
Edge Structure of Montmorillonite from Atomistic Simulations

1
Department of Geology, Kangwon National University, Chuncheon 24341, Korea
2
Division of Energy and Environmental Systems, Faculty of Engineering, Hokkaido University, N13 W8, Kita-ku, Sapporo 060-8628, Japan
*
Author to whom correspondence should be addressed.
Minerals 2016, 6(2), 25; https://doi.org/10.3390/min6020025
Submission received: 23 February 2016
/
Revised: 18 March 2016
/
Accepted: 18 March 2016
/
Published: 25 March 2016
(This article belongs to the Special Issue Mineral Surface Science and Nanogeoscience)
Abstract
:Classical molecular dynamics (MD) simulations have been performed to investigate the effects of substitutions in the octahedral sheet (Mg for Al) and layer charge on an atomistic model of the montmorillonite edge. The edge models considered substitutions in both the solvent accessible and inaccessible octahedral positions of the edge bond chain for a representative edge surface. The MD simulations based on CLAYFF, a fully-flexible forcefield widely used in the MD simulations of bulk clay minerals, predicted Mg–O bond distances at the edge and in bulk that agreed with those of the density functional theory (DFT) geometry optimizations and available experimental data. The DFT results for the edge surfaces indicated that substitutions in the solvent inaccessible positions of the edge bond chain are energetically favorable and an increase in layer charge and local substitution density coincided with the occurrence of five-coordinate, square pyramidal Mg and Al edge structures. Both computational methods predicted these square pyramidal structures, which are stabilized by water bridging H-bonds between the unsaturated bridging oxygen [(Al or Mg)–O–Si] and other surface O atoms. The MD simulations predict that the presence of Mg substitutions in the edge bond chain results in increased disorder of the edge Al polyhedra relative to the unsubstituted edge. In addition to the square pyramidal Al, these disordered structures include trigonal bipyramidal and tetrahedral Al at the edge and inverted Si tetrahedra. These simulation results represent the first test of the fully-flexible CLAYFF forcefield for classical MD simulations of the Na-monmorillonite edge and demonstrate the potential of combined classical MD simulations and DFT geometry-optimizations to elucidate the edge structure of 2:1 phyllosilicate minerals.
1. Introduction
Smectites are 2:1 dioctahedral phyllosilicates abundant in soils and sediments [1]. Smectites are natural nanominerals with layers ~1.0 nm thick composed of an octahedral sheet between two tetrahedral sheets. The basal surfaces of the tetrahedral sheets are inert, but isomorphic substitutions in the structure can impart a permanent negative charge to the layers. The hydration of interlayer cations that balance this permanent charge results in interlayer swelling and the creation of nanopores. This physical structure is responsible for the unique physicochemical properties of smectites that include high surface area, high cation exchange capacity, and low permeability. These microscopic properties affect a range of macroscopic geological and geochemical processes and have been exploited in engineered geologic barriers [2,3] and nanocomposite materials [4,5,6]. These applications have been developed based on the comprehensive experimental and theoretical investigations of the smectite interlayers. The edges of these layer-type minerals, although they represent a much smaller fraction of the total surface area, are no less important to the reactivity of smectites.
The edges of 2:1 phyllosilicates are a source of pH dependent charge in soils. These highly-reactive edge sites exhibit a strong influence on cation and anion retention [7], the stabilization of soil organic matter [8,9,10], and colloidal and rheological properties [11,12,13]. In addition, the edge represents the boundary that diffusing solutes must cross between interlayer nanopores and neighboring meso- and micropores [14] and the dissolution of clay nanoparticles has been observed to proceed predominantly from the edge surfaces [15,16]. With a role in such an extensive list of clay surface reactions, a comprehensive understanding of 2:1 phyllosilicate structure and reactivity cannot be attained without a detailed atomistic characterization of the edge. This detailed characterization has remained elusive due to the experimental difficulties of isolating the edges. Careful manipulation of experimental variables such as pH and ionic strength [17,18,19] or the choice of non- or low-swelling 2:1 clay mineral phases such as pyrophyllite, K-mica, and beidellite [20,21,22] have been of some assistance in isolating the edge, but by far, the most insightful approach has been through atomistic simulations.
Numerous quantum mechanical (QM) simulations have provided valuable insights regarding the relaxation of the edge structure [23,24], surface energy of various facets [24,25], acid-base reactivity [23,24,26,27,28], and dynamical properties of the edge-water interface [29,30,31]. These simulations of the two dominant edge structures typically employ pyrophyllite as a model mineral as it is a non-swelling, 2:1 dioctahedral phyllosilicate with no structural charge; pyrophyllite possesses only a strongly reactive edge surface. Pyrophyllite is a useful analog for the study of the 2:1 dioctahedral phyllosilicate edge surfaces although it is somewhat rare in soils. Much more common in the environment is montmorillonite (MMT), a smectite mineral with a relatively low layer charge (x = 0.2 to 0.6 per chemical formula unit), wherein isomorphic substitutions in the octahedral sheet are in excess of those in the tetrahedral sheet [32]. Recent atomistic simulations using density functional theory (DFT) have sought to address the gap between the idealized atomistic models of the edge and the actual structure of smectites by introducing isomorphic substitutions into the models [26,27,30,31]. These simulations have demonstrated the effects of isomorphic substitutions on the coordination of the edge cations, the interfacial water structure, and the acid dissociation constants (pKas) of the edge sites. Perhaps due to the computational costs of QM simulations, these previous studies have employed atomistic models that are small and considered only a single isomorphic substitution in isolation in the structure.
Molecular mechanical (MM) studies are rare for the edge surfaces compared to the QM studies and have typically employed a rigid clay structure [14,33,34]. MM simulations can address the limited length- and time-scales of QM methods, but the accuracy of MM simulations are strongly dependent on the choice of the potential-based forcefield that describes the interatomic interactions. No forcefield, however, has been extensively tested for the edge surfaces because of the absence of high-quality experimental data for the edge. The fully-flexible CLAYFF forcefield [35], which was derived from QM methods and spectroscopic data, has been used extensively in molecular mechanics simulations for clay mineral phases, particularly for the basal planes and interlayer nanopores. Recent classical molecular dynamics (MD) simulations [36] have demonstrated that CLAYFF predicts edge structures and surface energies consistent with those of QM studies of pyrophyllite. CLAYFF has also been used in simulations at the extended length- and time-scales that are currently inaccessible to QM methods to predict pyrophyllite nanoparticle structures and propose disordered edge structures [37,38]. Further investigation is required, but these studies have demonstrated that CLAYFF can be used in MM simulations of some clay edge surfaces. The validation of CLAYFF for atomistic simulations of more common 2:1 clay mineral edges that include isomorphic substitutions is a natural next step and would expand the already considerable list of model structures that can be simulated.
In this study, we report the results of MD simulations of a MMT edge with substitutions of Mg for Al in the octahedral sheet using the CLAYFF forcefield. The resultant MD edge structures are compared with those of DFT geometry-optimizations that examined the effects of layer charge and the position of the edge substitution on the edge structures. We provide a quantitative description of the octahedral sheet edge structures and infer a relationship between layer charge and disordered edge structures.
2. Materials and Methods
2.1. Molecular Mechanics: Na-Montmorillonite Edge
The atomistic model of the Na-MMT edge was derived from a previous model of the pyrophyllite edge. The details of this previous model are described elsewhere [36], but the general characteristics of the model will be briefly summarized and differences that distinguish the Na-MMT model from the pyrophyllite model will be described in detail. Crystal growth theory [39] can be applied to 2:1 dioctahedral phyllosilicates [40] to identify three periodic bond chains (PBCs)—one is unique and two are symmetrically equivalent—that parallel the clay mineral edge faces. In this paper, we have chosen the nomenclature of White and Zelazny [40] and will refer to the symmetrically equivalent PBCs as the AC edge and the unique PBC as the B edge. These PBCs are composed of linked tetrahedron-octahedron-tetrahedron (TOT) units. Previous simulations identified the AC edge as the 2:1 dioctahedral edge with the lowest surface energy [24,36], and we have chosen the AC edge as the representative edge of Na-MMT. The atomistic models of the Na-MMT AC edge began from a 4 × 4 × 2 (a × b × c) expansion of the pyrophyllite-1Tc unit cell of Lee and Guggenheim [41]. The octahedral sheet of this 2:1 dioctahedral phyllosilicate unit cell is trans-vacant (i.e., the trans-site, where the anionic positions (OH−) lie across the octahedron diagonal, is vacant; the two cis-sites, where the anionic positions are shared on an octahedral edge, are occupied; Figure 1). This triclinic supercell was cleaved along the (010) plane to create the AC edge face. A 60 Å vacuum space perpendicular to the AC edge was introduced to separate the surfaces across a macropore; the edge faces were also separated across the bulk mineral phase by approximately 30 Å. The unsaturated bonds of the edge surface were saturated in the manner of previous simulations of the pyrophyllite edge [36]—namely, through the dissociative chemisorption and physisorption of an integer number of water molecules to maintain the surface at the point of zero net proton charge (p.z.n.p.c.). Each solvent-accessible TOT unit at the edge surface requires two water molecules to be saturated. The simulation cell c dimension and relative clay layer position were adjusted to accommodate a hydrated bilayer. The simulation cell macropore and interlayer were hydrated with 1389 water molecules. The triclinic cell geometry was simplified to agree with the standard practice of using monoclinic simulation cells for Na-MMT. The initial simulation cell geometry was a = 20.64 Å, b = 98.86 Å, c = 30.69 Å, α = 90.00°, β = 100.46°, and γ = 90.00°.
Isomorphic substitutions of Al3+ by Mg2+ in the octahedral layer were chosen at random and subject to two constraints. The random assignment of substitutions is consistent with previous solid-state nuclear magnetic resonance (NMR) experiments and QM simulations that demonstrated that Mg2+ substitutions are randomly distributed within the octahedral layer [42,43]. The first constraint follows from these experimental and theoretical investigations and requires that no Mg–(OH)2–Mg pairs exist within the octahedral sheet. The second constraint is informed by the properties of synthetic MMTs [44,45,46,47]. Synthetic MMTs with only octahedral substitutions produced via hydrothermal synthesis are limited to a layer charge, x, of 0.20 to 0.40. The units of x are the number of moles of excess electron charge per chemical formula. We define the reference chemical formula for MMT as Mx[Al(2−x)Mgx]Si4O10(OH)2. Where M is the interlayer counter ion and the term in brackets represents the octahedral sheet. The octahedral substitution ratio (i.e., the number of substitutions per total octahedral sites) is one-half of the layer charge. Synthetic MMTs with layer charges greater than 0.40 begin to exhibit trioctahedral character in the octahedral sheet and isomorphic substitutions in the tetrahedral layers [46]. Thus, the second constraint, derived from the layer charge, limits the substitution ratio of the edge PBC (i.e., Mg edge substitutions: total no. of octahedral edge sites in the PBC) to less than 0.20. Random assignments of substitutions that violated either constraint were ignored and a new random assignment was made. The random assignment of isomorphic substitutions in the octahedral layer created two distinct edge surfaces. One of the interfaces contains no isomorphic substitutions in the two bond chains that define the edge of the upper and lower layer (Figure 1 and Figure 2, left edge surface); this interface and these edge bond chains are pyrophyllite-like. The second interface contains one isomorphic substitution in the edge PBC of each layer (i.e., substitution ratio: 1:8; Figure 1 and Figure 2, right edge surface). In the upper layer, this substitution is solvent inaccessible (i.e., a linking position of the PBC; Figure 1, MgPBC). In the lower layer, the isomorphic substitution is in a solvent accessible position in the PBC (i.e., a water molecule can sorb to the edge octahedra; Figure 1, MgSol). These two interfaces can be further distinguished by the substitution ratio of the PBC that is adjacent to the edge bond chain. The bulk bond chains that are adjacent to the substituted edge have a higher substitution ratio (0.375) than the bond chains adjacent to the unsubstituted edge (0.125 and 0.25). A total of 26 Mg substitutions were introduced into the octahedral layer; an equal number of Na ions were placed in the hydrated interlayer space to balance this permanent structural charge. This atomistic model of Na-MMT had a layer charge of 0.40. The chemical formula for the MMT without sorbed water molecules was (Mg26Al102)Si256O640(OH)128.
