The PARP1/ARTD1-Mediated Poly-ADP-Ribosylation and DNA Damage Repair in B Cell Diversification

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. PARylation is Essential for Cellular Responses to Environmental and Spontaneous Stresses
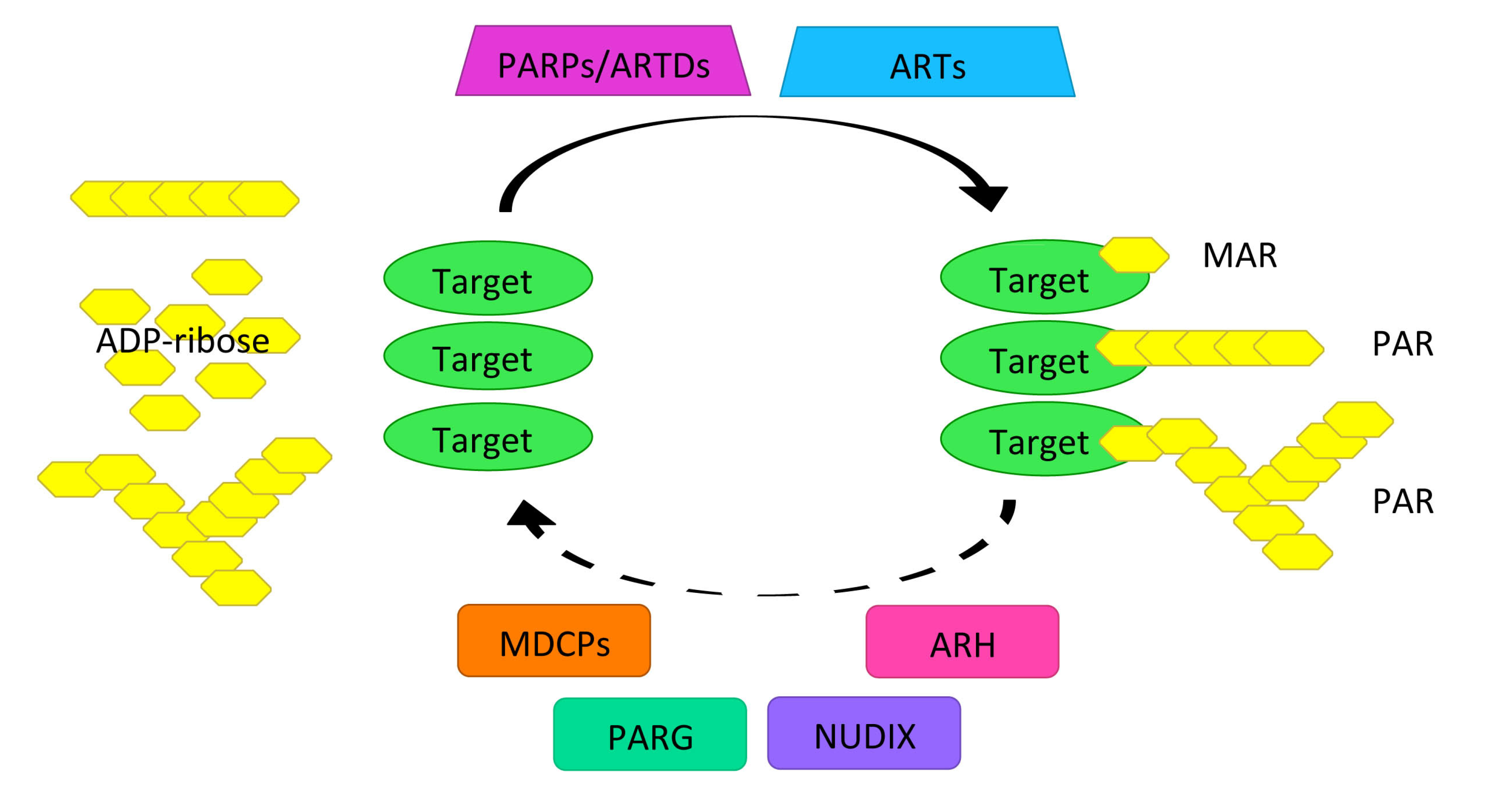
2.1. PARylation Homeostasis: PARPs/ARTDs, PARG, and Others
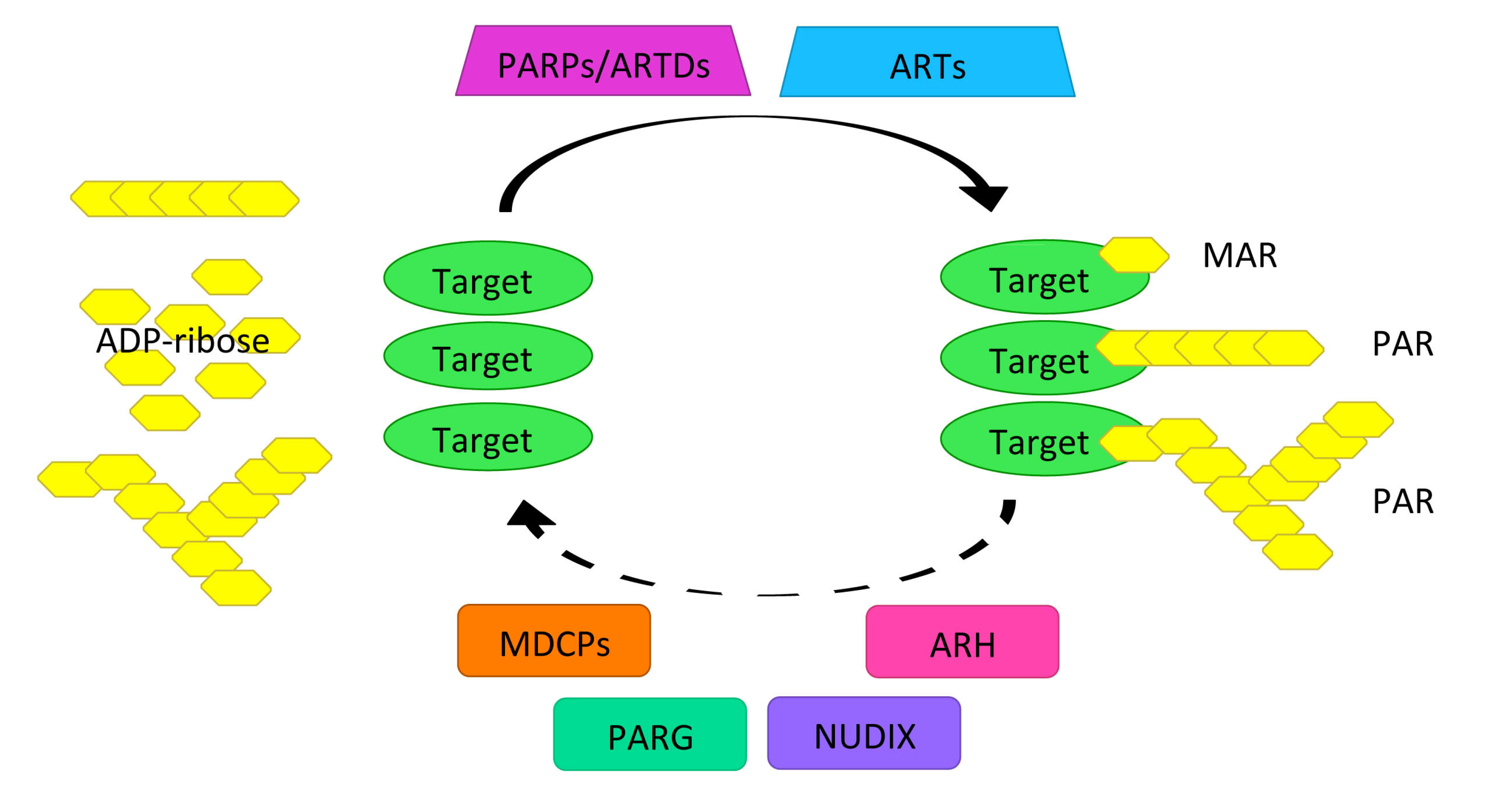
2.2. Role of PARylation as of a Platform for the DNA Damage Repair Protein Complex Assembly and Maintenance of Genomic Stability
2.3. PARylation in Cellular Responses to Programmed DNA Damage
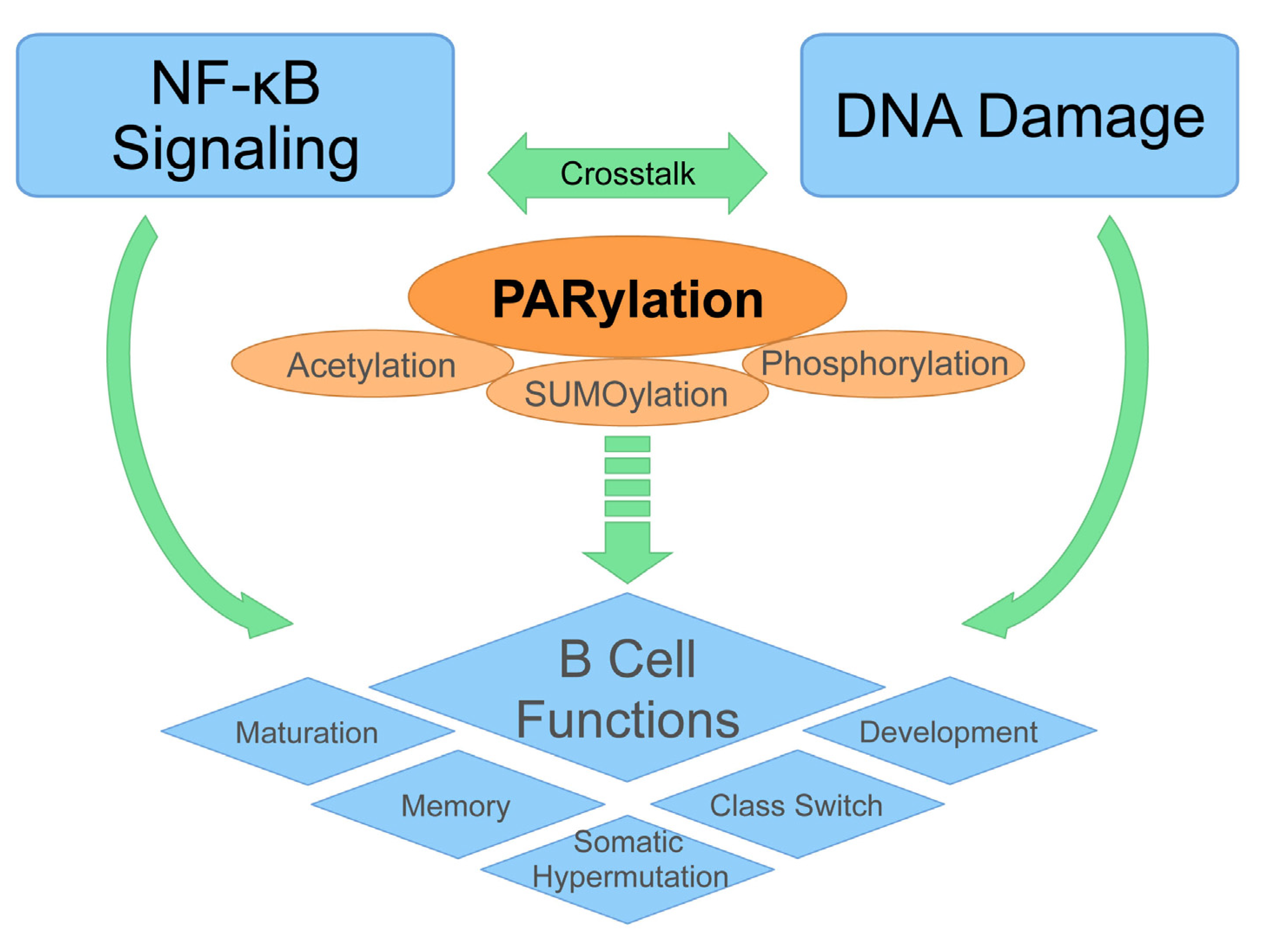
3. Emerging Role of PARylation in B Cell Function
3.1. DNA Damage, NF-κB, and B Cell Development
3.2. The Critical Role of PARylation in DNA Damage-Induced NF-κB Activation
3.3. Expanding Role of PARylation in B Cell Functions
4. Conclusion and Future Directions
Acknowledgements
Conflicts of Interest
References
- Hynes, N.E.; Ingham, P.W.; Lim, W.A.; Marshall, C.J.; Massague, J.; Pawson, T. Signalling change: Signal transduction through the decades. Nat. Rev. Mol. Cell Biol. 2013, 14, 393–398. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Kraus, W.L. The PARP Side of the Nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef]
- Ruf, A.; Mennissier de Murcia, J.; de Murcia, G.; Schulz, G.E. Structure of the catalytic fragment of poly(AD-ribose) polymerase from chicken. Proc. Natl. Acad. Sci. USA 1996, 93, 7481–7485. [Google Scholar] [CrossRef]
- Oliver, A.W.; Ame, J.C.; Roe, S.M.; Good, V.; de Murcia, G.; Pearl, L.H. Crystal structure of the catalytic fragment of murine poly(ADP-ribose) polymerase-2. Nucleic Acids Res. 2004, 32, 456–464. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- de Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dierich, A.; LeMeur, M.; et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar] [CrossRef]
- Masutani, M.; Nozaki, T.; Nishiyama, E.; Shimokawa, T.; Tachi, Y.; Suzuki, H.; Nakagama, H.; Wakabayashi, K.; Sugimura, T. Function of poly(ADP-ribose) polymerase in response to DNA damage: gene-disruption study in mice. Mol. Cell. Biochem. 1999, 193, 149–152. [Google Scholar] [CrossRef]
- Durkacz, B.W.; Omidiji, O.; Gray, D.A.; Shall, S. ADP-ribose)n participates in DNA excision repair. Nature 1980, 283, 593–596. [Google Scholar] [CrossRef]
- Shall, S.; de Murcia, G. Poly(ADP-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat. Res. 2000, 460, 1–15. [Google Scholar]
- Zhang, Y.; Wang, J.; Ding, M.; Yu, Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat. Methods 2013, 10, 981–984. [Google Scholar] [CrossRef]
