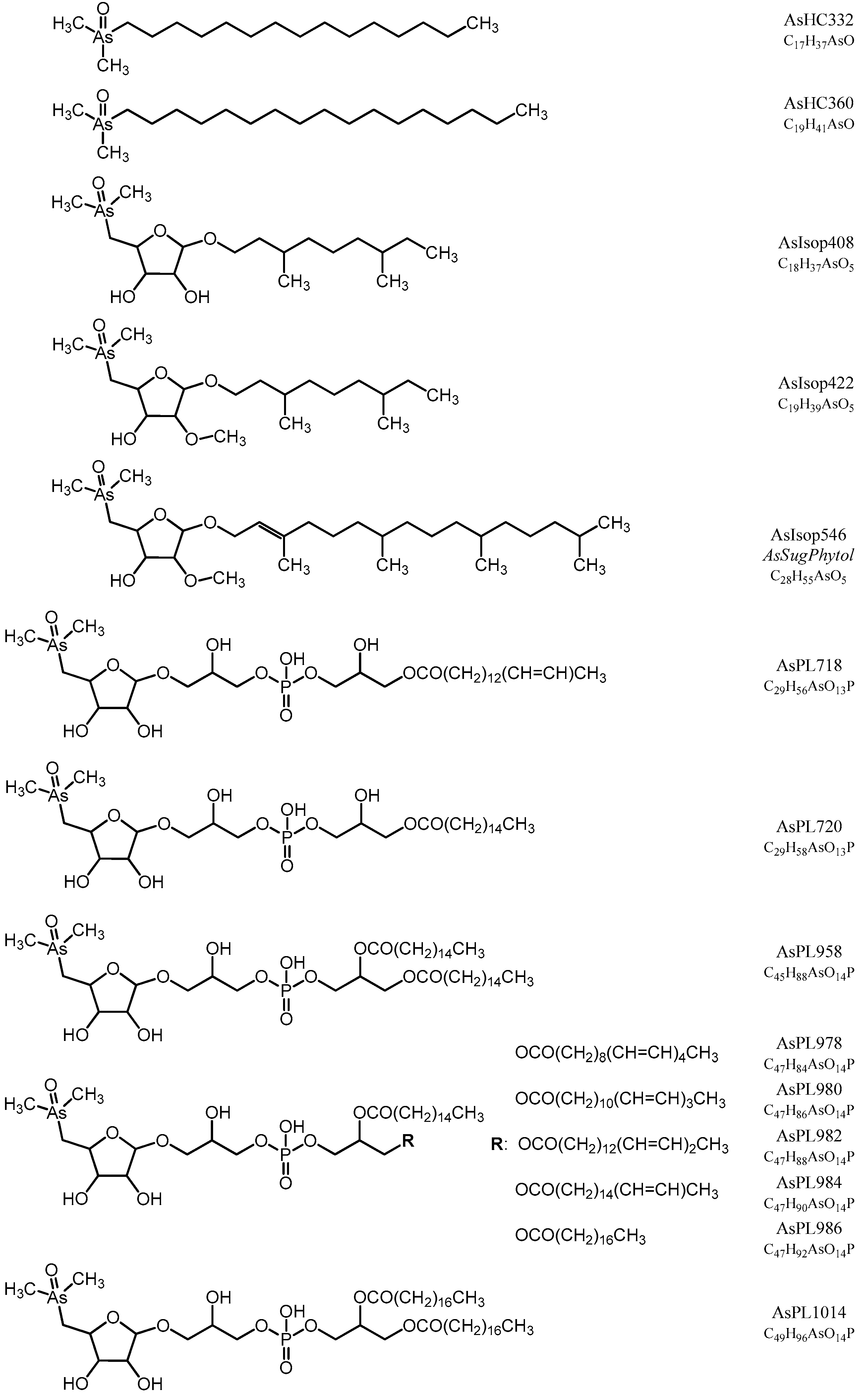
Figure S1) to propose the structures of the compounds. All determined arsenic species in this study are shown in
Figure 1 and
Figure 2.