The CLAYFF forcefield parameters [35] described the Coulomb and short-range interactions as well as the harmonic bonds of the hydroxyl groups in the atomistic model of Na-MMT. Minor adjustments were made to the partial Coulombic charges of the octahedral Mg atoms (qmgo = 1.3598 e) and the bridging O atoms with double substitution (qobss = −1.3116 e) to maintain the charge neutrality of the atomistic model. The aqueous phase was described by the extended simple point charge (SPC/E) water model [48] with harmonic bond stretching and angle bending terms [48,49]. Long-range electrostatic interactions and the attractive contribution to the van der Waals interaction were evaluated using the Ewald summation [50] with the repulsive contribution truncated at a distance of 9.0 Å. All Ewald summations were determined to an accuracy of 0.0001 kcal/mol.
All MD simulations were performed with the LAMMPS software package [51] installed on the supercomputer Hopper at the National Energy Research Scientific Computing Center (NERSC; Berkeley, CA, USA). The models were initiated with a series of short MD simulations in the microcanonical (NVE, T = 298.15 K) ensemble—where the number of atoms (N), simulation cell volume (V), and energy (E) were held constant. The temperature was controlled with a temperature-rescaling thermostat and the SHAKE algorithm for water molecule bonds and angles. The aqueous phase was equilibrated with a timestep progression of Δt = [0.001, 0.01, 0.2, 0.3, 0.4, 0.5 fs] for 40,000 steps each before the atoms of the MMT phase were relaxed from their rigid positions. The entire system was then simulated for an additional 10,000 steps with Δt = 0.25 and 0.5 fs. At this point, the ensemble was changed to isobaric-isothermal [NPT; (P = 1.0 atm, T = 298.15 K)]—where pressure (P) and temperature (T) were held constant. A chain of Nose-Hoover thermostats controlled the temperature and pressure for the next 100.0 ps of model equilibration with Δt = 0.5 fs. After these approximately 200 ps of equilibration, a production run of 3.5 ns was continued with this same simulation algorithm. The resultant edge structures from these 3.5 ns trajectories were assessed on the basis of the metal-oxygen pair distribution functions and the integral of these distribution functions that provides the metal-oxygen coordination number. The internal routines of the LAMMPS code were used to calculate these pair distribution functions. The charactersitic properties of the pair-distribution functions (i.e., metal-oxygen distances, rMeO; the first-shell minimum of the pair-distribution function, ρ; and the coordination numbers, CNs) were calculated from 10 time-averaged blocks of 200 ps duration. The hydrogen bonds (H-bonds) at the NaMMT edge-water interface were defined by the donor-acceptor separation distance and donor-hydrogen···acceptor angle criteria developed by Kumar, et al. [52] for the SPC/E water model (dH-bond ≤ 3.2 Å; ∠H-D···A ≤ 40°) and rendered with the open-source program, Visual Molecular Dynamics (VMD) [53].
2.2. Density Functional Theory (DFT) Computations
All DFT simulations were performed with the CASTEP code [54] using ultrasoft pseudopotentials [55] under the generalized gradient approximations [56]. The kinetic-energy cutoff for the planewave basis set was 600 eV. The primitive Brillouin zone was sampled using a 6 × 4 × 4 grid and one point in k space [57] for the pyrophyllite unit cell and supercells for the surface models, respectively. With the chosen kinetic-energy cutoff and k-point grid, the atomic forces converged to <<0.01 eV/Å. The geometry optimization permitted the coordinates of all ions to relax. Geometry optimizations were performed until the residual atomic force was ≤0.03 eV/Å and the root-mean-square stress of the bulk structure was ≤0.02 GPa.
A periodic slab model of the AC edge surface was created based on a 2 × 2 × 1 supercell of a geometry optimized pyrophyllite-1Tc unit cell from Lee and Guggenheim [41]. This slab model has two symmetric edge surfaces at the left and right of Figure 3. The AC edge structure was cleaved from the supercell in the same manner as the molecular mechanics simulations. The unsaturated bonds were healed via the same dissociative chemisorption and physisorption reactions with water molecules to create an edge surface at the p.z.n.p.c. The Si edge tetrahedra each possess one silanol (≡SiOH) group and the solvent accessible octahedron has an amphoteric [≡(Al or Mg)…OH2] group. A total of four H2O molecules per edge surface were used to saturate the solvent-accessible TOT units. The generic chemical formula for the surface slab models without sorbed water molecules was [Al(16−n)Mgn]Si32O80(OH)16, where n = 2 or 3 in our DFT simulations, corresponding to a layer charge of 0.25 or 0.375. The slab model was comprised of four PBCs (~18 Å thickness). The vacuum space perpendicular to the edge surface slab model was ~20 Å. The supercell dimensions for the DFT edge surface model were: 10.40 × 38.05 × 10.12 Å (a × b × c) with each edge surface ~1.0 nm2. The periodic images of the surface models were separated by a distance greater than 17.8 Å.
Isomorphic substitutions of Mg for Al were introduced into the PBC of each edge surface and, in some models, the bulk of the octahedral sheet. The effect of the substitution location in the edge PBC was investigated by considering two different models. In the first model, the isomorphic substitution occupied a solvent accessible octahedron in the edge PBC (Figure 3a, MgSol); the isomorphic substitution of the second model occupied a linking octahedral position in the PBC (Figure 3b, MgPBC). A single isomorphic substitution in each edge PBC results in a MMT with a layer charge of 0.25. The effect of layer charge was investigated by creating two additional models based on the originals. These two models introduced one additional isomorphic substitution in the bulk of the octahedral sheet (Figure 3b, MgBulk). The additional MgBulk substitution increased the layer charge of these models to 0.375, but the substitution ratio of the edge PBCs remained unchanged at 0.25. Although in excess of our defined constraint, this edge substitution ratio is the lowest possible for our DFT model geometry. The small size of these atomistic models imposes limits on the substitution ratios and layer charges that can be considered. Thus, we have chosen models that most closely agree with those of the classical mechanical simulations described above and are within the range of the octahedral substitution ratios of synthetic MMT.
The isomorphic substitution of Mg for Al results in a net negative charge in the super cell that was compensated through the application of a uniform positive background [58]. Although errors may arise from unwanted Coulombic charge interactions between periodic images within the uniform background, this error can be eliminated through the use of simulations cells with infinitively large volumes. In this study, we compared the total energy of the two models with variations only in the position of the substituted Mg in a fixed supercell volume. As the error associated with interaction between the periodic images does not depend on the atomic positions, this error was cancelled in the energy comparison.
For the edge surfaces with solvent accessible Mg, the adsorption energy of a water molecule physisorbed to the octahedral site (Hads) was calculated for the exposed Al and Mg sites (i.e., six-fold vs. five-fold Al or Mg) by
where E4s and E3s are the total energies of the surface with sorbed water coverage of 4 and 3 H2O/nm2, respectively, and is the total energy of an isolated H2O molecule in a 25 × 25 × 25 Å cell.
3. Results
3.1. Surface Mg
The characteristic properties of the cation-O pair distribution functions of the octahedral sheet obtained by MD simulations are summarized in Table 1. The Mg–O coordination number (CN) for the solvent accessible Mg (MgSol) was less than that of Mg in the linking PBC position (MgPBC) or bulk (MgBulk). The Mg substitutions in the linked PBC position and bulk maintained octahedral coordination (CN ≈ 6) throughout the simulation. The physisorbed water of MgSol was observed to exchange readily with the bulk water phase during the MD simulation. The ease with which water is exchanged from this site in the MD simulations is consistent with the low water adsorption energy for the solvent accessible Mg sites (Hads, −57 kJ/mol) calculated using DFT. This exchange resulted in a transient edge Mg that is in square pyramidal coordination with O.
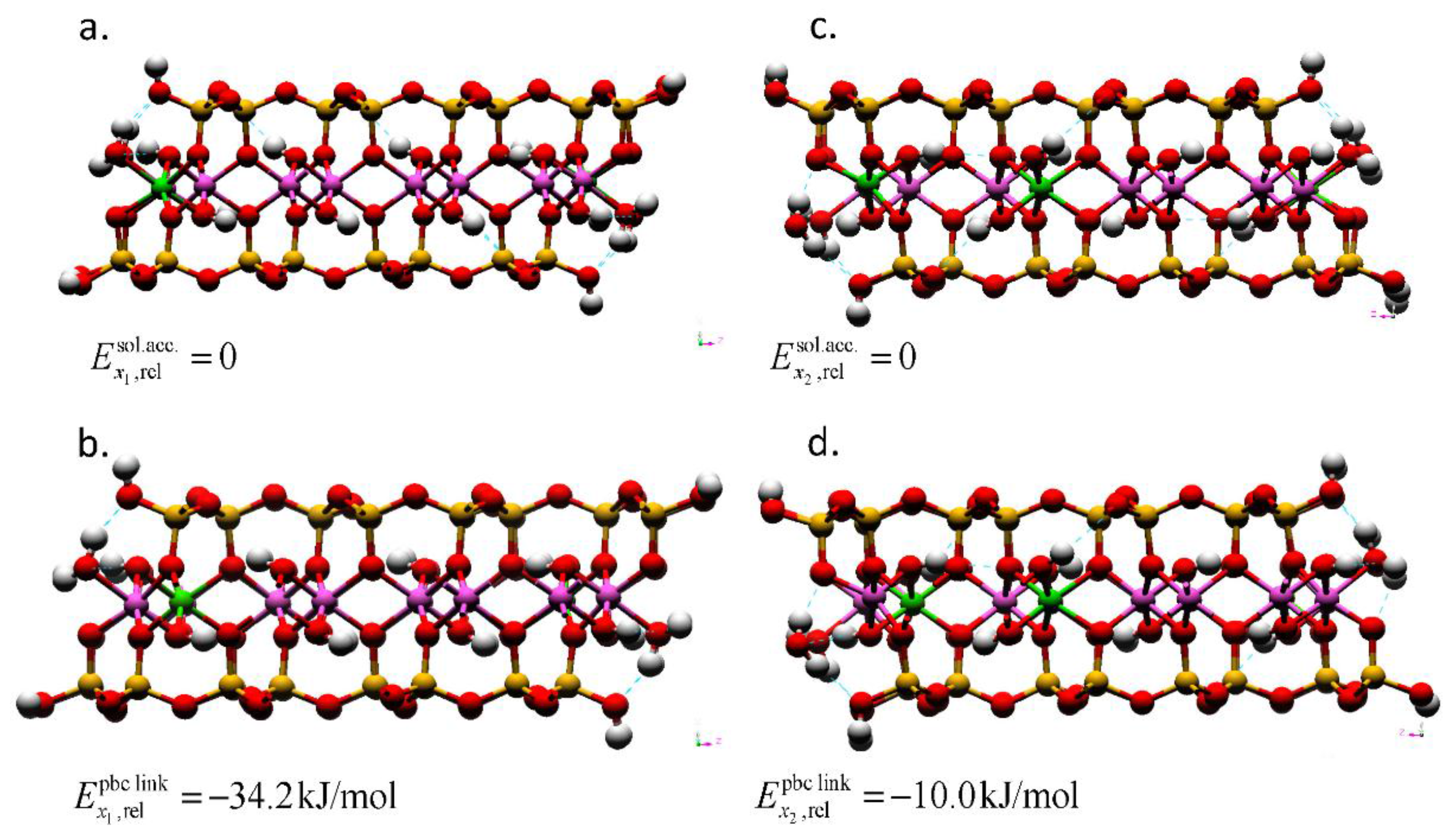
The Mg substitutions can occur at two different locations within the edge bond chain (MgSol and MgPBC). Using DFT, we compared the total energies of the different models to evaluate which substitution site is preferable (Figure 4). The MgPBC substitution possessed a lower total energy than the MgSol substitution at both layer charges considered. The energy difference indicates that, in isolation, the solvent accessible sites are not preferable for Mg substitution at the edge surface. However, the relative difference in the total energies between the models with solvent accessible and linking PBC substitutions decreased with an increase in layer charge (−34.2 to −10.0 kJ/mol). At the higher layer charge, the MgSol site that was closest to the MgBulk assumed a square pyramidal coordination (left side of Figure 4c), whereas the MgSol sites were in octahedral coordination at the edge most distant from the MgBulk (right side of Figure 4c) and at the lower layer charge (Figure 4a). This change in the coordination number and reduction in the energy difference imply that solvent accessible sites could be substituted at higher layer charges with a tendency to assume five-fold coordination. In the MD simulations, the model possessed a higher layer-charge and included a MgSol site. The CN of less than 6 at the MgSol site is rationalized based on the energy difference in our DFT results.
