
A Solid State NMR Investigation of Recent Marine Siliceous Sponge Spicules



Abstract
:
1. Introduction
2. Results
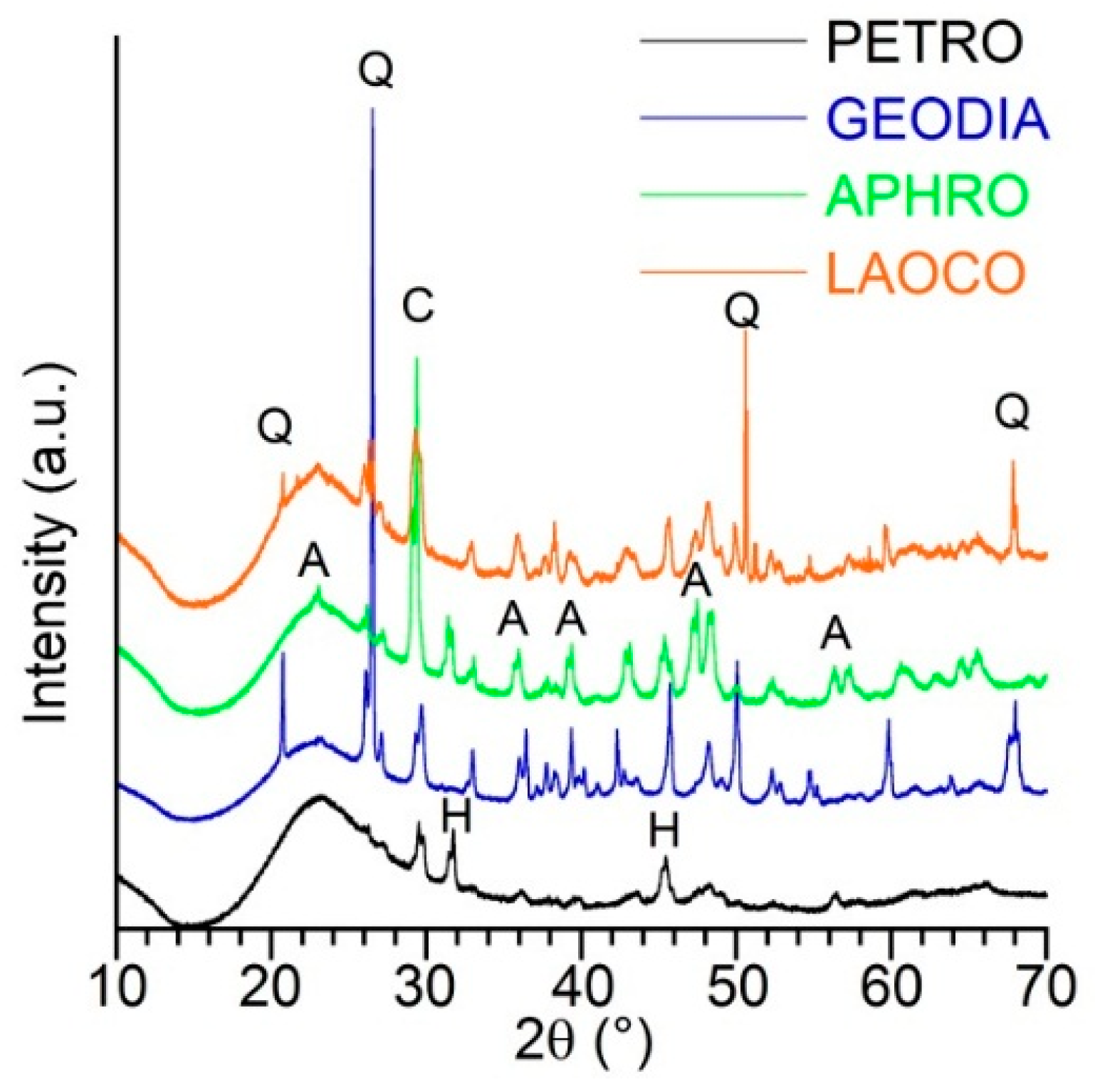
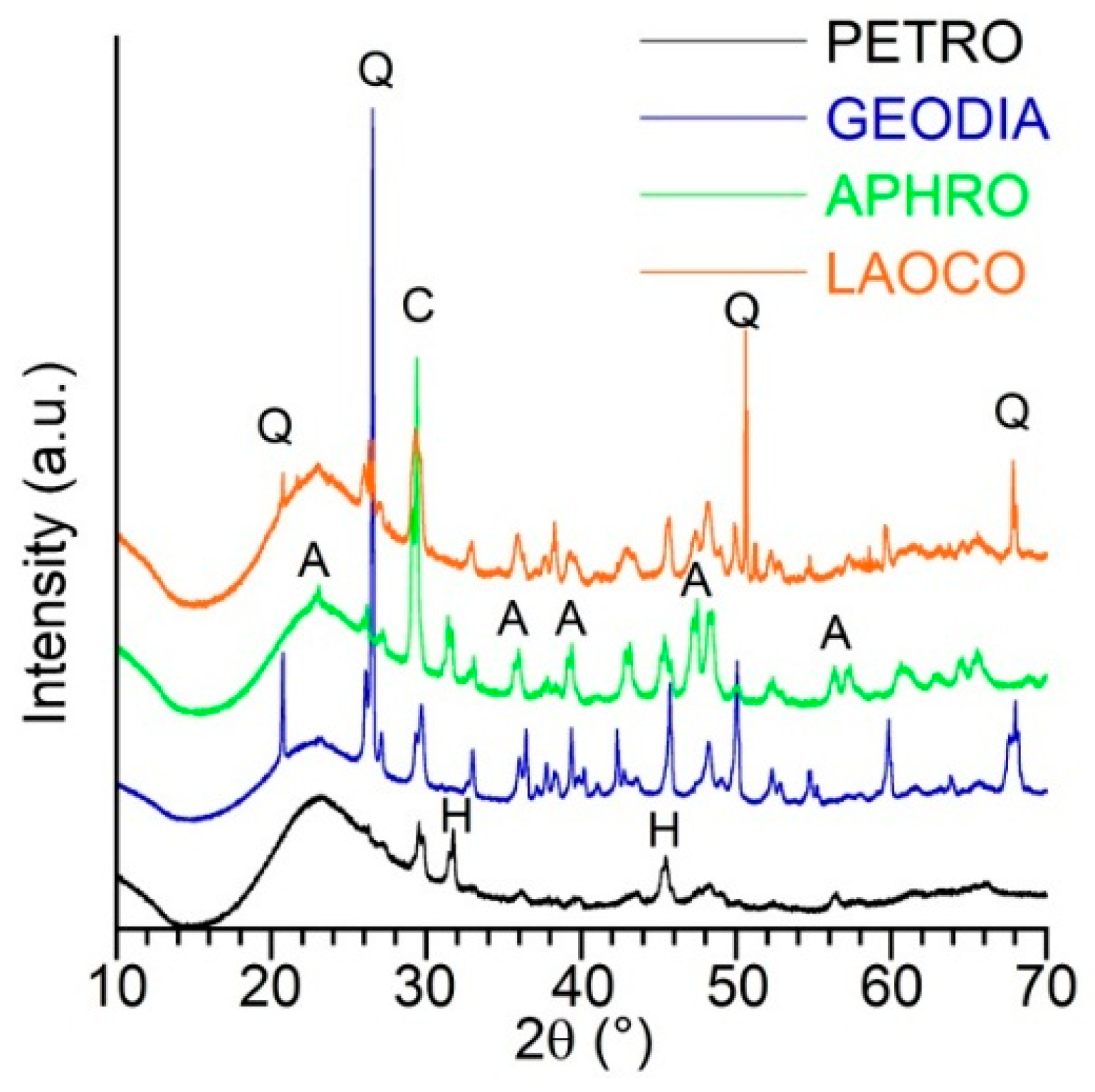
2.1. Sample Presentation and General Characterization
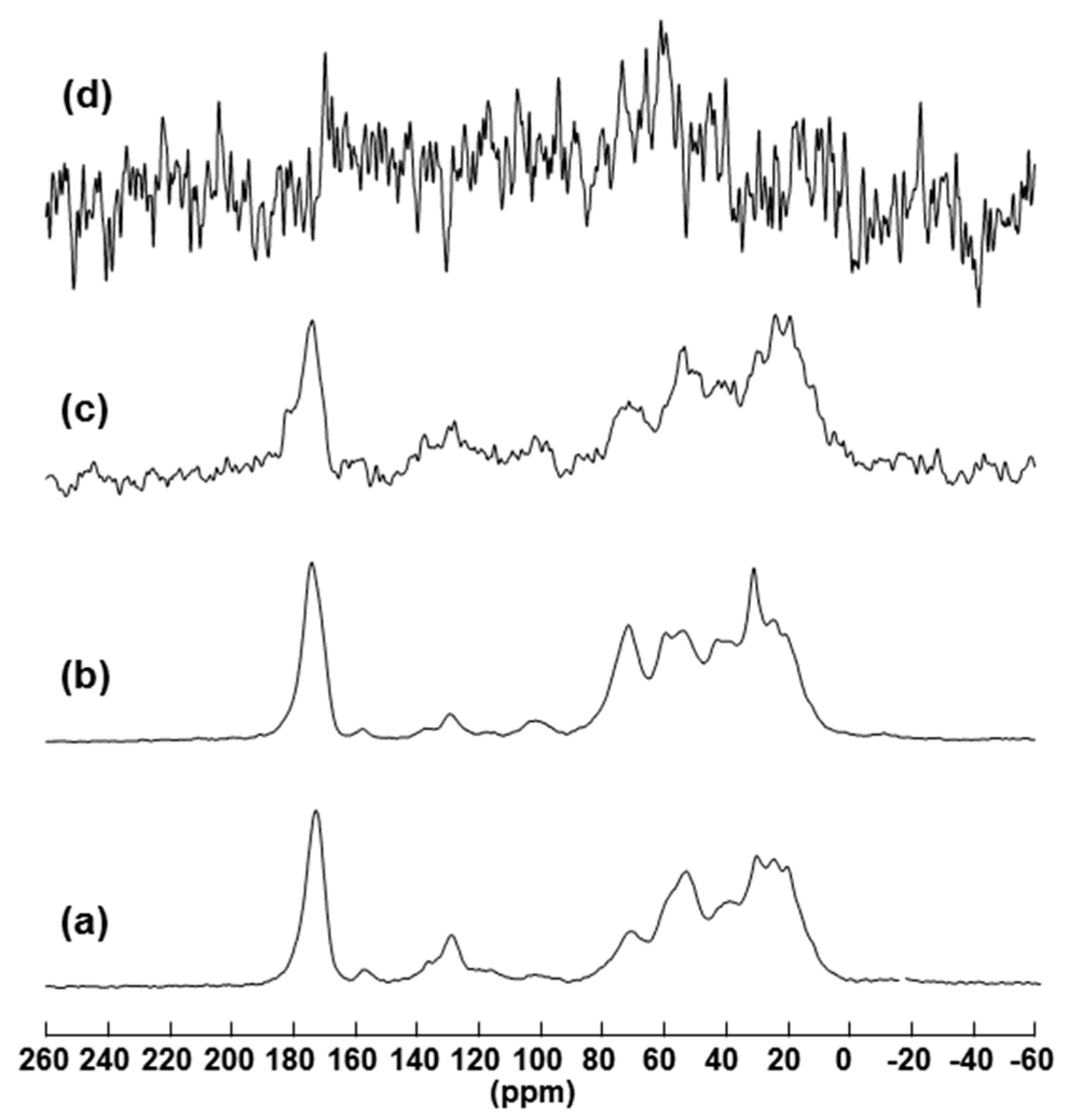