We used CRM 7405-a (Hijiki) and the in-house cultured reference material
Dunaliella tertiolecta as samples to assure quantitative results of arsenic species determinations. Total digestion and As determination of CRM 7405-a (Hijiki) provided good agreement with the certified value (35.8 ± 0.9 µg As/g); we measured 35.6 ± 1.0 µg As/g (
n = 6). The same CRM is also certified for inorganic arsenate (10.1 ± 0.5 µg As/g); we measured 8.5 ± 0.9 µg As/g (
n = 6), as sum of all aqueous fractions. The lower recovery was possibly related to the slightly different extraction procedure compared with that used in the original CRM determination in which As(V) was directly extracted from dry Hijiki with water at elevated temperatures [
36]. Concentrations of other arsenic species than As(V) are not certified in the CRM but can be compared with previously published values (
Table S1).
2.8. Analysis of Arsenic within Picocystis Cells by X-ray Spectroscopy
Bulk X-ray spectroscopy was performed at the Stanford Synchrotron Radiation Lightsource (SSRL) using beam line 7-3. The incident X-ray energy was obtained using a Si (220) double crystal monochromator, with the Stanford Positron Electron Accelerating Ring (SPEAR) storage ring containing 500 mA at 3.0 GeV in top-off mode. Samples were prepared by depositing a small aliquot of the centrifuged pellet onto a nucleopore filter, and the excess moisture of the sample was wicked away with a kimwipe below the filter. Samples were mounted in the beam with a motorized sample positioning system, and the sample position in the incident X-ray beam between each measurement was adjusted slightly to ensure that a fresh part of the sample was analyzed to minimize any beam-induced damage. The incident and transmitted X-ray intensities were measured with nitrogen-filled ion chambers. Energy calibration of the monochromator was monitored using an Au foil measured in transmission geometry between two ion chambers after the sample. Fluorescence detection of As K-alpha fluorescence was measured using a PIPS photodiode