The MD-calculated average Mg-O distances (Table 1: rpeak) for the substitutions in the linked PBC position are slightly longer, but not significantly so, than the Mg-O distances of the solvent accessible and bulk mineral substitutions. The extent of the Mg-O first shell (ρ) is more compact in the bulk than at the edge surface. This difference is attributed to the greater crystallinity of the bulk phase and surface relaxation. The Mg-O distances obtained by MD simulations are within 0.04 Å (≤2%) of the distances from our DFT geometry optimizations and both simulation results are in agreement with the experimental range (2.00–2.12 Å) reported for the bulk Mg-O distances in synthetic MMT [44] and hydrotalcite [59] and for aqueous Mg ions [60]. In general agreement with the MD results, the DFT-calculated average Mg–O bond distances were relatively constant regardless of the position of the substitution (i.e., MgSol, MgPBC, or MgBulk; Table 2) although an increase in the layer charge slightly decreased the MgSol-O distance. This decrease is a result of a contraction of the MgSol–O bonds associated with the mineral layer but the MgSol-OH2 distance that actually increased with increasing layer charge masks the extent of this contraction.
3.2. Surface Al
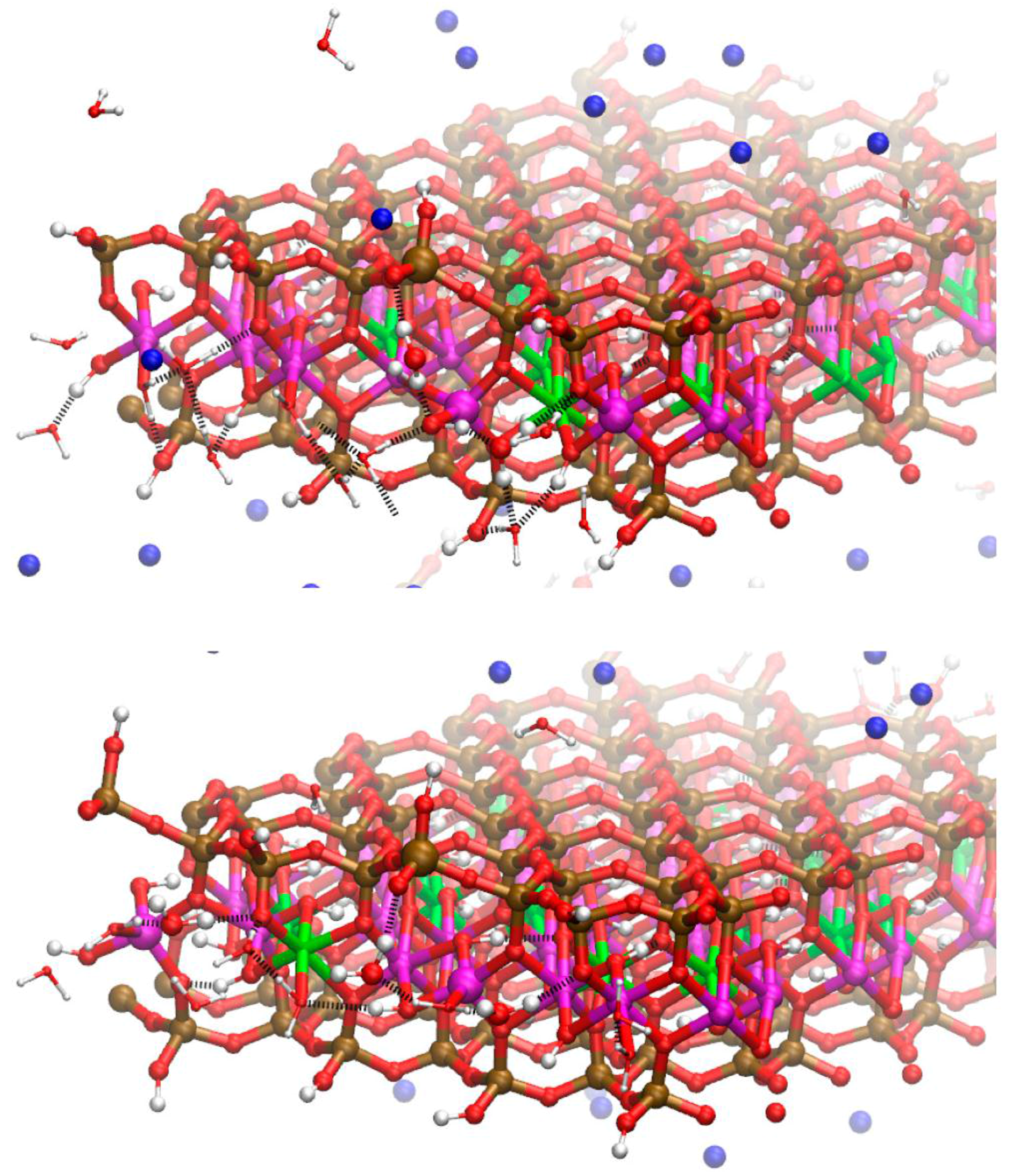
The MD simulations also showed that the presence of isomorphic substitutions in the edge PBCs affects the structure of the edge Al. The average Al-O distances and CNs of the unsubstituted edge are nearly indistinguishable from one another by position (i.e., AlSol, AlPBC, and AlBulk). In contrast, the average Al-O distance and CN of the substituted edge differ by the Al position. The Al-O distance and CN decrease proceeding from the bulk to the edge in the substituted edge. A comparison of the two edges reveals that the average Al-O distances in the solvent accessible (AlSol) and linked PBC positions (AlPBC) of the substituted edge (Table 1: substituted edge PBC) are less than the corresponding distances in the unsubstituted edge (Table 1: unsubstituted edge PBC). The decrease in the average Al-O distances in the substituted edge is a consequence of the greater number of edge Al in non-octahedral coordination with O, especially the solvent accessible Al. This interpretation of the Al-O CN can be confirmed visually in Figure 5 where square pyramidal, trigonal bipyramidal, and tetrahedral Al are present. These disordered edge Al structures have also been reported in previous MD simulations of pyrophyllite edges and nanoparticles [37,38].
The Al structural features shown by the MD simulations are consistent with our DFT results. In the DFT geometry optimizations, the effects of the Mg substitutions and increased layer charge on the Al-O distances are most evident when the AlSol-OH2 distances are considered (Table 2). For the pyrophyllite AC edge surfaces, which possess no isomorphic substitutions (x = 0), the AlSol-OH2 distances were 2.02–2.06 Å [24,61]. When the edge PBC includes a MgSol (Table 2: MgSol models), the AlSol-OH2 distance tends to increase with increasing layer charge. The greatest AlSol-OH2 distance was 2.10 Å at x = 0.375, when the solvent accessible Al was nearest the bulk Mg substitution. When the edge PBC substitution is in the linking position (Table 2: MgPBC models), a dramatic increase in the AlSol-OH2 distances occurred with increasing layer charge. The AlSol that share hydroxyl oxygens (oh) with the MgPBC have a greater AlSol-OH2 distance than the AlSol that share bridging oxygens (ob) with the MgPBC. The greatest AlSol-OH2 distance in the MgPBC models occurred at the AlSol that is nearest the bulk Mg substitution (3.20 Å), indicating non-octahedral coordination of edge Al as shown in the MD simulations.
In our MD simulations, the coordination number of the MgSol and MgPBC were 5.72 and 5.99—similar to the results of AlSol and AlPBC. The reduced coordination of the MgSol is attributed to the low water adsorption energy, Hads, (−57 kJ/mol). When we calculated the Hads for the solvent accessible Al sites using DFT, it was equivalent to that of the Mg sites (−57 kJ/mol). Thus, physisorbed water molecules associated with the solvent accessible octahedral sites of the AC edge are likely to be exchanged [29,30], regardless of whether the octahedral cation is Al or Mg.
3.3. Surface Si and H-Bonds
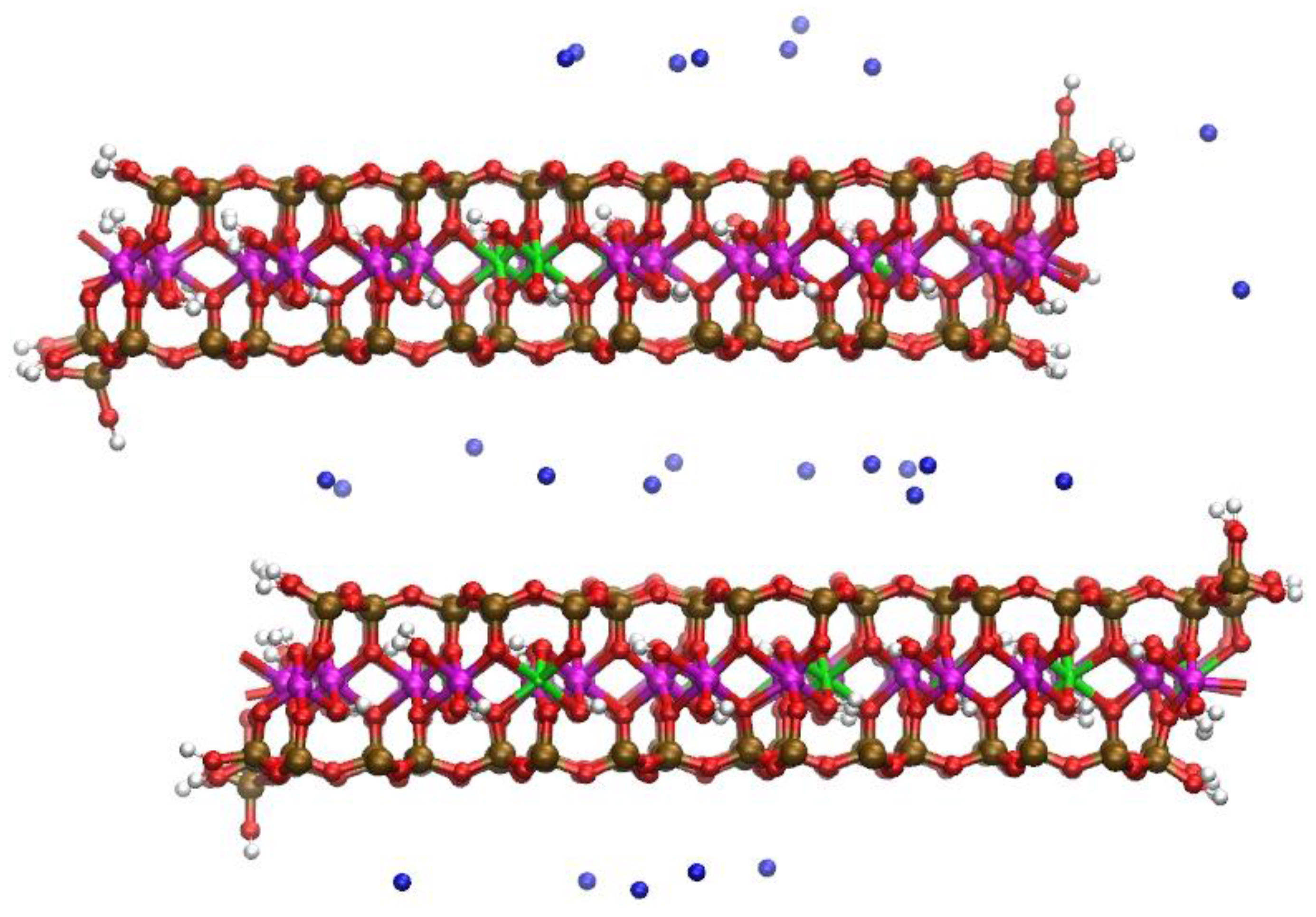
The MD simulations also predicted disorder in the Si tetrahedral sheet. The inverted Si tetrahedra of the AC edge (Figure 5 and Figure 6) develop as a consequence of the trigonal bipyramidal Al at the edge. These inverted Si are a feature of a historic alternative model of montmorillonite first proposed by Edelman and Favejee [62]. Both the pyrophyllite-like and the Mg-substituted edge faces in the MD simulations of the Na-MMT model possessed these Si-inversions (Figure 6). The inverted Si and non-octahedral Al structures were stabilized by water bridging H-bonds as recently reported for the pyrophyllite edge surface [37,38]. Although experiments have not yet provided direct evidence of an inverted Si in 2:1 dioctahedral phyllosilicates, the alternative EF structure can, in principle, explain some inconsistencies in smectite reactivity [63,64,65]. A recent atomic thermodynamics study based on DFT has suggested that these inverted Si at the edge of pyrophyllite are possible defect structures at the AC edge of 2:1 phyllosilicates [61].