- Jungmichel, S.; Rosenthal, F.; Altmeyer, M.; Lukas, J.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 2013, 52, 272–285. [Google Scholar] [CrossRef]
- Loseva, O.; Jemth, A.S.; Bryant, H.E.; Schuler, H.; Lehtio, L.; Karlberg, T.; Helleday, T. PARP-3 Is a Mono-ADP-ribosylase That Activates PARP-1 in the Absence of DNA. J. Biol. Chem. 2010, 285, 8054–8060. [Google Scholar]
- Kraus, W.L. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef]
- Potaman, V.N.; Shlyakhtenko, L.S.; Oussatcheva, E.A.; Lyubchenko, Y.L.; Soldatenkov, V.A. Specific Binding of Poly(ADP-ribose) Polymerase-1 to Cruciform Hairpins. J. Mol. Biol. 2005, 348, 609–615. [Google Scholar] [CrossRef]
- Cohen-Armon, M. PARP-1 activation in the ERK signaling pathway. Trends Pharmacol. Sci. 2007, 28, 556–560. [Google Scholar] [CrossRef]
- Krishnakumar, R.; Gamble, M.J.; Frizzell, K.M.; Berrocal, J.G.; Kininis, M.; Kraus, W.L. Reciprocal Binding of PARP-1 and Histone H1 at Promoters Specifies Transcriptional Outcomes. Science 2008, 319, 819–821. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.-W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef]
- Koh, D.W.; Dawson, T.M.; Dawson, V.L. Poly-ADP-ribosylation in health and disease. Cell. Mol. Life Sci. 2005, 62, 760–768. [Google Scholar] [CrossRef]
- Yu, S.-W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar]
- Rosenthal, F.; Feijs, K.L.; Frugier, E.; Bonalli, M.; Forst, A.H.; Imhof, R.; Winkler, H.C.; Fischer, D.; Caflisch, A.; Hassa, P.O.; et al. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 2013, 20, 502–507. [Google Scholar] [CrossRef]
- Jankevicius, G.; Hassler, M.; Golia, B.; Rybin, V.; Zacharias, M.; Timinszky, G.; Ladurner, A.G. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struct. Mol. Biol. 2013, 20, 508–514. [Google Scholar] [CrossRef]
- Min, W.; Wang, Z.Q. Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Front. Biosci. 2009, 14, 1619–1626. [Google Scholar] [CrossRef]
- Davidovic, L. Importance of Poly(ADP-Ribose) Glycohydrolase in the control of Poly(ADP-Ribose) metabolism. Exp. Cell Res. 2001, 268, 7–13. [Google Scholar] [CrossRef]
- Mortusewicz, O.; Fouquerel, E.; Ame, J.C.; Leonhardt, H.; Schreiber, V. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res. 2011, 39, 5045–5056. [Google Scholar] [CrossRef]
- Cortes, U.; Tong, W.M.; Coyle, D.L.; Meyer-Ficca, M.L.; Meyer, R.G.; Petrilli, V.; Herceg, Z.; Jacobson, E.L.; Jacobson, M.K.; Wang, Z.Q. Depletion of the 110-Kilodalton isoform of Poly(adp-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol. Cell. Biol. 2004, 24, 7163–7178. [Google Scholar] [CrossRef]