detector, when needed for samples containing lower As concentrations. Samples with high As concentrations were processed using the transmission data if possible to reduce the effect of sample self-absorption. Samples were mounted at 45 ° to the incident X-ray beam to minimize scattering. The spectra were collected from 200 eV below the arsenic K-edge to ~1200 eV above the edge, with a minimum counting time of 1 s per point below the edge, up to 30 s per point at the end of the scan. Each scan had a length of approximately 25 minutes. For each sample, approximately 5-20 replicates were measured, depending on the As concentration in the sample, to achieve the desired counting statistics and check for potential beam damage. Spectroscopy data were analyzed using standard methods with the SIXPACK software package [
37]. Calculations of the theoretical EXAFS were performed using the FEFF7 package [
38].
4. Discussion
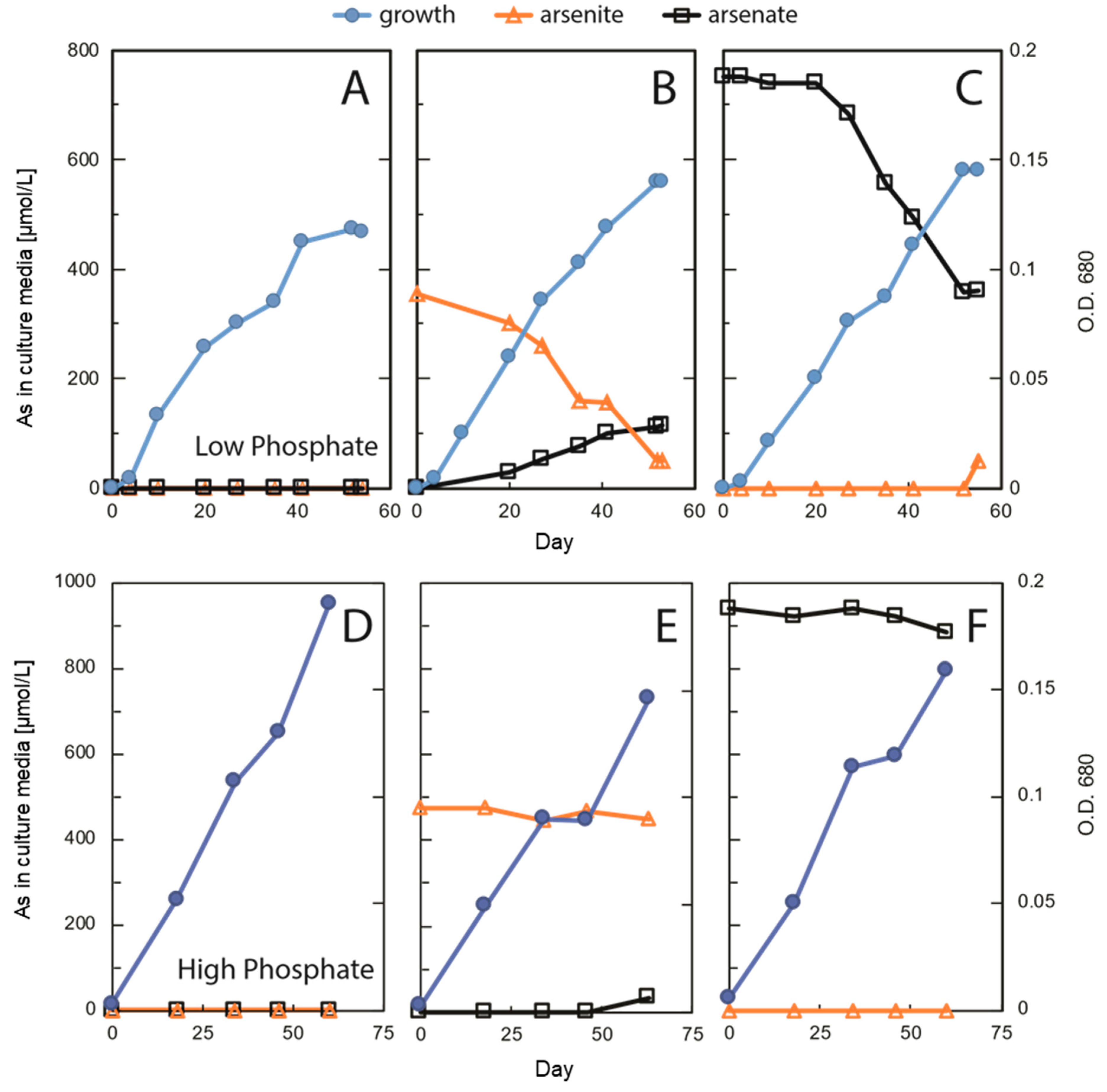
Laboratory incubation of
Picocystis strain ML always generated growth (
Figure 3), although not surprisingly, it grew more extensively under high-P conditions than at low-P (
Figure 3A,D). Growth at high-P was slightly retarded by inclusion of As(III) or As(V), but there was very little, if any, uptake or redox transformation of the oxyanions under these conditions (
Figure 3E,F and
Table 1). In contrast, under low-P removal of As(III) was evident as was its partial recovery in solution as As(V) (
Figure 3B). Presumably, the “missing” As under this condition was imported into the cells as As(V). In the As(V) culture medium, there was also a notable removal of As(V), but in contrast, there was little recovery in solution as As(III) (
Figure 3C). These results were reproducible (
Figure S2) and were previously shown to be light dependent [
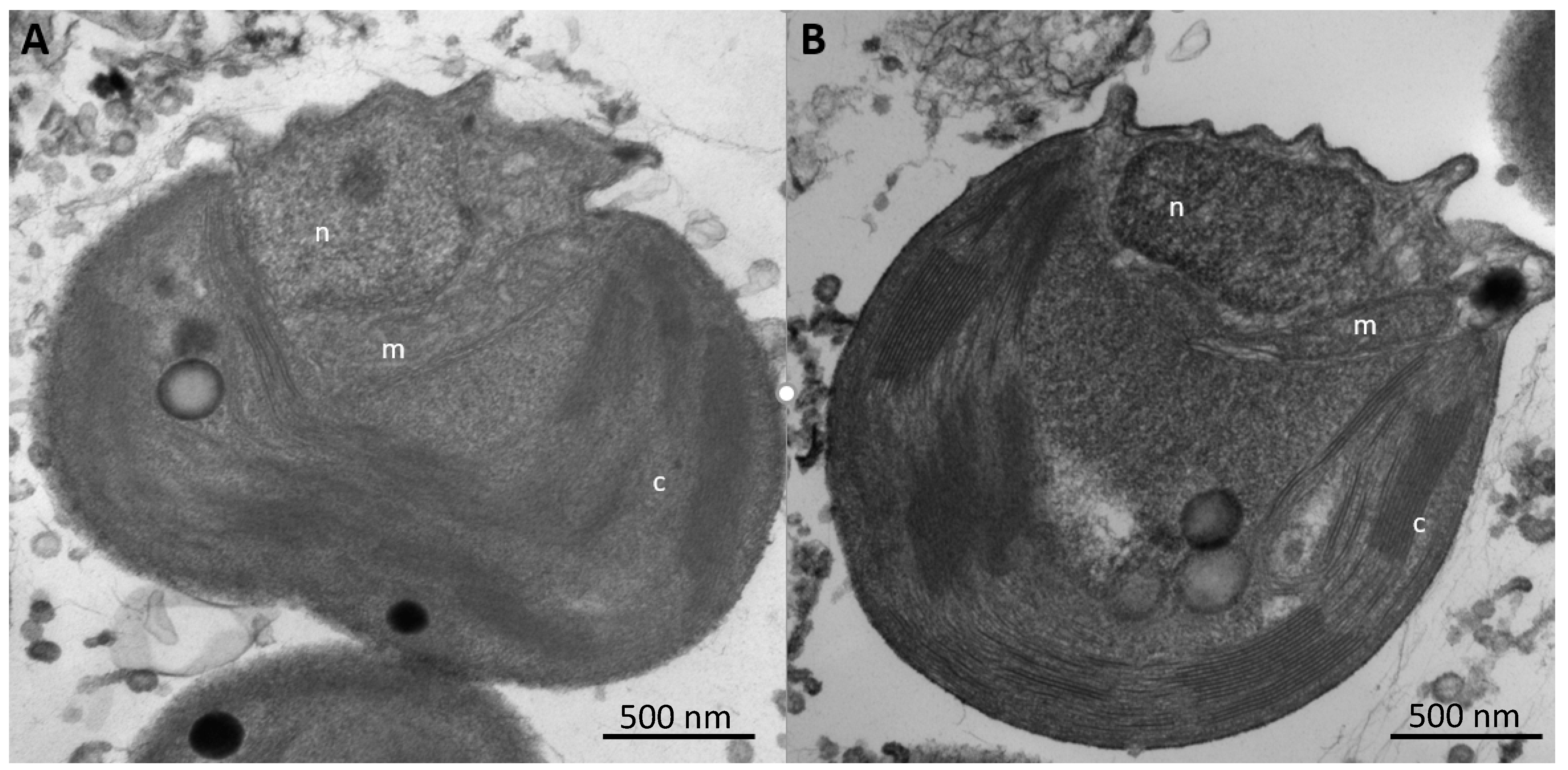
25]. In addition, we noted no changes in sub-cellular structures of cells under these different regimens, indicating that this organism was not experiencing any undue stress due to either P-limitation or As-importation (