Our MD simulations further showed that the H-bonds associated with the unsaturated bridging oxygens at the edge (i.e., ≣Al–O–Si≣ and ≣Mg–O–Si≣) differ depending on the identity of the edge cation. When the edge cation is Al, the average number of H-bonds between the bridging oxygen and water is 1.00. When a solvent accessible Mg is substituted at the edge, the average number of H-bonds between the water and the bridging oxygen was 1.64. This value should be interpreted carefully as only one bridging O bonded to MgSol exists in the model in contrast to the 10 bridging O bonded to AlSol. The average number of H-bonds indicates that this bridging O, when bonded to MgSol, is more likely to form two H-bonds with the water over the course of the MD trajectory than a single H-bond. This increase in the number of H-bonded water molecules at the site of the octahedral substitution agrees with that reported in a recent DFT MD simulation [31]. The square pyramidal Al and Mg observed in the geometry-optimized DFT models with layer charge x = 0.375 (Figure 4c,d) were also stabilized by water bridging H-bonds. The water bridge forms between the H atoms of water molecule and the unsaturated bridging O [≣(Al or Mg)–O–Si≣] and a silanol group (≣Si–OH). The square pyramidal Al is further stabilized through an additional hydrogen bond between the O of the water bridge and the exposed structural hydroxyl (≣Al2–OH).
4. Discussion
Previous QM simulations of a small 2:1 phyllosilicate edge model with Mg substitutions in the solvent accessible octahedral position have reported the existence of the square-pyramidal edge Mg in addition to octahedral Mg [30]. Our DFT geometry optimizations and classical MD simulations corroborate these previous findings and, contrary to the previous reports of the six-fold Al only being stable under the influence of an octahedral substitution, demonstrate that the solvent accessible Al can also take on a square pyramidal configuration. In the present DFT results, the square pyramidal Al develops when proximal to Mg substitutions in the linking PBC position and bulk mineral. Previous studies considered Mg substitutions only in one of the edge PBC and did not include bulk substitutions [30,31]; these edge PBC substitution ratios were less than or equal to the ratio in our models. Our larger slab model permitted us to consider substitutions both at interfaces and in bulk with a layer charge (x = 0.375) that more closely approaches the experimental layer charge constraint.
The five-coordinate Al and Mg surface structures developed in the model where the local charge deficit—due to isomorphic substitutions—would be greatest in the DFT model. These five-coordinate structures only developed at the solvent accessible cation closest to the bulk Mg substitution (Figure 4c); the solvent accessible cations isolated from the bulk substitution remained in octahedral coordination. Based on this co-occurrence, we infer that a relationship between disordered edge structures and local charge deficits exists. Our classical MD simulations qualitatively support this inference and predict that the presence of octahedral substitutions results in disordered Al edge structures. The disorder in the substituted edges of the MD model was more extensive than in the DFT geometry optimization. We attribute this increased disorder to the Mg in the edge PBC and the octahedral substitutions in the adjacent bulk-side bond chains. The substitution ratio of these adjacent bond chains is greater at the substituted edge (0.375) than at the pyrophyllite-like edges (0.25 and 0.125). Both the MD and DFT simulation results support the inference that disordered edge structures are related to excess negative charge at the edge.
Our classical MD simulations and DFT geometry optimization results provide a basis from which future atomistic simulations can be used to explore the structure and reactivity of the lateral edges of montmorillonite and to interpret experimental results. Synthetic MMTs with a permanent structural charge originating from only the octahedral sheet are limited to layer charges less than 0.4. When the edge substitution ratio of an atomistic model of the Na-MMT AC edge is similarly constrained, our results have demonstrated that the CLAYFF forcefield may be used in molecular mechanics investigations of the MMT AC edge-water interface at the p.z.n.p.c. The presence of octahedral edge substitutions have already been incorporated into some models of the acid-base chemistry of montmorillonite [66]. Acid dissociation constants that have been determined from DFT simulations indicate that solvent accessible Mg substitutions possess extremely high pKas and increase the pKa of neighboring silanol groups [26,27]. The effects of Mg substitutions on the pKa of neighboring Al sites have not been reported but are worthy of consideration based on our present findings that Mg substitutions in the linking PBC position are energetically favored and increased layer charge can result in disordered edge structures. Our models of the Mg-substituted smectite edge also hold promise for future molecular mechanics simulations that examine surface phenomena such as cation complexation and diffusion across the boundary between meso- and nano-pores.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, Information/Communication Teechnology (ICT) and Future Planning (NRF-2013-R1A1A1004657 and NRF-2015R1A4A1041105). Aric G. Newton received additional funding for this research through the project “Multidisciplinary investigation on radiocesium fate and transport for safety assessment for interim storage and disposal of heterogeneous waste”, under the Initiatives for Atomic Energy Basic and Generic Strategic Research by the Ministry of Education, Culture, Sports, Science and Technology of Japan. A portion of the computations were performed using resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author Contributions
Aric G. Newton performed the molecular dynamics (MD) simulations; Kideok D. Kwon performed the density functional theory (DFT) geometry optimizations; and Aric G. Newton, Kideok D. Kwon, and Dae-kyo Cheong wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Sposito, G.; Skipper, N.T.; Sutton, R.; Park, S.H.; Soper, A.K.; Greathouse, J.A. Surface geochemistry of the clay minerals. Proc. Natl. Acad. Sci. USA 1999, 96, 3358–3364. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.W.; Wang, Y. Radionuclide interaction with clays in dilute and heavily compacted systems: A critical review. Environ. Sci. Technol. 2012, 46, 1981–1994. [Google Scholar] [CrossRef] [PubMed]