- Gao, H.; Coyle, D.L.; Meyer-Ficca, M.L.; Meyer, R.G.; Jacobson, E.L.; Wang, Z.-Q.; Jacobson, M.K. Altered poly(ADP-ribose) metabolism impairs cellular responses to genotoxic stress in a hypomorphic mutant of poly(ADP-ribose) glycohydrolase. Exp. Cell Res. 2007, 313, 984–996. [Google Scholar] [CrossRef]
- Masutani, M.; Nakagama, H.; Sugimura, T. Poly(ADP-ribose) and carcinogenesis. Genes Chromosom. Cancer 2003, 38, 339–348. [Google Scholar] [CrossRef]
- Min, W.; Cortes, U.; Herceg, Z.; Tong, W.M.; Wang, Z.Q. Deletion of the nuclear isoform of poly(ADP-ribose) glycohydrolase (PARG) reveals its function in DNA repair, genomic stability and tumorigenesis. Carcinogenesis 2010, 31, 2058–2065. [Google Scholar] [CrossRef]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef]
- Messner, S.; Altmeyer, M.; Zhao, H.; Pozivil, A.; Roschitzki, B.; Gehrig, P.; Rutishauser, D.; Huang, D.; Caflisch, A.; Hottiger, M.O. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010, 38, 6350–6362. [Google Scholar] [CrossRef]
- Poirier, G.G.; de Murcia, G.; Jongstra-Bilen, J.; Niedergang, C.; Mandel, P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427. [Google Scholar] [CrossRef]
- Realini, C.A.; Althaus, F.R. Histone shuttling by poly(ADP-ribosylation). J. Biol. Chem. 1992, 267, 18858–18865. [Google Scholar]
- Ahel, D.; Horejsi, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-ribose)-Dependent Regulation of DNA Repair by the Chromatin Remodeling Enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar] [CrossRef]
- Polo, S.E.; Kaidi, A.; Baskcomb, L.; Galanty, Y.; Jackson, S.P. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010, 29, 3130–3139. [Google Scholar] [CrossRef]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiácovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef]
- Okano, S.; Lan, L.; Caldecott, K.W.; Mori, T.; Yasui, A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 2003, 23, 3974–3981. [Google Scholar] [CrossRef]
- Mortusewicz, O.; Ame, J.C.; Schreiber, V.; Leonhardt, H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007, 35, 7665–7675. [Google Scholar] [CrossRef]
- Spagnolo, L.; Barbeau, J.; Curtin, N.J.; Morris, E.P.; Pearl, L.H. Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res. 2012, 40, 4168–4177. [Google Scholar] [CrossRef]
- Haince, J.F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007, 282, 16441–16453. [Google Scholar]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-κB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef]