Figure 4). These observations suggest extensive As uptake into the cells under low-P vs. high-P conditions, as verified quantitatively by the extractions (
Table 1). It is also evident that cells were not seriously inconvenienced by this internal As accumulation under the former conditions. Possibly, some of the imported As(V) was employed to substitute for phosphate in various biopolymers, such as membrane associated arsenolipids. This was shown to occur for the bacterium
Agrobacterium tumefaciens under highly complex cellular regulation [
41,
42]. It was also observed for the eukaryote thermoacidophile
Cyanidioschyzon sp., which carried out a variety of arsenic bio-transformations of As-oxyanions, including redox, methylation, and volatilization [
43]. Little is known about arsenic metabolism in unicellular eukaryotic algae as compared with prokaryotes, let alone by a
Cyanidioschyzon sp. isolated from arsenic-rich hot springs.
Picocystis strain ML, by contrast, is a broadly adaptable haloalkaliphile [
26] that is the main photoautotrophic picoplankton inhabiting Mono Lake, an environment particularly rich in arsenic oxyanions (~200 µM).
Based on our observations (
Figure 2) we assume that
Picocystis strain ML contains the genetic machinery for arsenic uptake (e.g., phosphate transporters; aquaglycerol porins) and for redox transformations (e.g.,
arsC; aioBA) expressed more highly under P-limitation. Such expression occurred in the cyanobacterium
Synechocystis [
44]. A draft genome of strain ML was published [
27] but not assembled. Moreover, because the sequences were scattered amongst hundreds of contigs, searches for specifically annotated arsenic genes were impossible to discern at this early stage. What is clear is that strain ML will actively oxidize As(III) to As(V) under P-limitation and will actively acquire As(V) from the aqueous medium under these conditions (
Figure 3B,C). This would likely be for its incorporation into macromolecules that under normal P-replete conditions will employ phosphate.