- Pusch, R.; Carlsson, T. The physical state of pore water of Na smectite used as barrier component. Eng. Geol. 1985, 21, 257–265. [Google Scholar] [CrossRef]
- Boulet, P.; Coveney, P.V.; Stackhouse, S. Simulation of hydrated Li+-, Na+- and K+-montmorillonite/polymer nanocomposites using large-scale molecular dynamics. Chem. Phys. Lett. 2004, 389, 261–267. [Google Scholar] [CrossRef]
- Liu, X.D.; Lu, X.C.; Wang, R.C.; Zhou, H.Q.; Xu, S.J. Interlayer structure and dynamics of alkylammonium-intercalated smectites with and without water: A molecular dynamics study. Clays Clay Miner. 2007, 55, 554–564. [Google Scholar] [CrossRef]
- Yu, Y.R.; Yang, X.N. Molecular simulation of swelling and interlayer structure for organoclay in supercritical CO2. Phys. Chem. Chem. Phys. 2011, 13, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Sposito, G. The Chemistry of Soils, 2nd ed.; Oxford University Press: Oxford, UK; New York, NY, USA, 2008; p. 329. [Google Scholar]
- Lal, R. Soil carbon sequestration impacts on global climate change and food security. Science 2004, 304, 1623–1627. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, F.J. Humus Chemistry: Genesis, Composition, Reactions, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1994. [Google Scholar]
- Wan, J.; Tyliszczak, T.; Tokunaga, T.K. Organic carbon distribution, speciation, and elemental correlations within soil micro aggregates: Applications of STXM and NEXAFS spectroscopy. Geochim. Cosmochim. Acta 2007, 71, 5439–5449. [Google Scholar] [CrossRef]
- Ramos-Tejada, M.D.; Ontiveros, A.; Plaza, R.D.C.; Delgado, A.V.; Duran, J.D.G. A rheological approach to the stability of humic acid/clay colloidal suspensions. Rheol. Acta 2003, 42, 148–157. [Google Scholar] [CrossRef]
- Rand, B.; Pekenc, E.; Goodwin, J.W.; Smith, R.W. Investigation into the existence of edge-face coagulated structures in Na-montmorillonite suspensions. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Ph. 1980, 76, 225–235. [Google Scholar] [CrossRef]
- Tombacz, E.; Szekeres, M. Colloidal behavior of aqueous montmorillonite suspensions: The specific role of pH in the presence of indifferent electrolytes. Appl. Clay Sci. 2004, 27, 75–94. [Google Scholar] [CrossRef]
- Rotenberg, B.; Marry, V.; Dufreche, J.F.; Giffaut, E.; Turq, P. A multiscale approach to ion diffusion in clays: Building a two-state diffusion-reaction scheme from microscopic dynamics. J. Colloid Interface Sci. 2007, 309, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Bickmore, B.R.; Bosbach, D.; Hochella, M.F.; Charlet, L.; Rufe, E. In situ atomic force microscopy study of hectorite and nontronite dissolution: Implications for phyllosilicate edge surface structures and dissolution mechanisms. Am. Mineral. 2001, 86, 411–423. [Google Scholar] [CrossRef]
- Bosbach, D.; Charlet, L.; Bickmore, B.; Hochella, M.F. The dissolution of hectorite: In-situ, real-time observations using atomic force microscopy. Am. Mineral. 2000, 85, 1209–1216. [Google Scholar] [CrossRef]
- Morton, J.D.; Semrau, J.D.; Hayes, K.F. An X-ray absorption spectroscopy study of the structure and reversibility of copper adsorbed to montmorillonite clay. Geochim. Cosmochim. Acta 2001, 65, 2709–2722. [Google Scholar] [CrossRef]
- Papelis, C.; Hayes, K.F. Distinguishing between interlayer and external sorption sites of clay minerals using X-ray absorption spectroscopy. Colloids Surf. A 1996, 107, 89–96. [Google Scholar] [CrossRef]
- Strawn, D.G.; Palmer, N.E.; Furnare, L.J.; Goodell, C.; Amonette, J.E.; Kukkadapu, R.K. Copper sorption mechanisms on smectites. Clays Clay Miner. 2004, 52, 321–333. [Google Scholar] [CrossRef]
- Brouwer, E.; Baeyens, B.; Maes, A.; Cremers, A. Cesium and rubidium ion equilibria in illite clay. J. Phys. Chem. 1983, 87, 1213–1219. [Google Scholar] [CrossRef]
- Elzinga, E.J.; Sparks, D.L. Nickel sorption mechanisms in a pyrophyllite-montmorillonite mixture. J. Colloid Interface Sci. 1999, 213, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Zachara, J.M.; Smith, S.C.; Liu, C.X.; McKinley, J.P.; Serne, R.J.; Gassman, P.L. Sorption of Cs+ to micaceous subsurface sediments from the Hanford site, USA. Geochim. Cosmochim. Acta 2002, 66, 193–211. [Google Scholar] [CrossRef]
- Bickmore, B.R.; Rosso, K.M.; Nagy, K.L.; Cygan, R.T.; Tadanier, C.J. Ab initio determination of edge surface structures for dioctahedral 2:1 phyllosilicates: Implications for acid-base reactivity. Clays Clay Miner. 2003, 51, 359–371. [Google Scholar] [CrossRef]
- Churakov, S.V. Ab initio study of sorption on pyrophyllite: Structure and acidity of the edge sites. J. Phys. Chem. B 2006, 110, 4135–4146. [Google Scholar] [CrossRef] [PubMed]
- Lavikainen, L.P.; Tanskanen, J.T.; Schatz, T.; Kasa, S.; Pakkanen, T.A. Montmorillonite interlayer surface chemistry: Effect of magnesium ion substitution on cation adsorption. Theor. Chem. Acc. 2015, 134. [Google Scholar] [CrossRef]
- Liu, X.; Lu, X.; Sprik, M.; Cheng, J.; Meijer, E.J.; Wang, R. Acidity of edge surface sites of montmorillonite and kaolinite. Geochim. Cosmochim. Acta 2013, 117, 180–190. [Google Scholar] [CrossRef]
- Liu, X.D.; Cheng, J.; Sprik, M.; Lu, X.C.; Wang, R.C. Surface acidity of 2:1-type dioctahedral clay minerals from first principles molecular dynamics simulations. Geochim. Cosmochim. Acta 2014, 140, 410–417. [Google Scholar] [CrossRef]
- Tazi, S.; Rotenberg, B.; Salanne, M.; Sprik, M.; Sulpizi, M. Absolute acidity of clay edge sites from ab-initio simulations. Geochim. Cosmochim. Acta 2012, 94, 1–11. [Google Scholar] [CrossRef]
- Churakov, S.V. Structure and dynamics of the water films confined between edges of pyrophyllite: A first principle study. Geochim. Cosmochim. Acta 2007, 71, 1130–1144. [Google Scholar] [CrossRef]
- Liu, X.; Lu, X.; Meijer, E.J.; Wang, R.; Zhou, H. Atomic-scale structures of interfaces between phyllosilicate edges and water. Geochim. Cosmochim. Acta 2012, 81, 56–68. [Google Scholar] [CrossRef]
- Suter, J.L.; Kabalan, L.; Khader, M.; Coveney, P.V. Ab initio molecular dynamics study of the interlayer and micropore structure of aqueous montmorillonite clays. Geochim. Cosmochim. Acta 2015, 169, 17–29. [Google Scholar] [CrossRef]
- Sposito, G. The Surface Chemistry of Natural Particles; Oxford University Press: Oxford, UK; New York, NY, USA, 2004; p. 242. [Google Scholar]
- Churakov, S.V.; Dähn, R. Zinc adsorption on clays inferred from atomistic simulations and EXAFS spectroscopy. Environ. Sci. Technol. 2012, 46, 5713–5719. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, M.; Karnland, O. Donnan equilibrium in Na-montmorillonite from a molecular dynamics perspective. Geochim. Cosmochim. Acta 2012, 77, 266–274. [Google Scholar] [CrossRef]