- Tramontano, F.; Malanga, M.; Quesada, P. Differential contribution of poly(ADP-ribose)polymerase-1 and -2 (PARP-1 and -2) to the poly(ADP-ribosyl)ation reaction in rat primary spermatocytes. Mol. Hum. Reprod. 2007, 13, 821–828. [Google Scholar] [CrossRef]
- Dantzer, F.; Mark, M.; Quenet, D.; Scherthan, H.; Huber, A.; Liebe, B.; Monaco, L.; Chicheportiche, A.; Sassone-Corsi, P.; de Murcia, G.; et al. Poly(ADP-ribose) polymerase-2 contributes to the fidelity of male meiosis I and spermiogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 14854–14859. [Google Scholar] [CrossRef]
- Meyer-Ficca, M.L.; Lonchar, J.D.; Ihara, M.; Meistrich, M.L.; Austin, C.A.; Meyer, R.G. Poly(ADP-Ribose) Polymerases PARP1 and PARP2 modulate topoisomerase II Beta (TOP2B) function during chromatin condensation in mouse spermiogenesis. Biol. Reprod. 2011, 84, 900–909. [Google Scholar] [CrossRef]
- Yang, F.; Baumann, C.; De La Fuente, R. Persistence of histone H2AX phosphorylation after meiotic chromosome synapsis and abnormal centromere cohesion in poly (ADP-ribose) polymerase (Parp-1) null oocytes. Develop. Biol. 2009, 331, 326–338. [Google Scholar] [CrossRef]
- Meyer-Ficca, M.L.; Lonchar, J.; Credidio, C.; Ihara, M.; Li, Y.; Wang, Z.Q.; Meyer, R.G. Disruption of Poly(ADP-Ribose) homeostasis affects spermiogenesis and sperm chromatin integrity in mice. Biol. Reprod. 2009, 81, 46–55. [Google Scholar] [CrossRef]
- Turlure, F.; Devroe, E.; Silver, P.A.; Engelman, A. Human cell proteins and human immunodeficiency virus DNA integration. Front. Biosci. 2004, 9, 3187–3208. [Google Scholar] [CrossRef]
- Yoder, K.E.; Bushman, F.D. Repair of gaps in retroviral DNA integration intermediates. J. Virol. 2000, 74, 11191–11200. [Google Scholar] [CrossRef]
- Gäken, J.A.; Tavassoli, M.; Gan, S.U.; Vallian, S.; Giddings, I.; Darling, D.C.; Galea-Lauri, J.; Thomas, M.G.; Abedi, H.; Schreiber, V.; et al. Efficient retroviral infection of mammalian cells is blocked by inhibition of poly(ADP-ribose) polymerase activity. J. Virol. 1996, 70, 3992–4000. [Google Scholar]
- Kameoka, M.; Tanaka, Y.; Ota, K.; Itaya, A.; Yoshihara, K. Poly (ADP-ribose) polymerase is involved in PMA-induced activation of HIV-1 in U1 cells by modulating the LTR function. Biochem. Biophys. Res. Commun. 1999, 262, 285–289. [Google Scholar] [CrossRef]
- Kameoka, M.; Nukuzuma, S.; Itaya, A.; Tanaka, Y.; Ota, K.; Inada, Y.; Ikuta, K.; Yoshihara, K. Poly(ADP-ribose)polymerase-1 is required for integration of the human immunodeficiency virus type 1 genome near centromeric alphoid DNA in human and murine cells. Biochem. Biophys. Res. Commun. 2005, 334, 412–417. [Google Scholar] [CrossRef]