As expected from our above observations, uptake and incorporation of As(V) into cells of strain ML occurred, as evident in the extracted fractions (
Table 1). Uptake was most extensive under conditions of P-limitation for both the As(III) [which was oxidized to As(V)] and As(V) amendments, and the lipophilic arsenic portions exceeded those for the P-replete conditions. An unexpected finding was the extensive amount of As recovered from the cells under the low-P conditions. In these cases, the recovered arsenic accounted for 0.35% and 13.3% of the cell dry weight, under the As(III)- and As(V)-treated conditions, respectively. Comparably high As levels were previously only reported in As-hyper-accumulators such as terrestrial ferns containing up to ca 2.7% of their dry mass mainly as inorganic arsenite [
45,
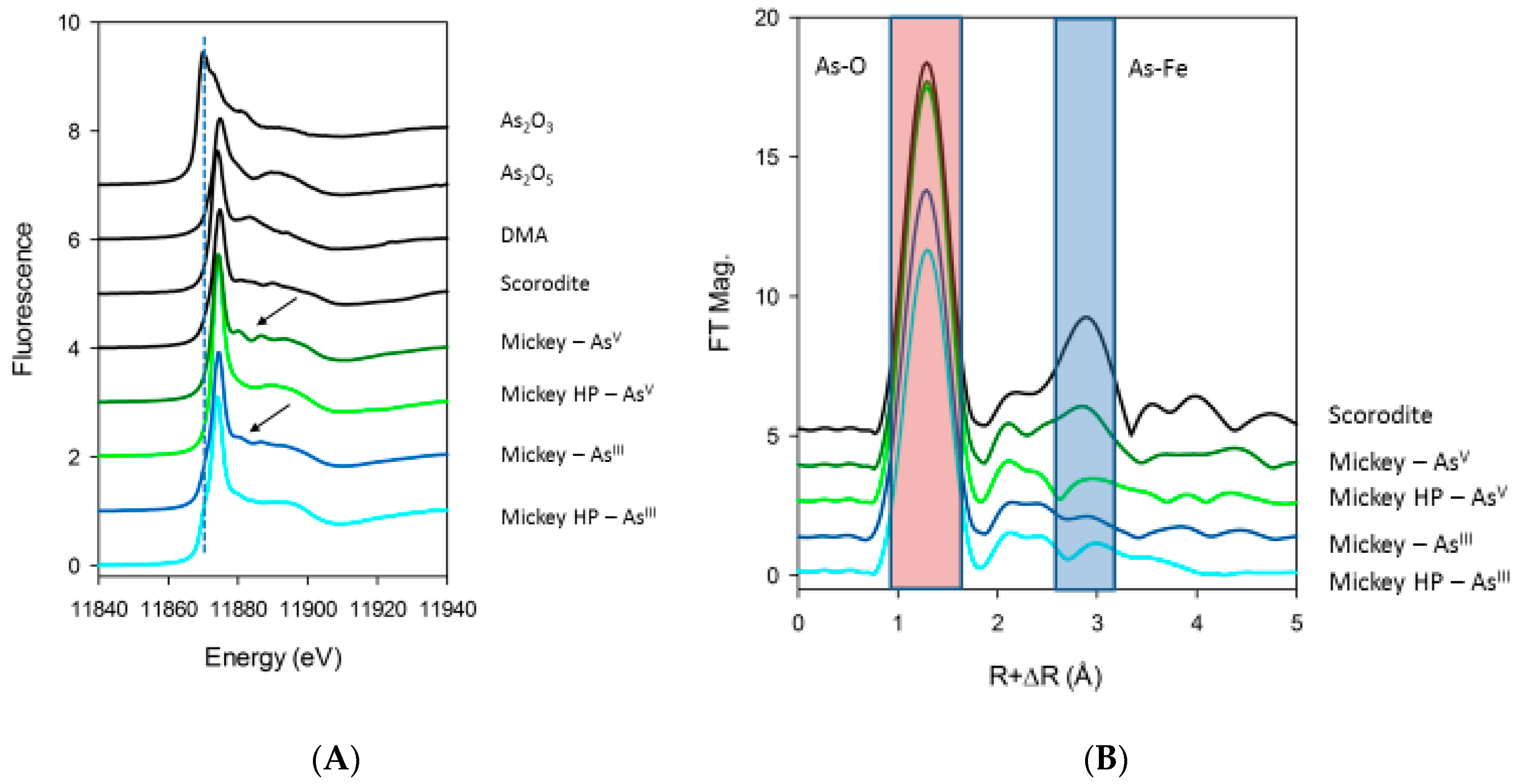
46]. Most of the here detected algal arsenic was first classified as recalcitrant but ultimately was solubilized as As(V) after TFA extraction, leaving behind smaller amounts of non-extractable arsenic. We initially suspected that this unusually large amount of recalcitrant arsenic was incorporated into a biopolymer, but X-ray spectroscopy revealed it to be primarily a precipitate composed of mixed As-Fe oxide minerals like scorodite (
Figure 6) with a smaller quantity of cellular organoarsenic compounds entrained during precipitation. We do not know the specific function of this complex mixture, if any. We speculate that it is an adaptation to the presence of an excessive amount of internal As, which overwhelms the cells’ capacity for methylation and eventual export. It is possible that
Picocystis strain ML stores this fraction of imported As(V) as a generally inaccessible, innocuous precipitate. Perhaps cells can make use of this faction under sustained nutrient limitation, which if true would make it akin to a luxury uptake of nutrients (i.e., storage) like that for phosphorus and nitrogen [
47].