- Cygan, R.T.; Liang, J.J.; Kalinichev, A.G. Molecular models of hydroxide, oxyhydroxide, and clay phases and the development of a general force field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Newton, A.G.; Sposito, G. Molecular dynamics simulations of pyrophyllite edge surfaces: Structure, surface energies, and solvent accessibility. Clays Clay Miner. 2015, 65, 278–290. [Google Scholar] [CrossRef]
- Newton, A.G.; Sposito, G. Molecular simulations of disordered edge structures on 2:1 clay mineral nanoparticles. J. Colloid Interface Sci. 2016. under review. [Google Scholar]
- Newton, A.G. Atomistic Simulations of the Dioctahedral 2:1 Phyllosilicate Edge—Water Interface: Structure, Dynamic Properties, and Cation Complexes. Ph.D. Thesis, University of California, Berkeley, Berkeley, CA, USA, 2012. [Google Scholar]
- Hartman, P. Crystal Growth: An Introduction; North-Holland Pub. Co.: Amsterdam, The Netherlands; Elsevier: New York, NY, USA, 1973; p. 531. [Google Scholar]
- White, G.N.; Zelazny, L.W. Analysis and implications of the edge structure of dioctahedral phyllosilicates. Clays Clay Miner. 1988, 36, 141–146. [Google Scholar] [CrossRef]
- Lee, J.H.; Guggenheim, S. Single-crystal X-ray refinement of pyrophyllite-1Tc. Am. Mineral. 1981, 66, 350–357. [Google Scholar]
- Sainz-Diaz, C.I.; Timon, V.; Botella, V.; Artacho, E.; Hernandez-Laguna, A. Quantum mechanical calculations of dioctahedral 2:1 phyllosilicates: Effect of octahedral cation distributions in pyrophyllite, illite, and smectite. Am. Mineral. 2002, 87, 958–965. [Google Scholar] [CrossRef]
- Cadars, S.; Guegan, R.; Garaga, M.N.; Bourrat, X.; le Forestier, L.; Fayon, F.; Huynh, T.V.; Allier, T.; Nour, Z.; Massiot, D. New insights into the molecular structures, compositions, and cation distributions in synthetic and natural montmorillonite clays. Chem. Mater. 2012, 24, 4376–4389. [Google Scholar] [CrossRef] [Green Version]
- Reinholdt, M.; Brendlé, J.; Tuilier, M.-H.; Kaliaguine, S.; Ambroise, E. Hydrothermal synthesis and characterization of Ni-Al montmorillonite-like phyllosilicates. Nanomaterials 2013, 3, 48–69. [Google Scholar] [CrossRef]
- Reinholdt, M.; Miehe-Brendle, J.; Delmotte, L.; le Dred, R.; Tuilier, M.H. Synthesis and characterization of montmorillonite-type phyllosilicates in a fluoride medium. Clay Miner. 2005, 40, 177–190. [Google Scholar] [CrossRef]
- Reinholdt, M.; Miehe-Brendle, J.; Delmotte, L.; Tuilier, M.H.; le Dred, R.; Cortes, R.; Flank, A.M. Fluorine route synthesis of montmorillonites containing Mg or Zn and characterization by XRD, thermal analysis, MAS NMR, and EXAFS spectroscopy. Eur. J. Inorg. Chem. 2001, 11, 2831–2841. [Google Scholar] [CrossRef]
- Lantenois, S.; Champallier, R.; Bény, J.M.; Muller, F. Hydrothermal synthesis and characterization of dioctahedral smectites: A montmorillonites series. Appl. Clay Sci. 2008, 38, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Lopez-Lemus, J.; Chapela, G.A.; Alejandre, J. Effect of flexibility on surface tension and coexisting densities of water. J. Chem. Phys. 2008, 128. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, N.; Goddard, W.A. Acceleration of convergence for lattice sums. J. Phys. Chem. 1989, 93, 7320–7327. [Google Scholar] [CrossRef]
- Plimpton, S.J. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Kumar, R.; Schmidt, J.R.; Skinner, J.L. Hydrogen bonding definitions and dynamics in liquid water. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B Condens. Matter Mater. Phys. 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Leslie, M.; Gillan, M.J. The energy and elastic dipole tensor of defects in ionic-crystals calculated by the supercell method. J. Phys. C Solid State Phys. 1985, 18, 973–982. [Google Scholar] [CrossRef]
- Wedepohl, K.H. Handbook of Geochemistry; Springer-Verlag: Berlin, Germany; Heidelberg, Germany; New York, NY, USA, 1978. [Google Scholar]
- Ohtaki, H.; Radnai, T. Structure and dynamics of hydrated ions. Chem. Rev. 1993, 93, 1157–1204. [Google Scholar] [CrossRef]
- Kwon, K.; Newton, A.G. Structure and stability of pyrophyllite edge surfaces: Effect of temperature and water chemical potential. Geochim. Cosmochim. Acta 2016. under review. [Google Scholar]
- Edelman, C.H.; Favejee, J.C.L. On the crystal structure of montmorillonite and halloysite. Z. Kristallogr. 1940, 102, 417–431. [Google Scholar] [CrossRef]
- Monasterio, F.E.; Dias, M.L.; Pita, V.J.R.R.; Erdmann, E.; Destefanis, H.A. Effect of the organic groups of difunctional silanes on the preparation of coated clays for olefin polymer modification. Clay Miner. 2010, 45, 489–502. [Google Scholar] [CrossRef]
- Pusch, R.; Karnland, O. Physico chemical stability of smectite clays. Eng. Geol. 1996, 41, 73–85. [Google Scholar] [CrossRef]
- Sterte, J.; Shabtai, J. Cross-linked smecites. V. Synthesis and properties of hydroxy-silicoaluminum cross-linked montmorillonites and fluorhectorites. Clays Clay Miner. 1987, 35, 429–439. [Google Scholar] [CrossRef]
- Tournassat, C.; Ferrage, E.; Poinsignon, C.; Charlet, L. The titration of clay minerals: II. Structure-based model and implications for clay reactivity. J. Colloid Interface Sci. 2004, 273, 234–246. [Google Scholar] [CrossRef] [PubMed]
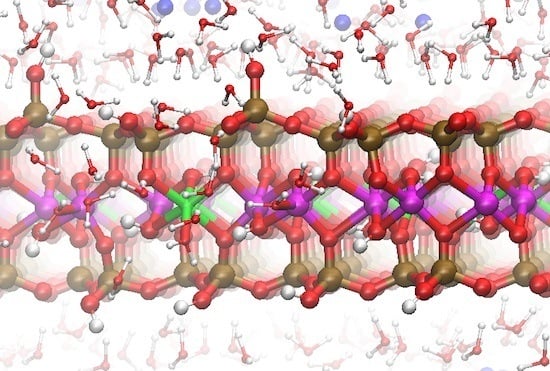
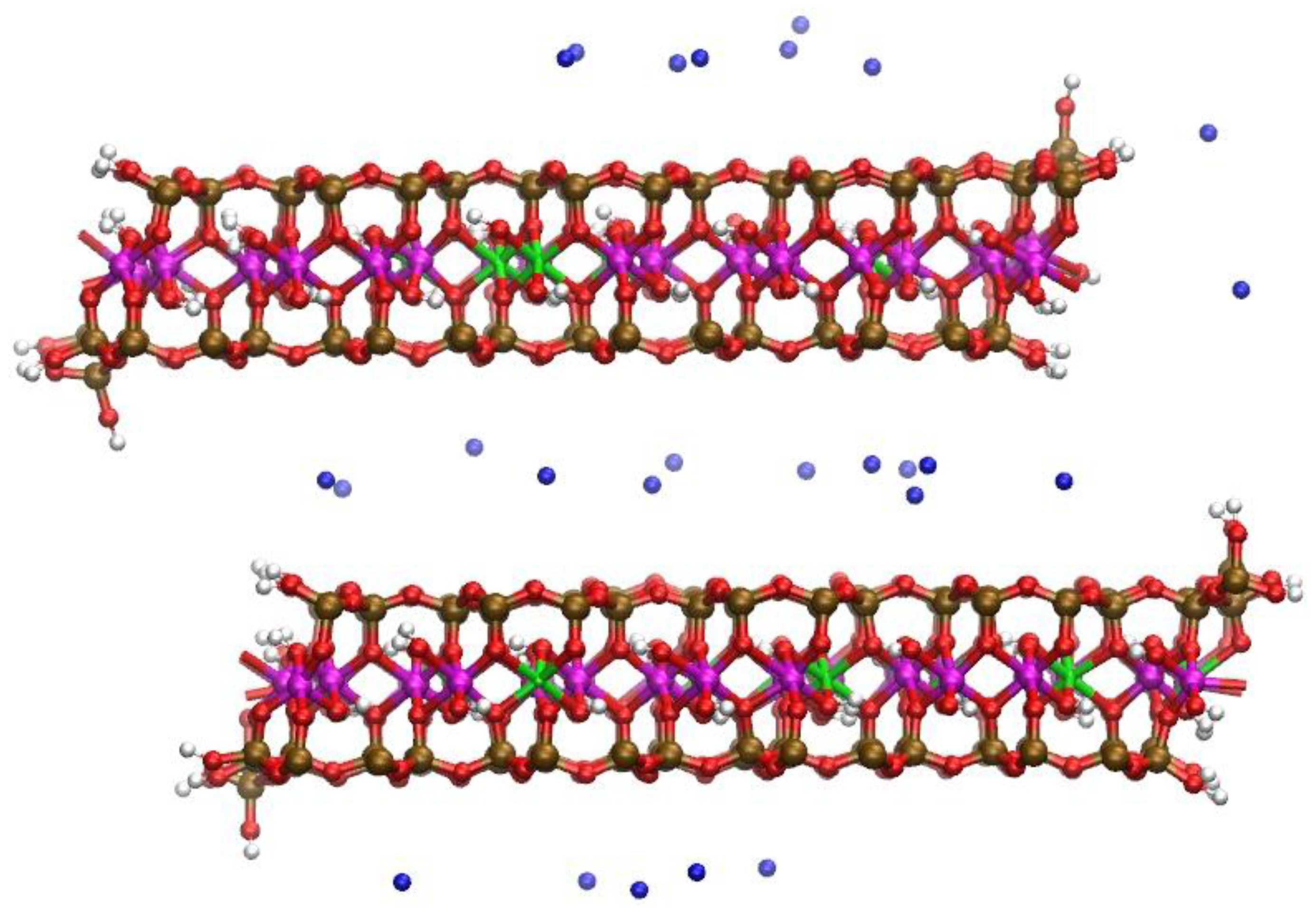
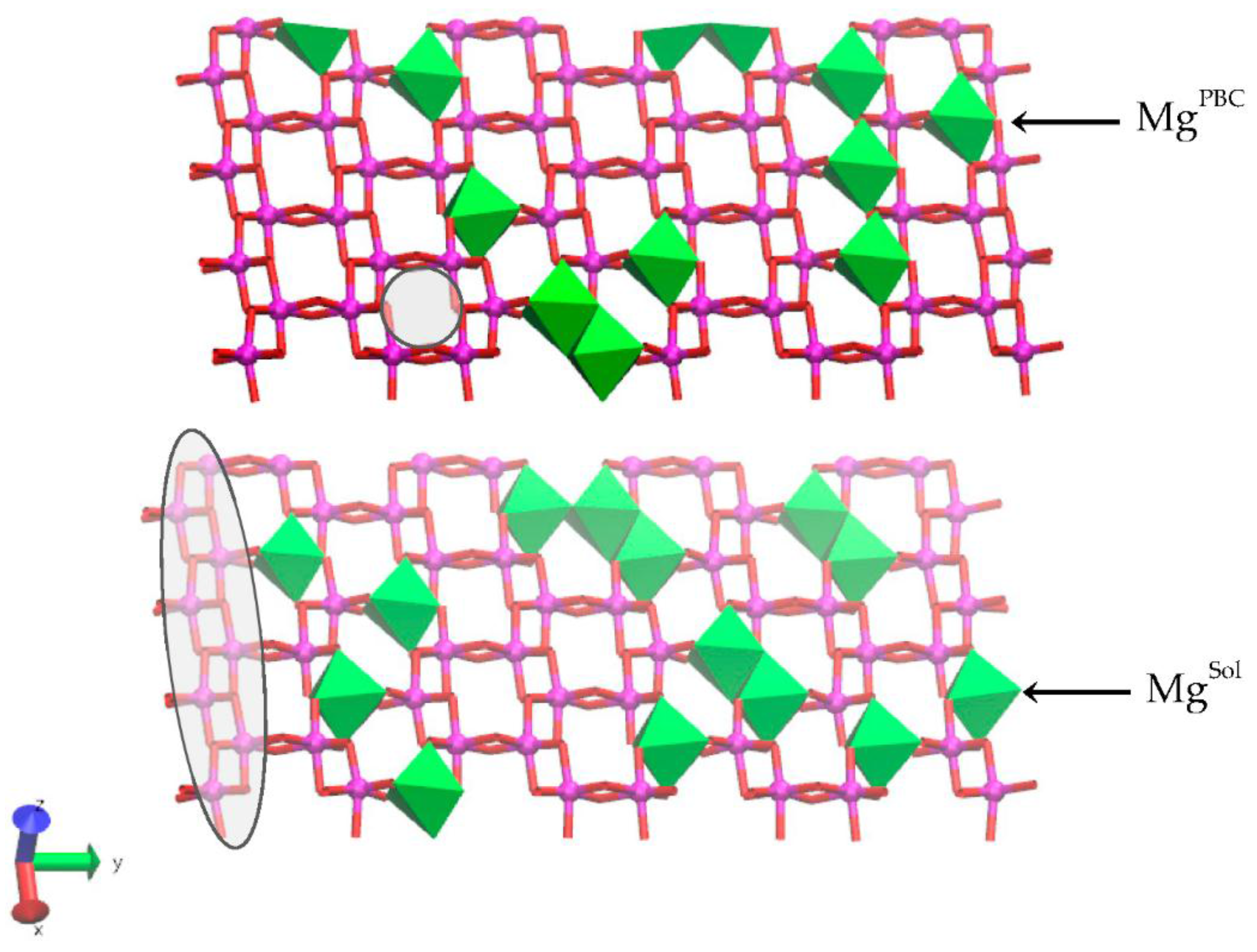
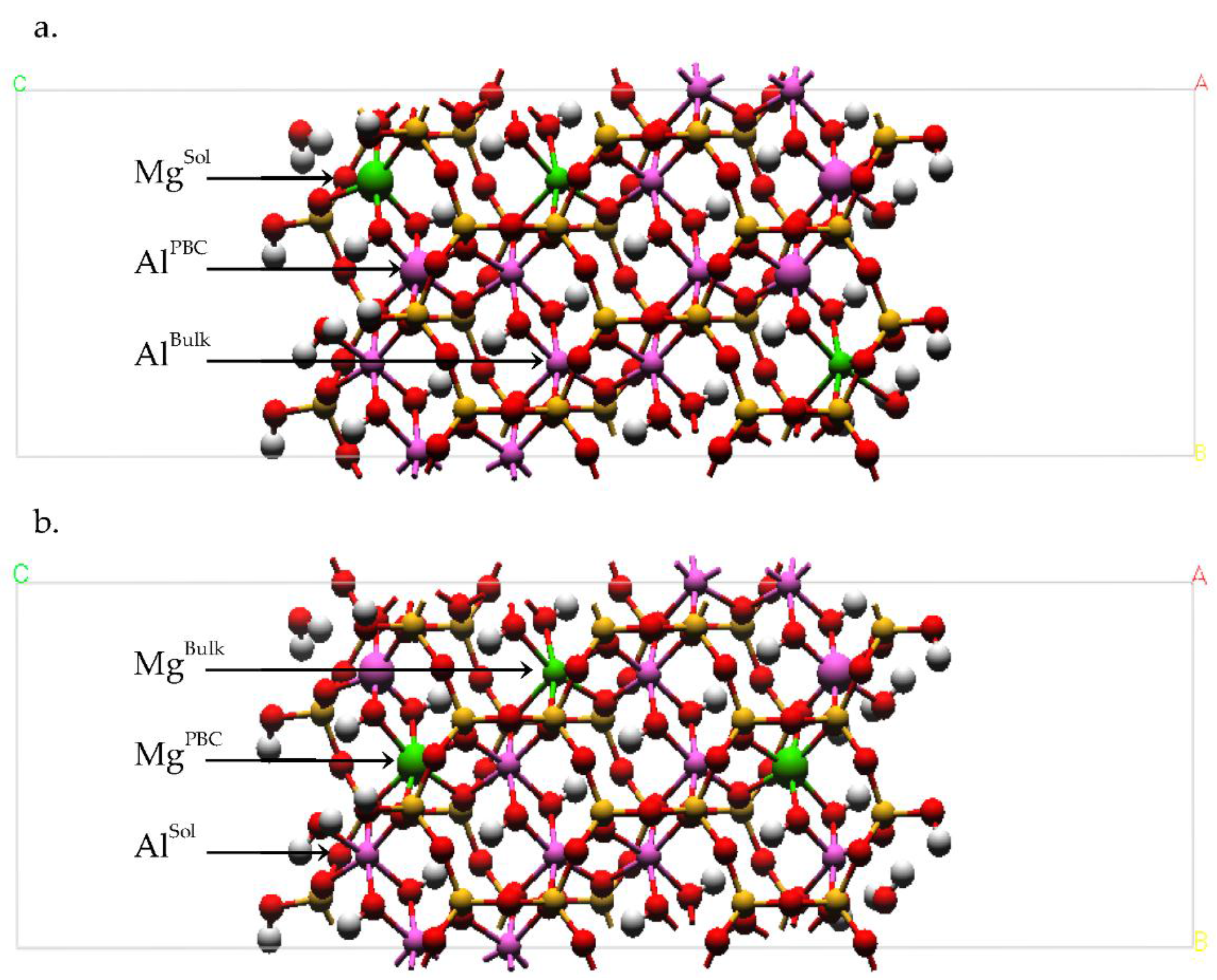
Figure 1.
The upper and lower octahedral sheets of the Na-montmorillonite (Na-MMT) layers showing the locations of random Mg substitutions for Al. The solvent accessible Mg substitutions (MgSol) are in the lower octahedral sheet at right; the solvent inaccessible Mg substitution in the linking PBC position is in the upper octahedral sheet at right. A representative trans-vacant site is shown in the shaded circle of the upper sheet. The octahedra of a periodic bond chain (PBC) that parallels the edge surface are highlighted in the shaded ellipse. The Al atoms are magenta spheres, the Mg atoms are green octahedra, and the O atoms in octahedral coordination with the Mg and Al are red.
Figure 1.
The upper and lower octahedral sheets of the Na-montmorillonite (Na-MMT) layers showing the locations of random Mg substitutions for Al. The solvent accessible Mg substitutions (MgSol) are in the lower octahedral sheet at right; the solvent inaccessible Mg substitution in the linking PBC position is in the upper octahedral sheet at right. A representative trans-vacant site is shown in the shaded circle of the upper sheet. The octahedra of a periodic bond chain (PBC) that parallels the edge surface are highlighted in the shaded ellipse. The Al atoms are magenta spheres, the Mg atoms are green octahedra, and the O atoms in octahedral coordination with the Mg and Al are red.