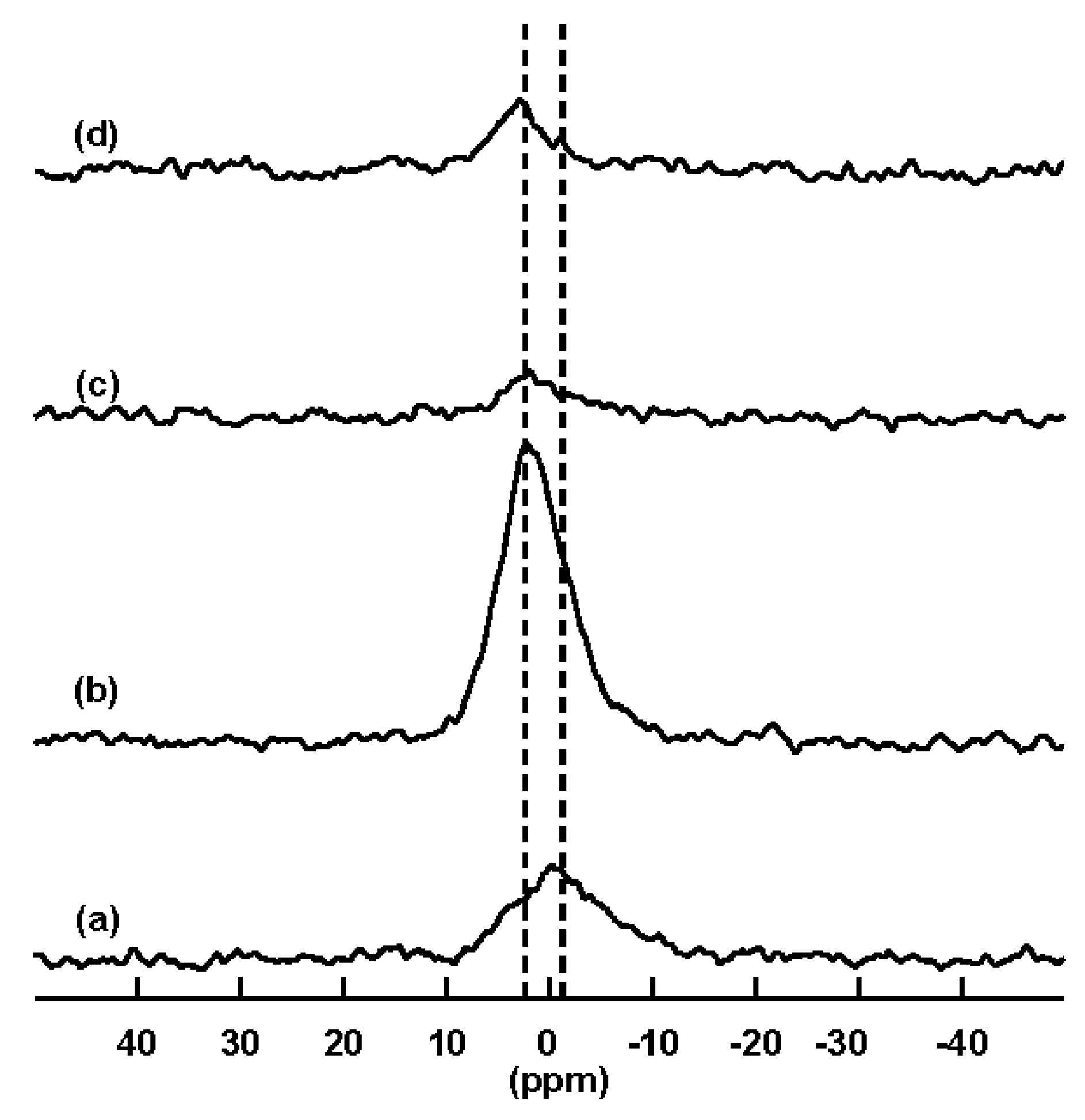
2.2. Solid State Nuclear Magnetic Resonance (NMR) Characterization
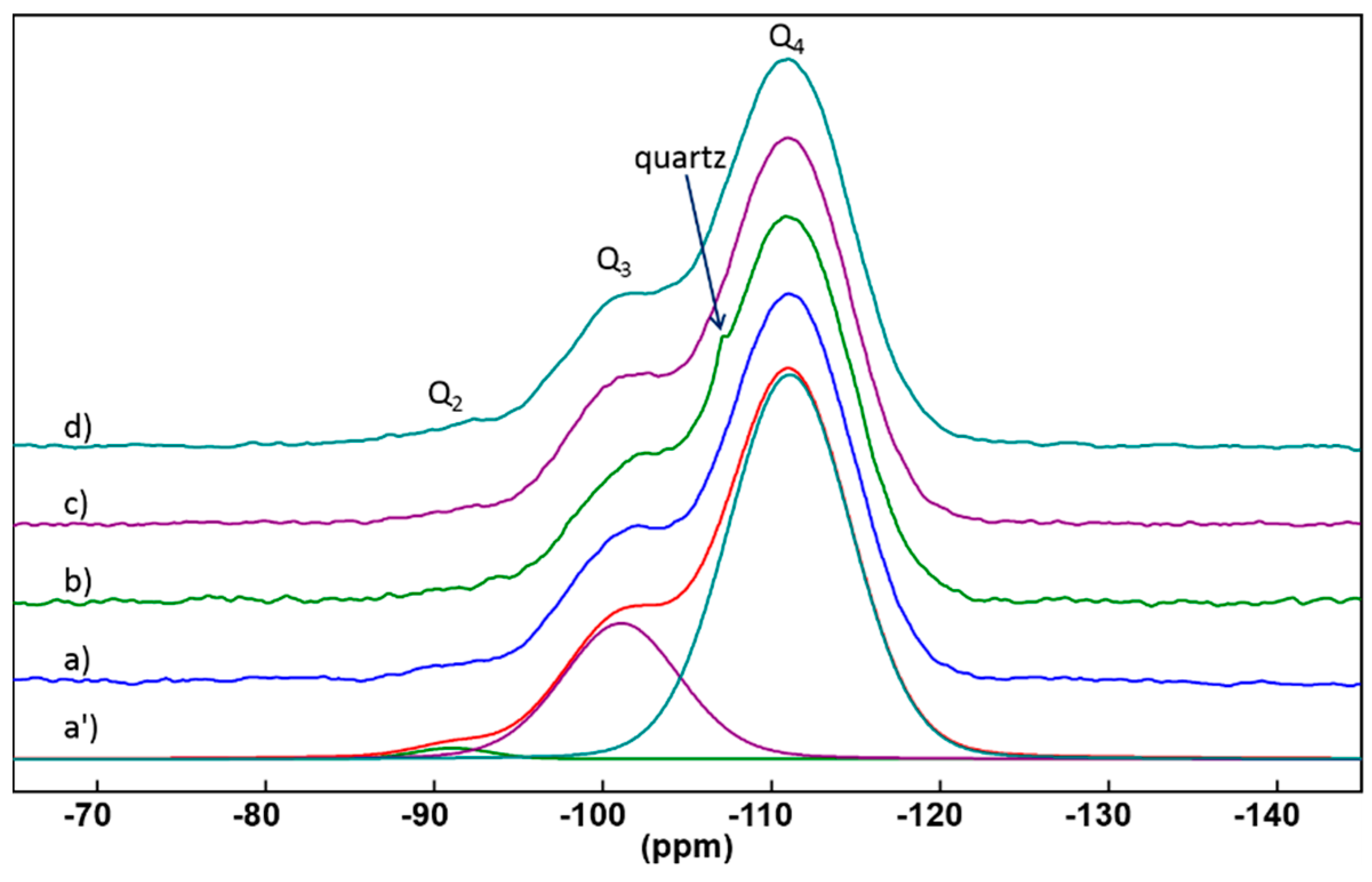
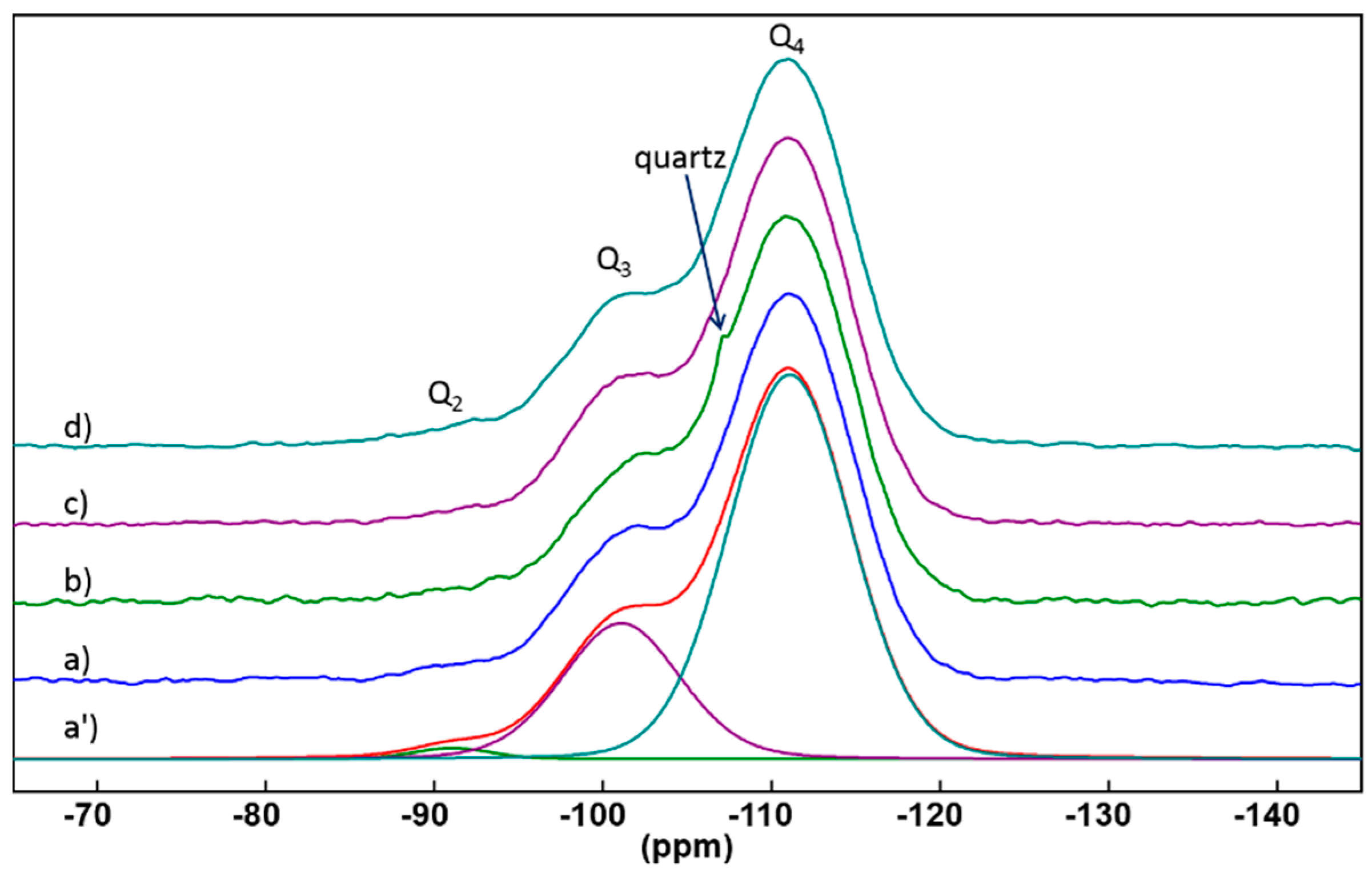
2.2.1. 29Si Solid State NMR
2.2.2. 13C Solid State NMR
2.2.3. 31P Solid State NMR
3. Discussion
4. Materials and Methods