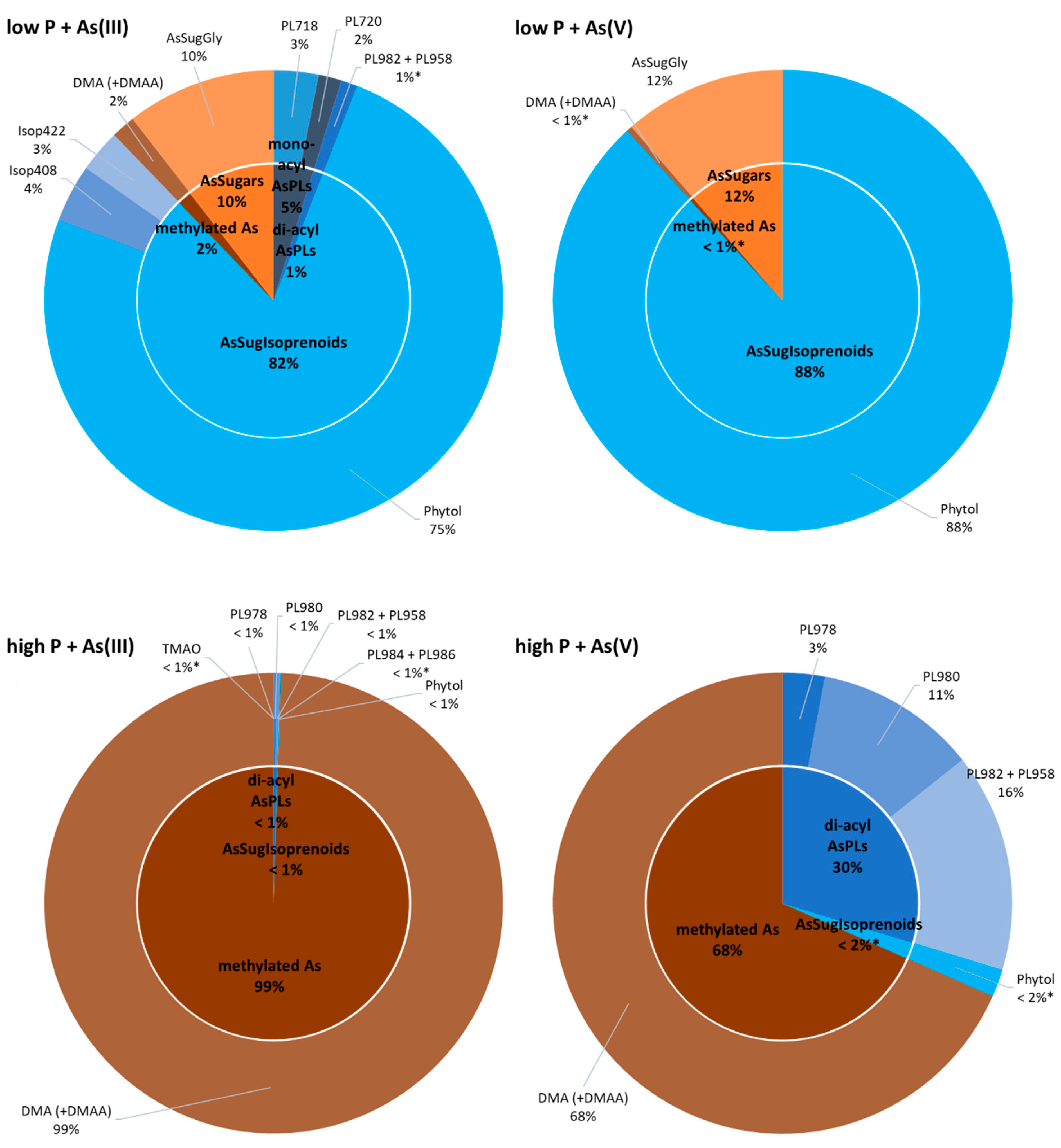
There were broad differences in the composition of organoarsenic species extracted from strain ML under the different incubation regimens (
Figure 5). At low-P, most of the extractable As was in the form of arsenolipids, which includes isoprenoyl 2-
O-metyl arsenosugars, forming the bulk of material recovered, 82% and 88%, from the As(III) and As(V) amended samples, respectively. In the case of the As(III) amendment, most of the arsenic was imported after its external oxidation to As(V), as occurs for
A. tumefaciens under P-limitation [
42]. There was only a small proportion of water-soluble organoarsenic species detected. The opposite situation was clearly evident under P-replete conditions, most strikingly for the As(III) amendment, where arsenolipids made up less than 1% of the extracted organoarsenicals, the remainder being water-soluble As compounds. This also held true for the As(V) amendment, but here arsenolipids represented a much larger percentage fraction (~32%) of the extractable arsenic. We conclude that the imported As(V) likely fulfills some of the functions of phosphate under the conditions of P-limitation. However, the presence of significant amounts of arsenolipids in the high-P/As(V) amendment implies that they can also fulfill a basic cellular function, such as incorporation into the cell’s membrane structure as already suggested by Ender and coworkers [
48]. Their near-absence in the P-replete/As(III) amendment suggests that this function is not mandatory for cell growth, and that synthesis of an As(III)-oxidizing protein (e.g., AioBA) is a waste of cellular energy, and its expression is repressed. Curiously, strain ML formed the phytyl 2-