- Ha, H.C.; Juluri, K.; Zhou, Y.; Leung, S.; Hermankova, M.; Snyder, S.H. Poly(ADP-ribose) polymerase-1 is required for efficient HIV-1 integration. Proc. Natl. Acad. Sci. USA 2001, 98, 3364–3368. [Google Scholar]
- Baekelandt, V.; Claeys, A.; Cherepanov, P.; De Clercq, E.; De Strooper, B.; Nuttin, B.; Debyser, Z. DNA-Dependent protein kinase is not required for efficient lentivirus integration. J. Virol. 2000, 74, 11278–11285. [Google Scholar] [CrossRef]
- Siva, A.C.; Bushman, F. Poly(ADP-Ribose) Polymerase 1 is not strictly required for infection of murine cells by retroviruses. J. Virol. 2002, 76, 11904–11910. [Google Scholar] [CrossRef]
- Ariumi, Y.; Turelli, P.; Masutani, M.; Trono, D. DNA damage sensors ATM, ATR, DNA-PKcs, and PARP-1 are dispensable for human immunodeficiency virus Type 1 integration. J. Virol. 2005, 79, 2973–2978. [Google Scholar] [CrossRef]
- Jacelon, C.S.; Hanson, A. Older adults’ participation in the development of smart environments: An integrated review of the literature. Geriatr. Nurs. 2013, 34, 116–121. [Google Scholar] [CrossRef]
- Boboila, C.; Alt, F.W.; Schwer, B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012, 116, 1–49. [Google Scholar] [CrossRef]
- Puebla-Osorio, N.; Zhu, C. DNA damage and repair during lymphoid development: Antigen receptor diversity, Genomic integrity and lymphomagenesis. Immunol. Res. 2008, 41, 103–122. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Kaileh, M.; Sen, R. NF-kappaB function in B lymphocytes. Immunol. Res. 2012, 246, 254–271. [Google Scholar]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, a000182. [Google Scholar]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Wan, F.; Anderson, D.E.; Barnitz, R.A.; Snow, A.; Bidere, N.; Zheng, L.; Hegde, V.; Lam, L.T.; Staudt, L.M.; Levens, D.; et al. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell 2007, 131, 927–939. [Google Scholar] [CrossRef]
- Fu, K.; Sun, X.; Zheng, W.; Wier, E.M.; Hodgson, A.; Tran, D.Q.; Richard, S.; Wan, F. Sam68 modulates the promoter specificity of NF-kappaB and mediates expression of CD25 in activated T cells. Nat. Commun. 2013, 4, 1909. [Google Scholar] [CrossRef]
- Wan, F.; Lenardo, M.J. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb. Perspect. Biol. 2009, 1, a000067. [Google Scholar]
- Wan, F.; Lenardo, M.J. The nuclear signaling of NF-kappaB: Current knowledge, New insights, and future perspectives. Cell Res. 2010, 20, 24–33. [Google Scholar] [CrossRef]
- Claudio, E.; Saret, S.; Wang, H.; Siebenlist, U. Cell-autonomous role for NF-kappa B in immature bone marrow B cells. J. Immunol. 2009, 182, 3406–3413. [Google Scholar] [CrossRef]