4.1. Main Techniques of Characterization
4.2. Solid State NMR Studies
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mann, S. Molecular tectonics in biomineralization and biomimetic materials chemistry. Nature 1993, 365, 499–505. [Google Scholar] [CrossRef]
- Coradin, T.; Lopez, P.J.; Gautier, C.; Livage, J. From biogenic to biomimetic silica. C. R. Palevol 2004, 3, 443–452. [Google Scholar] [CrossRef]
- Patwardhan, S.V. Biomimetic and bioinspired silica: Recent developments and applications. Chem. Commun. 2011, 47, 7567–7582. [Google Scholar] [CrossRef] [PubMed]
- Ragueneau, O.; Tréguer, P.; Leynaert, A.; Anderson, R.F.; Brzezinski, M.A.; DeMaster, D.J.; Dugdale, R.C.; Dymond, J.; Fischer, G.; François, R.; et al. A review of the Si cycle in the modern ocean: Recent progress and missing gaps in the application of biogenic opal as a paleoproductivity proxy. Glob. Planet Chang. 2000, 26, 317–365. [Google Scholar] [CrossRef]
- Boury-Esnault, N. Le rôle de la silice dans la biosphère: L’exemple des spongiaires. C. R. Chim. 2008, 11, 261–267. (In French) [Google Scholar] [CrossRef]
- Hooper, N.A.V.; Soest, R.W.M. (Eds.) Systema Porifera: A Guide to the Classification of Sponges; Kluwer Academic Publishers: New York, NY, USA, 2002.