O-methyl arsenosugar of particular interest in our study, under all added inorganic As incubation conditions. Upregulation of this As-containing potential membrane lipid was obvious under low-P conditions, resulting in ca 30-fold increase in cellular concentrations compared to high-P treatments independent of As-oxyanion amendment (
Table S3). Sufficient P supply led to an upregulation of arsenosugar phospholipids (likely incorporated into biological membranes) and, at the same time, complete downregulation of arsenosugars (potential energy storage molecules); the opposite was observed under P-starvation. These observations emphasize the distinct adaptive capacity of
Picocystis strain ML towards dynamic metabolism of organoarsenic species depending on prevalent environmental challenges.
Owing to the harshness of the lake’s water chemistry (high salinity, high pH, high toxic element concentrations), the ecosystem is simple as there is low diversity in the phytoplankton (primarily
Picocystis) and zooplankton (primarily
Artemia) communities, and no vertebrates (i.e., fish) are present. Direct trophic transfer was previously demonstrated between
Picocystis and
Artemia in feeding experiments [
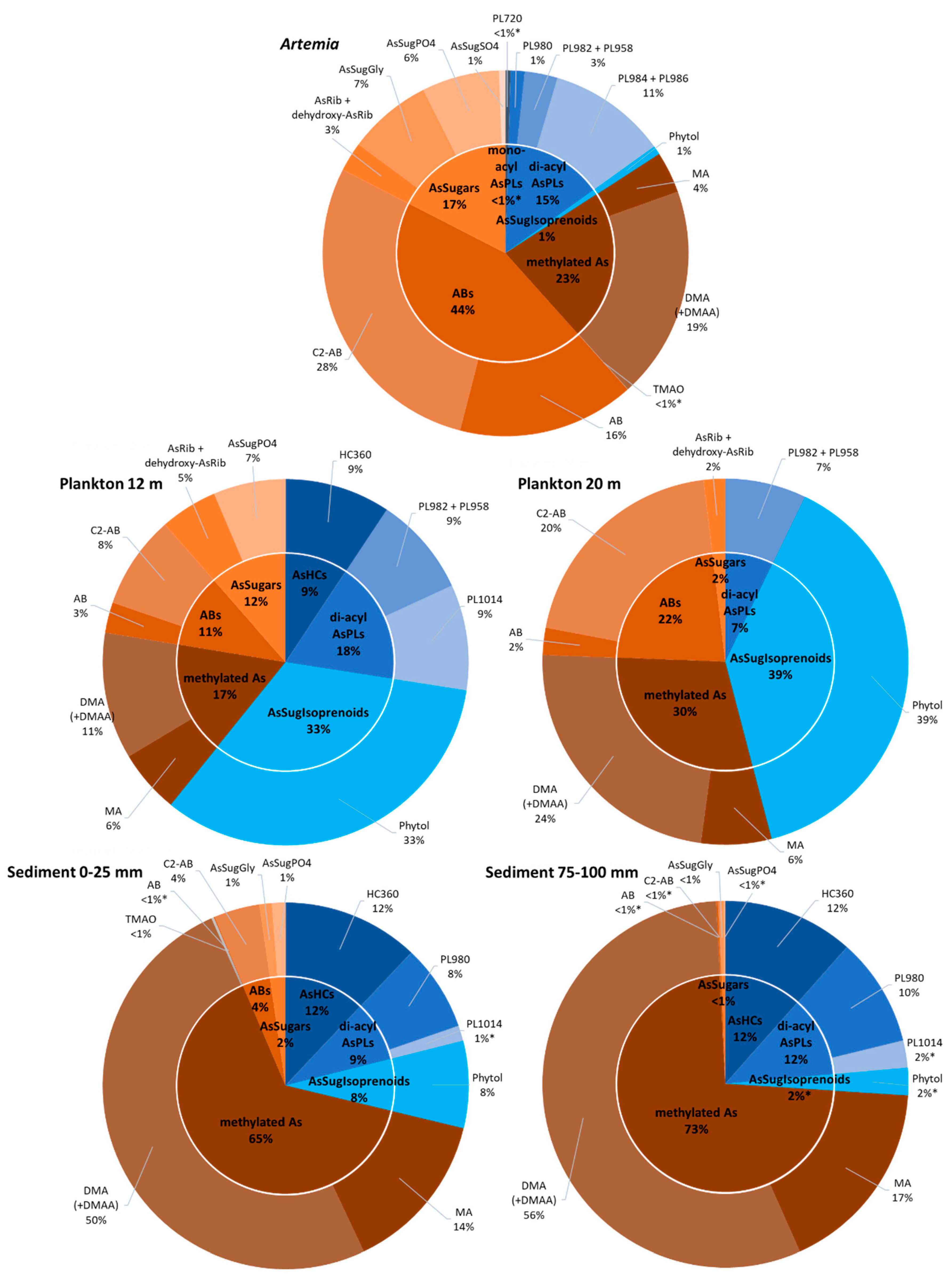
26]. Arsenolipids were present in all the samples recovered from Mono Lake (
Table 1 and
Table S3 and
Figure 7). Overall, they made up the largest percentage (~60%) of the organoarsenic recovered from the 12 m plankton sample, with the rest composed of water-soluble As, including arsenobetaine. Arsenobetaine is an analog of glycine betaine, and both molecules function as compatible solutes for osmotic balance in this hypersaline environment. The percentage of arsenolipids declined to ~45% in the 20 m plankton sample, while the amount of arsenobetaine doubled, probably due to adaptation to the higher salinity prevalent beneath the thermocline/pycnocline caused by a continuity of lake-wide meromixis (e.g., [