- Verkoczy, L.; Ait-Azzouzene, D.; Skog, P.; Martensson, A.; Lang, J.; Duong, B.; Nemazee, D. A role for nuclear factor kappa B/rel transcription factors in the regulation of the recombinase activator genes. Immunity 2005, 22, 519–531. [Google Scholar] [CrossRef]
- Cadera, E.J.; Wan, F.; Amin, R.H.; Nolla, H.; Lenardo, M.J.; Schlissel, M.S. NF-kappaB activity marks cells engaged in receptor editing. J. Exp. Med. 2009, 206, 1803–1816. [Google Scholar] [CrossRef]
- Allman, D.; Pillai, S. Peripheral B cell subsets. Curr. Opin. Immunol. 2008, 20, 149–157. [Google Scholar] [CrossRef]
- Srinivasan, L.; Sasaki, Y.; Calado, D.P.; Zhang, B.; Paik, J.H.; DePinho, R.A.; Kutok, J.L.; Kearney, J.F.; Otipoby, K.L.; Rajewsky, K. PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009, 139, 573–586. [Google Scholar] [CrossRef]
- Gerondakis, S.; Grumont, R.J.; Banerjee, A. Regulating B-cell activation and survival in response to TLR signals. Immunol. Cell Biol. 2007, 85, 471–475. [Google Scholar] [CrossRef]
- Jain, A.; Ma, C.A.; Liu, S.; Brown, M.; Cohen, J.; Strober, W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat. Immunol. 2001, 2, 223–228. [Google Scholar] [CrossRef]
- Jain, A.; Ma, C.A.; Lopez-Granados, E.; Means, G.; Brady, W.; Orange, J.S.; Liu, S.; Holland, S.; Derry, J.M. Specific NEMO mutations impair CD40-mediated c-Rel activation and B cell terminal differentiation. J. Clin. Invest. 2004, 114, 1593–1602. [Google Scholar]
- Stilmann, M.; Hinz, M.; Arslan, S.C.; Zimmer, A.; Schreiber, V.; Scheidereit, C. A Nuclear Poly(ADP-Ribose)-Dependent signalosome confers DNA damage-induced IĸB kinase activation. Mol. Cell 2009, 36, 365–378. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol. Chem. 1999, 380, 953–959. [Google Scholar]
- Hassa, P.O. The Enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar] [CrossRef]
- Veuger, S.J.; Hunter, J.E.; Durkacz, B.W. Ionizing radiation-induced NF-κB activation requires PARP-1 function to confer radioresistance. Oncogene 2008, 28, 832–842. [Google Scholar] [CrossRef]
- Mabb, A.M.; Wuerzberger-Davis, S.M.; Miyamoto, S. PIASy mediates NEMO sumoylation and NF-κB activation in response to genotoxic stress. Nat. Cell Biol. 2006, 8, 986–993. [Google Scholar] [CrossRef]
- Helmink, B.A.; Sleckman, B.P. The response to and repair of RAG-mediated DNA double-strand breaks. Annu. Rev. Immunol. 2012, 30, 175–202. [Google Scholar] [CrossRef]
- Schatz, D.G.; Ji, Y. Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 2011, 11, 251–263. [Google Scholar] [CrossRef]