- Pisera, A. Some aspects of silica deposition in lithistid demosponge desmas. Microsc. Res. Tech. 2003, 62, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Cha, J.; Stucky, G.D.; Morse, D.E. Silicatein alpha: Cathepsin L-like protein in sponge biosilica. Proc. Natl. Acad. Sci. USA 1998, 95, 6234–6238. [Google Scholar] [CrossRef] [PubMed]
- Müller, W.E.G.; Wang, X.; Kropf, K.; Ushijima, H.; Geurtsen, W.; Eckert, C.; Nawaz Tahir, M.; Tremel, W.; Boreiko, A.; Schlossmacher, U.; et al. Bioorganic/inorganic hybrid composition of sponge spicules: Matrix of the giant spicules and of the comitalia of the deep sea hexactinellid Monorhaphis. J. Struct. Biol. 2008, 161, 188–203. [Google Scholar] [CrossRef] [PubMed]
- Sumper, M.; Brunner, E. Learning from diatoms: Nature’s tools for the production of nanostructured silica. Adv. Funct. Mater. 2006, 16, 17–26. [Google Scholar] [CrossRef]
- Wang, X.; Schröder, H.C.; Wang, K.; Kaandorp, J.A.; Müller, W.E.G. Genetic, biological and structural hierarchies during sponge spicule formation: From soft sol-gels to solid 3D silica composite structures. Soft Mater. 2012, 8, 9501–9518. [Google Scholar] [CrossRef]
- Coradin, T.; Lopez, P.J. Biogenic silica patterning: Simple chemistry or subtle biology? Chembiochem 2003, 4, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Mann, S.; Perry, C.C.; Williams, R.J.P.; Fyfe, C.A.; Gobbi, G.C.; Kennedy, G.J. The characterization of the nature of silica in biological systems. J. Chem. Soc. Chem. Commun. 1983, 4, 168–170. [Google Scholar] [CrossRef]
- Gendron-Badou, A.; Coradin, T.; Maquet, J.; Frohlich, F.; Livage, J. Spectroscopic characterization of biogenic silica. J. Non Cryst. Solids 2003, 316, 331–337. [Google Scholar] [CrossRef]
- Abramson, L.; Wirick, S.; Lee, C.; Jacobsen, C.; Brandes, J.A. The use of soft X-ray spectromicroscopy to investigate the distribution and composition of organic matter in a diatom frustule and a biomimetic analog. Deep Sea Res. II 2009, 56, 1369–1380. [Google Scholar] [CrossRef]
- Sandford, F. Physical and chemical analysis of the siliceous skeletons in six sponges of two groups (Demospongiae and Hexactinellida). Microsc. Res. Tech. 2003, 62, 336–355. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, C.; Coelho, C.; Baccile, N.; Gervais, C.; Azaïs, T.; Babonneau, F. Advanced solid state NMR techniques for the characterization of sol–gel-derived materials. Acc. Chem. Res. 2007, 40, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Bertermann, R.; Kröger, N.; Tacke, R. Solid-state 29Si MAS NMR studies of diatoms: Structural characterization of biosilica deposits. Anal. Bioanal. Chem. 2003, 375, 630–634. [Google Scholar] [PubMed]
- Tesson, B.; Masse, S.; Laurent, G.; Maquet, J.; Livage, J.; Martin-Jezequel, V.; Coradin, T. Contribution of multi-nuclear solid state NMR to the characterization of the Thalassiosira pseudonana diatom cell wall. Anal. Bioanal. Chem. 2008, 390, 1889–1898. [Google Scholar] [CrossRef] [PubMed]