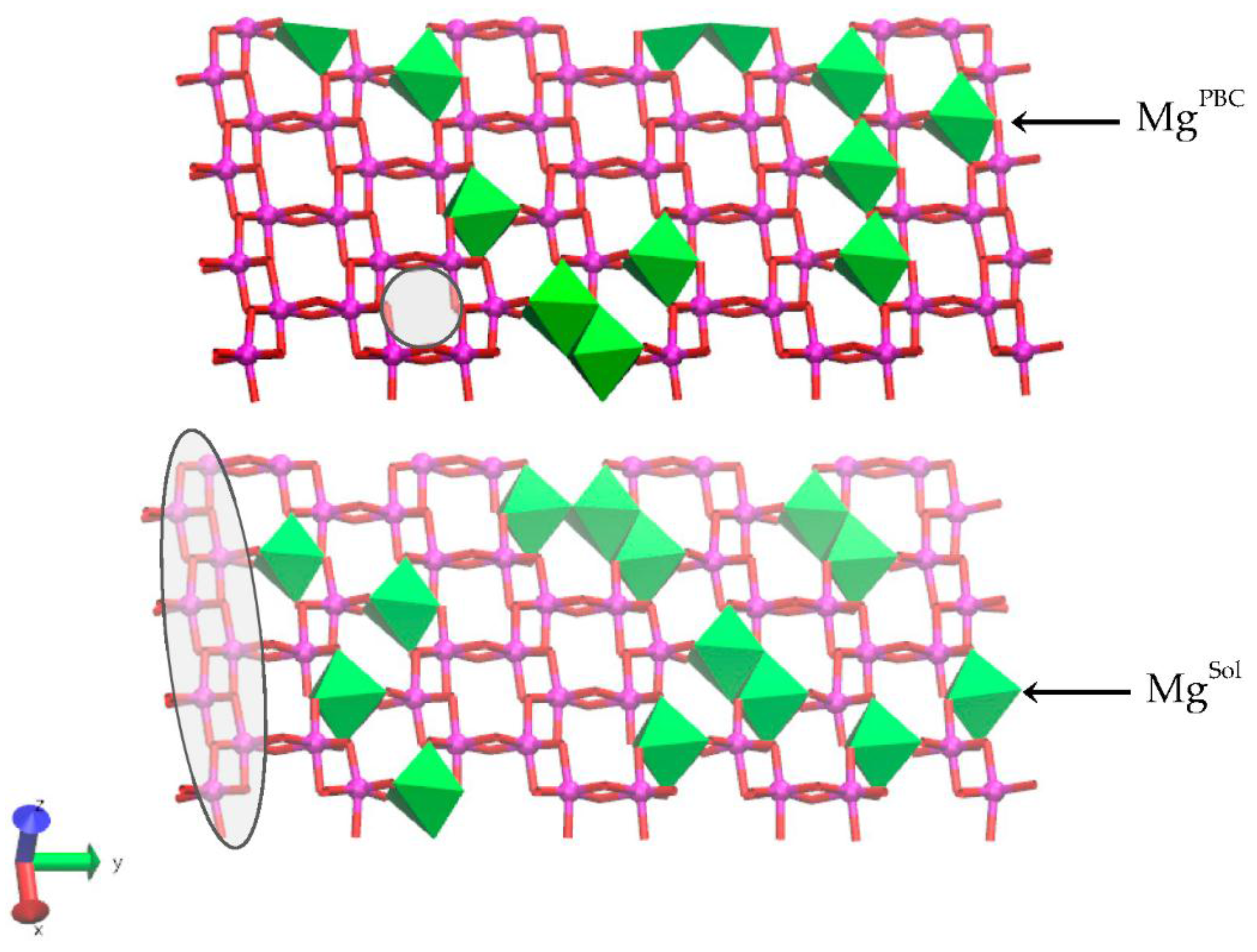
Figure 2.
Simulation cell and initial atomic configuration of Na-MMT AC edge at t = 0. The Al atoms are magenta; Mg atoms are green; Si atoms are ochre, O atoms are red, H atoms are white, and Na atoms are blue.
Figure 2.
Simulation cell and initial atomic configuration of Na-MMT AC edge at t = 0. The Al atoms are magenta; Mg atoms are green; Si atoms are ochre, O atoms are red, H atoms are white, and Na atoms are blue.
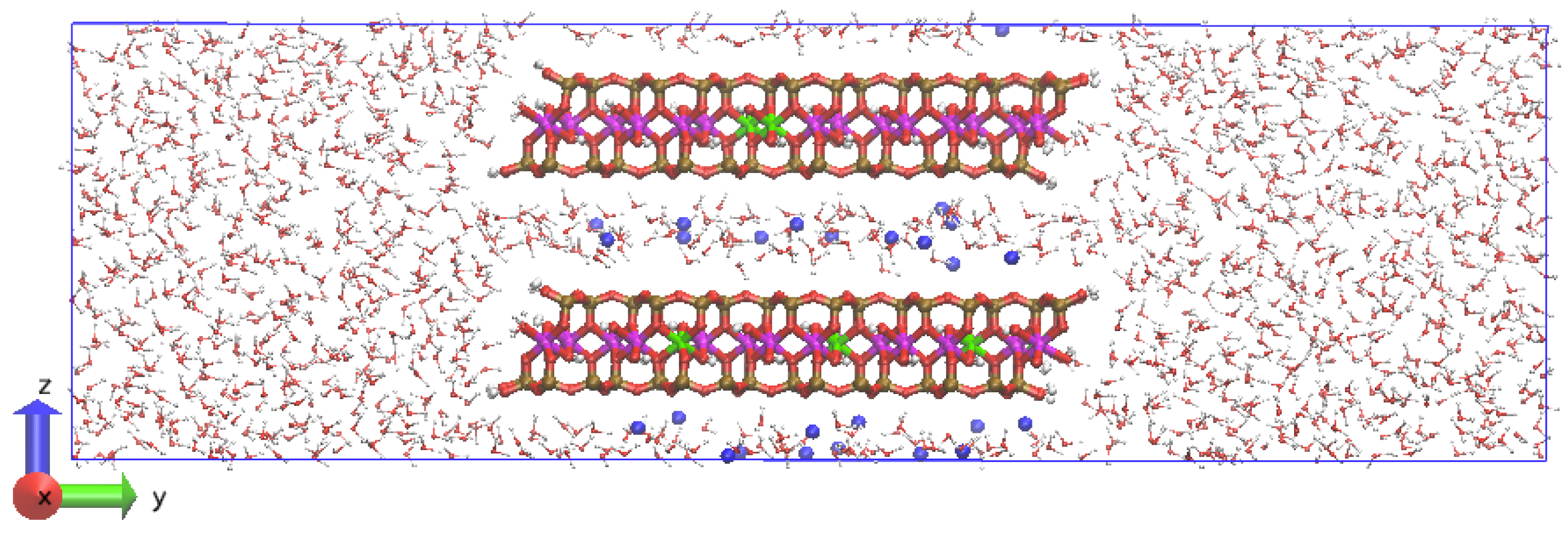
Figure 3.
Plan view of the (a) solvent accessible Mg (MgSol) and the (b) linking PBC Mg (MgPBC) models with a layer charge (x) of 0.375 from the DFT geometry optimizations. The solvent accessible (MgSol and AlSol) and linking PBC cation positions (MgPBC and AlPBC) in the edge bond chain are defined. The bulk Mg substitution (MgBulk) is also defined. In models with x = 0.25, the MgBulk atoms are replaced with an Al atom (AlBulk). Representative cation pairs for the hydroxyl sharing (Table 2: Alsol-OH2, oh) and bridging oxygen (Table 2: Alsol-OH2, ob) pairs are depicted as enlarged cations at left and right in the edge PBCs, respectively. Atom legend is the same as Figure 2.
Figure 3.
Plan view of the (a) solvent accessible Mg (MgSol) and the (b) linking PBC Mg (MgPBC) models with a layer charge (x) of 0.375 from the DFT geometry optimizations. The solvent accessible (MgSol and AlSol) and linking PBC cation positions (MgPBC and AlPBC) in the edge bond chain are defined. The bulk Mg substitution (MgBulk) is also defined. In models with x = 0.25, the MgBulk atoms are replaced with an Al atom (AlBulk). Representative cation pairs for the hydroxyl sharing (Table 2: Alsol-OH2, oh) and bridging oxygen (Table 2: Alsol-OH2, ob) pairs are depicted as enlarged cations at left and right in the edge PBCs, respectively. Atom legend is the same as Figure 2.

Figure 4.
Montmorillonite AC edge-surface models geometry-optimized by DFT with layer charges x1 = 0.25 and x2 = 0.375. The Mg substitutions for Al are in (a) solvent accessible and (b) the linking PBC positions for the models with a layer charge of x = 0.25. For the models with a layer charge of x = 0.375; the (c) solvent accessible and (d) linking PBC position models also show the location of the bulk isomorphic substitution. The total energies of the models with the solvent accessible substitution are taken as the reference (). The Mg substitutions at the right edge interfaces are obscured by the edge Al atom in these profiles. The hydrogen bonds are depicted as dotted blue lines. The atom legend is the same as Figure 2.
Figure 4.
Montmorillonite AC edge-surface models geometry-optimized by DFT with layer charges x1 = 0.25 and x2 = 0.375. The Mg substitutions for Al are in (a) solvent accessible and (b) the linking PBC positions for the models with a layer charge of x = 0.25. For the models with a layer charge of x = 0.375; the (c) solvent accessible and (d) linking PBC position models also show the location of the bulk isomorphic substitution. The total energies of the models with the solvent accessible substitution are taken as the reference (). The Mg substitutions at the right edge interfaces are obscured by the edge Al atom in these profiles. The hydrogen bonds are depicted as dotted blue lines. The atom legend is the same as Figure 2.
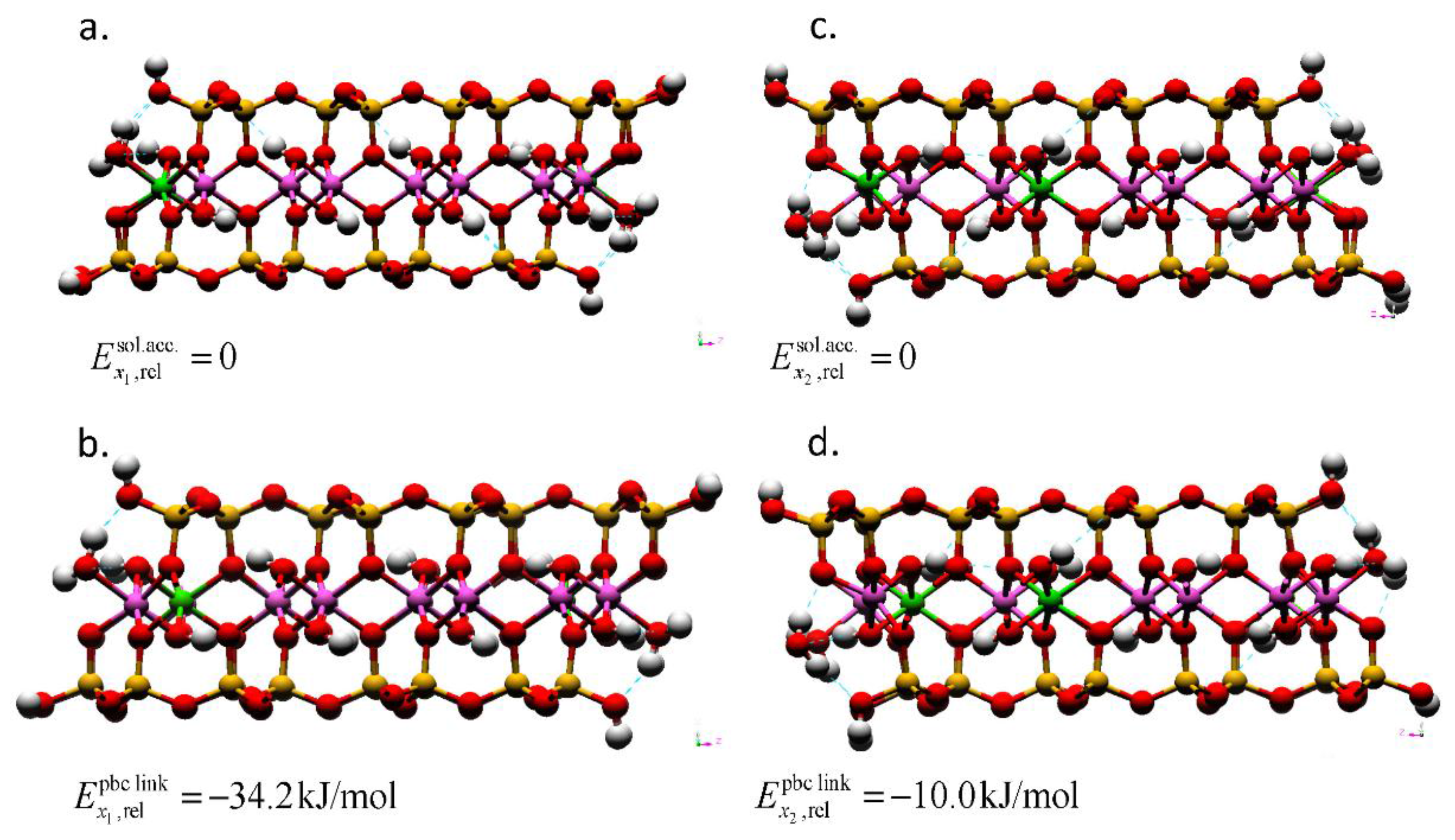
Figure 5.