- Ambrose, H.E.; Willimott, S.; Beswick, R.W.; Dantzer, F.; de Murcia, J.M.; Yelamos, J.; Wagner, S.D. Poly(ADP-ribose) polymerase-1 (Parp-1)-deficient mice demonstrate abnormal antibody responses. Immunol. 2009, 127, 178–186. [Google Scholar] [CrossRef]
- Robert, I. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J. Exp. Med. 2009, 206, 1047–1056. [Google Scholar] [CrossRef]
- Robert, I.; Karicheva, O.; Martin, B.R. S.; Schreiber, V.; Dantzer, F. Functional aspects of PARylation in induced and programmed DNA repair processes: Preserving genome integrity and modulating physiological events. Mol. Aspects Med. 2013, 34, 1–15. [Google Scholar] [CrossRef]
- Yelamos, J.; Schreiber, V.; Dantzer, F. Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol. Med. 2008, 14, 169–178. [Google Scholar] [CrossRef]
- Nicolas, L.; Martinez, C.; Baro, C.; Rodriguez, M.; Baroja-Mazo, A.; Sole, F.; Flores, J.M.; Ampurdanes, C.; Dantzer, F.; Martin-Caballero, J.; et al. Loss of poly(ADP-ribose) polymerase-2 leads to rapid development of spontaneous T-cell lymphomas in p53-deficient mice. Oncogene 2010, 29, 2877–2883. [Google Scholar] [CrossRef]
- Yelamos, J.; Monreal, Y.; Saenz, L.; Aguado, E.; Schreiber, V.; Mota, R.; Fuente, T.; Minguela, A.; Parrilla, P.; de Murcia, G.; et al. PARP-2 deficiency affects the survival of CD4+CD8+ double-positive thymocytes. EMBO J. 2006, 25, 4350–4360. [Google Scholar] [CrossRef]
- Arakawa, H. Requirement of the Activation-Induced Deaminase (AID) gene for Immunoglobulin gene conversion. Science 2002, 295, 1301–1306. [Google Scholar] [CrossRef]
- Tang, E.S.; Martin, A. Immunoglobulin gene conversion: Synthesizing antibody diversification and DNA repair. DNA Repair 2007, 6, 1557–1571. [Google Scholar] [CrossRef]
- Paddock, M.N.; Bauman, A.T.; Higdon, R.; Kolker, E.; Takeda, S.; Scharenberg, A.M. Competition between PARP-1 and Ku70 control the decision between high-fidelity and mutagenic DNA repair. DNA Repair 2011, 10, 338–343. [Google Scholar] [CrossRef]
- Paddock, M.N.; Buelow, B.D.; Takeda, S.; Scharenberg, A.M. The BRCT domain of PARP-1 is required for immunoglobulin gene conversion. PLoS Biol. 2010, 8, e1000428. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef]
- Mansour, W.Y.; Borgmann, K.; Petersen, C.; Dikomey, E.; Dahm-Daphi, J. The absence of Ku but not defects in classical non-homologous end-joining is required to trigger PARP1-dependent end-joining. DNA Repair 2013, 12, 1134–1142. [Google Scholar] [CrossRef]
- Ma, J.L.; Kim, E.M.; Haber, J.E.; Lee, S.E. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol. Cell. Biol. 2003, 23, 8820–8828. [Google Scholar] [CrossRef]