- Beterman, R.; Tacke, R. Solid-state 29Si VACP/MAS NMR studies of silicon-accumulating plants: Structural characterization of biosilica deposits. Ziet. Naturforsch. B 2000, 55, 459–461. [Google Scholar]
- Michel, F.M.; MacDonald, J.; Feng, J.; Phillips, B.L.; Ehm, L.; Tarabrella, C.; Parise, J.B.; Reeder, R.J. Structural characteristics of synthetic amorphous calcium carbonate. Chem. Mater. 2008, 20, 4720–4728. [Google Scholar] [CrossRef]
- Nassif, N.; Pinna, N.; Gehrke, N.; Antonietti, M.; Jäger, C.; Cölfen, H. Amorphous layer around aragonite platelets in nacre. PNAS 2005, 102, 12653–12655. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Wielgosz-Collin, G.; Njinkoué, J.M.; Velosaotsy, N.E.; Kornprobst, J.M.; Gouygou, J.P.; Vacelet, J.; Barnathan, G. New trends in phospholipid class composition of marine sponges. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 150, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Masse, S.; Laurent, G.; Chuburu, F.; Cadiou, C.; Deschamps, I.; Coradin, T. Modification of the stöber process by a polyazamacrocycle leading to unusual core-shell silica nanoparticles. Langmuir 2008, 24, 4026–4031. [Google Scholar] [CrossRef] [PubMed]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 42, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling one- and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Si (wt %) | C (wt %) | H (wt %) | N (wt %) | P (wt %) |
|---|---|---|---|---|---|
| Petrosidae (PETRO) | 33.99 | 6.60 | 1.61 | 1.55 | 0.11 |
| Geodia (GEODIA) | 28.76 | 6.81 | 1.39 | 1.23 | 0.18 |
| Aphrocallistes (APHRO) | 33.82 | 2.94 | 0.88 | 0.26 | 0.05 |
| Laocoetis perion (LAOCO) | 34.65 | 2.35 | 0.80 | 0.14 | 0.03 |
| Sample | Q2 (%, ±1%) | Q3 (%, ±1%) | Q4 (%, ±1%) | D (±0.01) |
|---|---|---|---|---|
| Petrosidae (PETRO) | 1 | 26 | 73 | 0.93 |
| Geodia (GEODIA) | 1 | 24 | 75 | 0.94 |
| Aphrocallistes (APHRO) | 1 | 26 | 73 | 0.93 |
| Laocoetis perion (LAOCO) | 2 | 25 | 73 | 0.93 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masse, S.; Pisera, A.; Laurent, G.; Coradin, T. A Solid State NMR Investigation of Recent Marine Siliceous Sponge Spicules. Minerals 2016, 6, 21. https://doi.org/10.3390/min6010021
Masse S, Pisera A, Laurent G, Coradin T. A Solid State NMR Investigation of Recent Marine Siliceous Sponge Spicules. Minerals. 2016; 6(1):21. https://doi.org/10.3390/min6010021
Chicago/Turabian StyleMasse, Sylvie, Andrzej Pisera, Guillaume Laurent, and Thibaud Coradin. 2016. "A Solid State NMR Investigation of Recent Marine Siliceous Sponge Spicules" Minerals 6, no. 1: 21. https://doi.org/10.3390/min6010021
APA StyleMasse, S., Pisera, A., Laurent, G., & Coradin, T. (2016). A Solid State NMR Investigation of Recent Marine Siliceous Sponge Spicules. Minerals, 6(1), 21. https://doi.org/10.3390/min6010021