49]). Arsenolipids comprised only 16% of the organoarsenic extracted from
Artemia, while there was a significant arsenobetaine content (44%). This likely reflected the osmoregulatory strategy of this zooplankter, the main consumer of
Picocystis, thereby gratuitously obtaining a “salt-out” adaptation capacity by feeding. Arsenolipids were still present in the sediment samples, although at lower percentage levels than that displayed in the phytoplankton, presumably due to microbial degradation in the water column and within the sediment itself, as suggested by Glabonjat et al. [
22,
50]. This is better reflected by the greatly diminished levels of arsenobetaine, which presumably lends itself more readily to anaerobic biodegradation than arsenolipids, analogous to that for glycine betaine [
51].
Isoprenoyl 2-
O-methyl arsenosugars were present (<10%) in the sediment samples and as such now document the occurrence of these unusual compounds, in a new environment besides the oceans [
15,
52] and the Great Salt Lake [
22]. Considering that isoprenoyl 2-
O-methyl arsenosugars are the only such structures known outside the RNA molecule [
15], it is interesting to speculate that they could have had a broader role in the origin of life or geo-biological evolution. The occurrence of these phytol lipids in other environments, such as freshwaters and soils, would indicate a universality and perhaps determine if its relationship to RNA bears any evolutionary significance or is merely coincidental. Considering the surprisingly widespread global occurrence of
Picocystis strains like
P. salinarium [
53], a closer examination of whether this phytyl 2-
O-methyl arsenosugar has a broader physiological role than membrane anchoring would be worth investigating.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}