- Nussenzweig, A.; Nussenzweig, M.C. Origin of chromosomal translocations in lymphoid cancer. Cell 2010, 141, 27–38. [Google Scholar] [CrossRef]
- Pavri, R.; Nussenzweig, M.C. AID Targeting in Antibody Diversity. Adv. Immunol. 2011, 110, 1–26. [Google Scholar] [CrossRef]
- Di Noia, J.M.; Neuberger, M.S. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22. [Google Scholar] [CrossRef]
- Xu, Z.; Zan, H.; Pone, E.J.; Mai, T.; Casali, P. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat. Rev. Immunol. 2012, 12, 517–531. [Google Scholar] [CrossRef]
- Dantzer, F.; de La Rubia, G.; Menissier-De Murcia, J.; Hostomsky, Z.; de Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar]
- Liu, Y.; Kadyrov, F.A.; Modrich, P. PARP-1 enhances the mismatch-dependence of 5'-directed excision in human mismatch repair in vitro. DNA Repair 2011, 10, 1145–1153. [Google Scholar] [CrossRef]
- Jacobs, H.; Fukita, Y.; van der Horst, G.T.; de Boer, J.; Weeda, G.; Essers, J.; de Wind, N.; Engelward, B.P.; Samson, L.; Verbeek, S.; et al. Hypermutation of immunoglobulin genes in memory B cells of DNA repair-deficient mice. J. Exp. Med. 1998, 187, 1735–1743. [Google Scholar] [CrossRef]
- Küppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Nussenzweig, M.C. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 79–103. [Google Scholar] [CrossRef]
- Shockett, P.; Stavnezer, J. Inhibitors of poly(ADP-ribose) polymerase increase antibody class switching. J. Immunol. 1993, 151, 6962–6976. [Google Scholar]
- Lim, K.H.; Yang, Y.; Staudt, L.M. Pathogenetic importance and therapeutic implications of NF-kappaB in lymphoid malignancies. Immunol. Rev. 2012, 246, 359–378. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lasola, J.J.M.; Hodgson, A.; Sun, X.; Wan, F. The PARP1/ARTD1-Mediated Poly-ADP-Ribosylation and DNA Damage Repair in B Cell Diversification. Antibodies 2014, 3, 37-55. https://doi.org/10.3390/antib3010037
Lasola JJM, Hodgson A, Sun X, Wan F. The PARP1/ARTD1-Mediated Poly-ADP-Ribosylation and DNA Damage Repair in B Cell Diversification. Antibodies. 2014; 3(1):37-55. https://doi.org/10.3390/antib3010037
Chicago/Turabian StyleLasola, Jackline J.M., Andrea Hodgson, Xin Sun, and Fengyi Wan. 2014. "The PARP1/ARTD1-Mediated Poly-ADP-Ribosylation and DNA Damage Repair in B Cell Diversification" Antibodies 3, no. 1: 37-55. https://doi.org/10.3390/antib3010037
APA StyleLasola, J. J. M., Hodgson, A., Sun, X., & Wan, F. (2014). The PARP1/ARTD1-Mediated Poly-ADP-Ribosylation and DNA Damage Repair in B Cell Diversification. Antibodies, 3(1), 37-55. https://doi.org/10.3390/antib3010037