Snapshot of the molecular dynamics (MD) simulations showing disorder in the edge Al coordination on the Mg-substituted edges of the Na-MMT model. Non-octahedral edge Al are present in the top layer as a tetrahedron and square pyramid and, in the lower layer, as two trigonal bipyramids and a square pyramid. These non-octahedral Al are depicted as enlarged spheres. The bridging water molecules that stabilize these disordered edge structures through H-bonds are also enlarged. Near surface H-bonds shown as dashed black lines. Bulk water molecules removed for clarity. Atom legend is the same as Figure 2.
Figure 5.
Snapshot of the molecular dynamics (MD) simulations showing disorder in the edge Al coordination on the Mg-substituted edges of the Na-MMT model. Non-octahedral edge Al are present in the top layer as a tetrahedron and square pyramid and, in the lower layer, as two trigonal bipyramids and a square pyramid. These non-octahedral Al are depicted as enlarged spheres. The bridging water molecules that stabilize these disordered edge structures through H-bonds are also enlarged. Near surface H-bonds shown as dashed black lines. Bulk water molecules removed for clarity. Atom legend is the same as Figure 2.
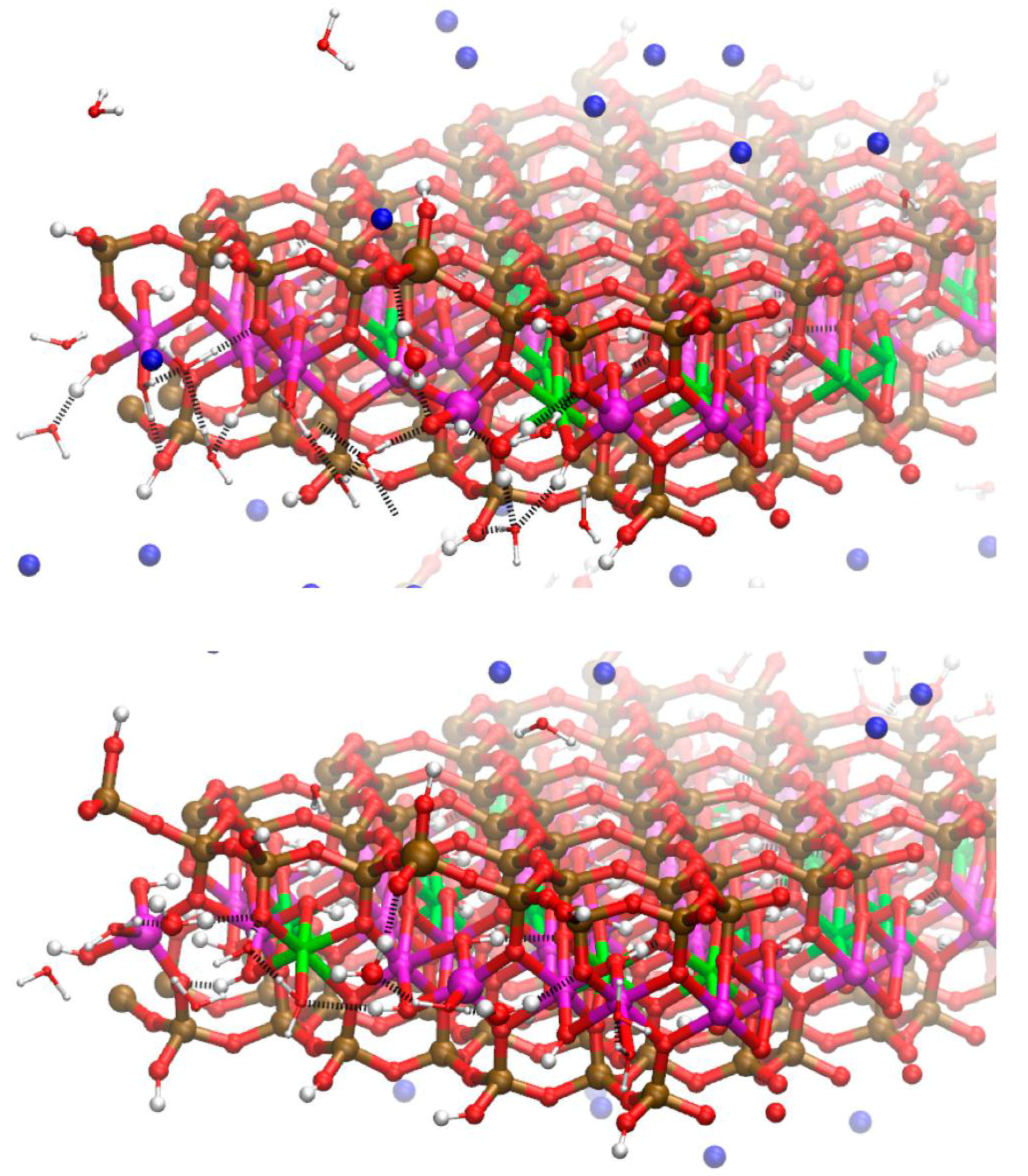
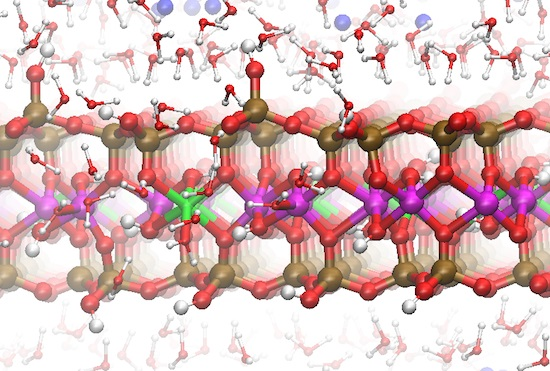
Figure 6.
Snapshot of the Na-MMT MD simulations shown in profile at t ~3.5 ns; water molecules removed for clarity. Disordered structures have developed at the AC edge interfaces. The Al atoms are magenta; Mg atoms are green; Si atoms are ochre, O atoms are red, H atoms are white, and Na atoms are blue.
Figure 6.
Snapshot of the Na-MMT MD simulations shown in profile at t ~3.5 ns; water molecules removed for clarity. Disordered structures have developed at the AC edge interfaces. The Al atoms are magenta; Mg atoms are green; Si atoms are ochre, O atoms are red, H atoms are white, and Na atoms are blue.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Characteristic properties of the cation-oxygen (α-β) pair distribution functions of the octahedral sheet of the Na-MMT model from molecular dynamics (MD) simulations. The pair distribution functions are summarized by the distance to the first peak (rpeak), coordination number (CN), and the first minimum (ρ) of the cation-O pair distribution for the substituted edge PBC, unsubstituted edge PBC, and bulk positions within the model. The standard deviations for each property (+/−) are provided.
| α-β Pair | rpeak, Å | +/− | CN | +/− | ρ, Å | +/− |
|---|---|---|---|---|---|---|
| Substituted Edge PBC | ||||||
| MgSol-O | 2.10 | 0.003 | 5.72 | 0.054 | 2.88 | 0.033 |
| MgPBC-O | 2.12 | 0.002 | 5.99 | 0.004 | 2.60 | 0.038 |
| AlSol-O | 1.87 | 0.003 | 5.25 | 0.051 | 2.86 | 0.052 |
| AlPBC-O | 1.90 | 0.004 | 5.85 | 0.006 | 2.91 | 0.017 |
| Unsubstituted Edge PBC | ||||||
| AlSol-O | 1.92 | 0.005 | 5.81 | 0.040 | 2.89 | 0.083 |
| AlPBC-O | 1.94 | 0.003 | 6.00 | 0.001 | 2.88 | 0.052 |
| Bulk | ||||||
| MgBulk-O | 2.10 | 0.001 | 5.98 | 0.001 | 2.34 | 0.002 |
| AlBulk-O | 1.93 | 0.002 | 5.98 | 0.001 | 2.47 | 0.004 |
Table 2.
Interatomic distances (in Å) of the octahedral sheets obtained by DFT for the MgSol model and MgPBC model at each layer charge (x = 0.25 or 0.375). The model with x = 0.375 possesses two unique edges (Figure 3): one edge with an adjacent bond chain that is unsubstituted by Mg (unsub MgBulk) and the other with the adjacent bond chain substituted by Mg (sub MgBulk). The solvent accessible, linking PBC, and bulk sites are designated by MgSol and AlSol; MgPBC and AlPBC; and MgBulk, and AlBulk respectively. The AlSol-OH2 distances are delineated based on the connection to the MgPBC through a hydroxyl oxygen (oh) or bridging oxygen (ob).
| α-β Pair | α-β Interatomic Distance, Å | |
|---|---|---|
| Layer Charge, x = 0.25 | Layer Charge, x = 0.375 Unsub/Sub MgBulk | |
| MgSol Model | ||
| MgSol-O | 2.09 | 2.06 |
| MgSol-OH2 | 2.25 | 2.40/3.06 |
| MgBulk-O | - | 2.07 |
| AlSol-O | 1.91 | 1.92 |
| AlSol-OH2 | 2.07 | 2.08/2.10 |
| AlPBC-O | 1.92 | 1.92 |
| AlBulk-O | 1.92 | 1.92 |
| MgPBC Model | ||
| MgPBC-O | 2.08 | 2.08 |
| MgBulk-O | - | 2.07 |
| AlSol-O | 1.92 | 1.90 |
| AlSol-OH2, oh | 2.17 | 2.28/3.20 |
| AlSol-OH2, ob | 2.09 | 2.12/2.10 |
| AlPBC-O | 1.92 | 1.92 |
| AlBulk-O | 1.92 | 1.92 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Newton, A.G.; Kwon, K.D.; Cheong, D.-K. Edge Structure of Montmorillonite from Atomistic Simulations. Minerals 2016, 6, 25. https://doi.org/10.3390/min6020025
AMA Style
Newton AG, Kwon KD, Cheong D-K. Edge Structure of Montmorillonite from Atomistic Simulations. Minerals. 2016; 6(2):25. https://doi.org/10.3390/min6020025
Chicago/Turabian StyleNewton, Aric G., Kideok D. Kwon, and Dae-Kyo Cheong. 2016. "Edge Structure of Montmorillonite from Atomistic Simulations" Minerals 6, no. 2: 25. https://doi.org/10.3390/min6020025
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.