Antibody Drug Conjugates as Cancer Therapeutics

Abstract
:1. Introduction
2. General Characteristics of ADCs
3. ADC Targets and Mabs


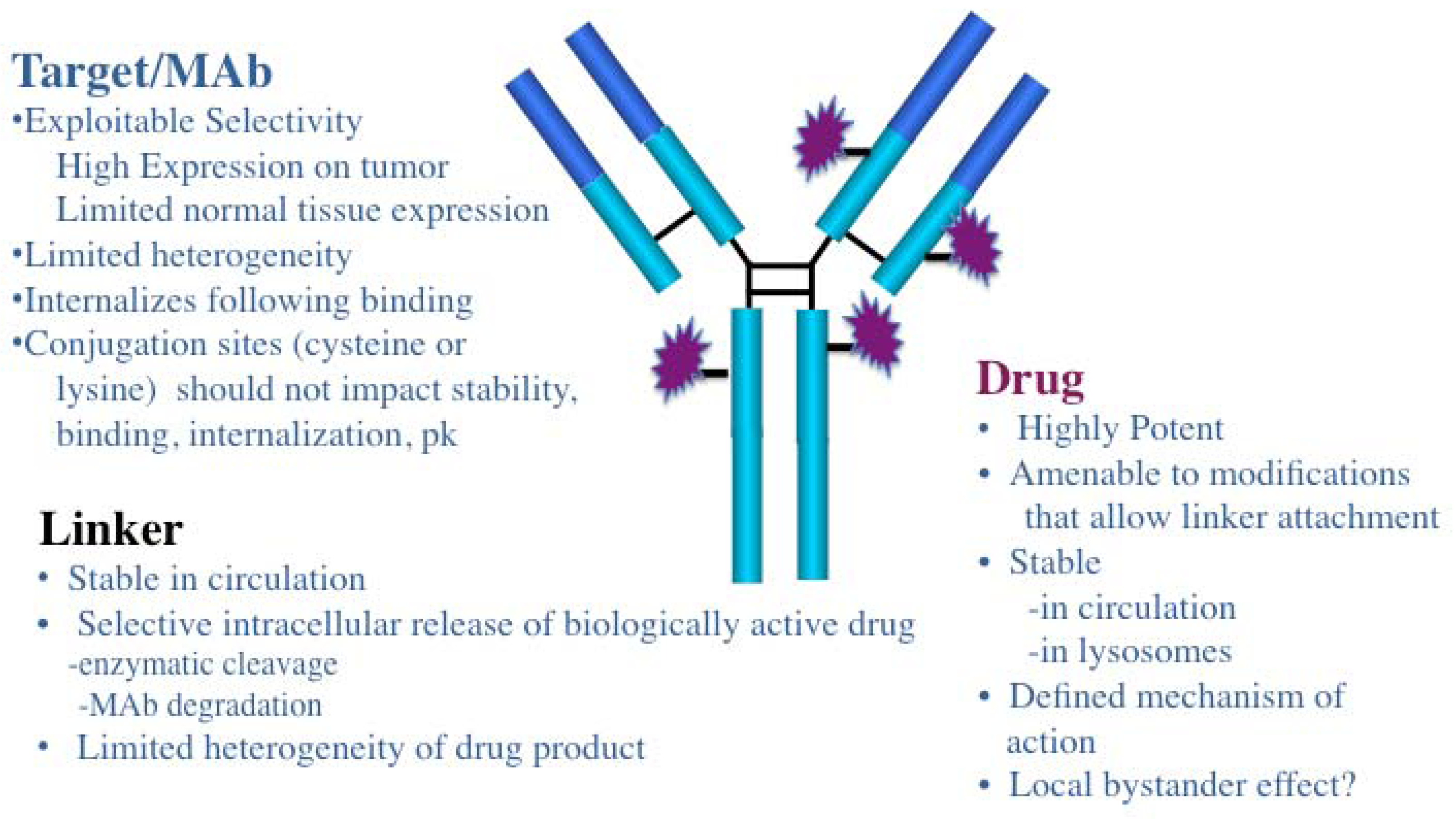{kind=link}
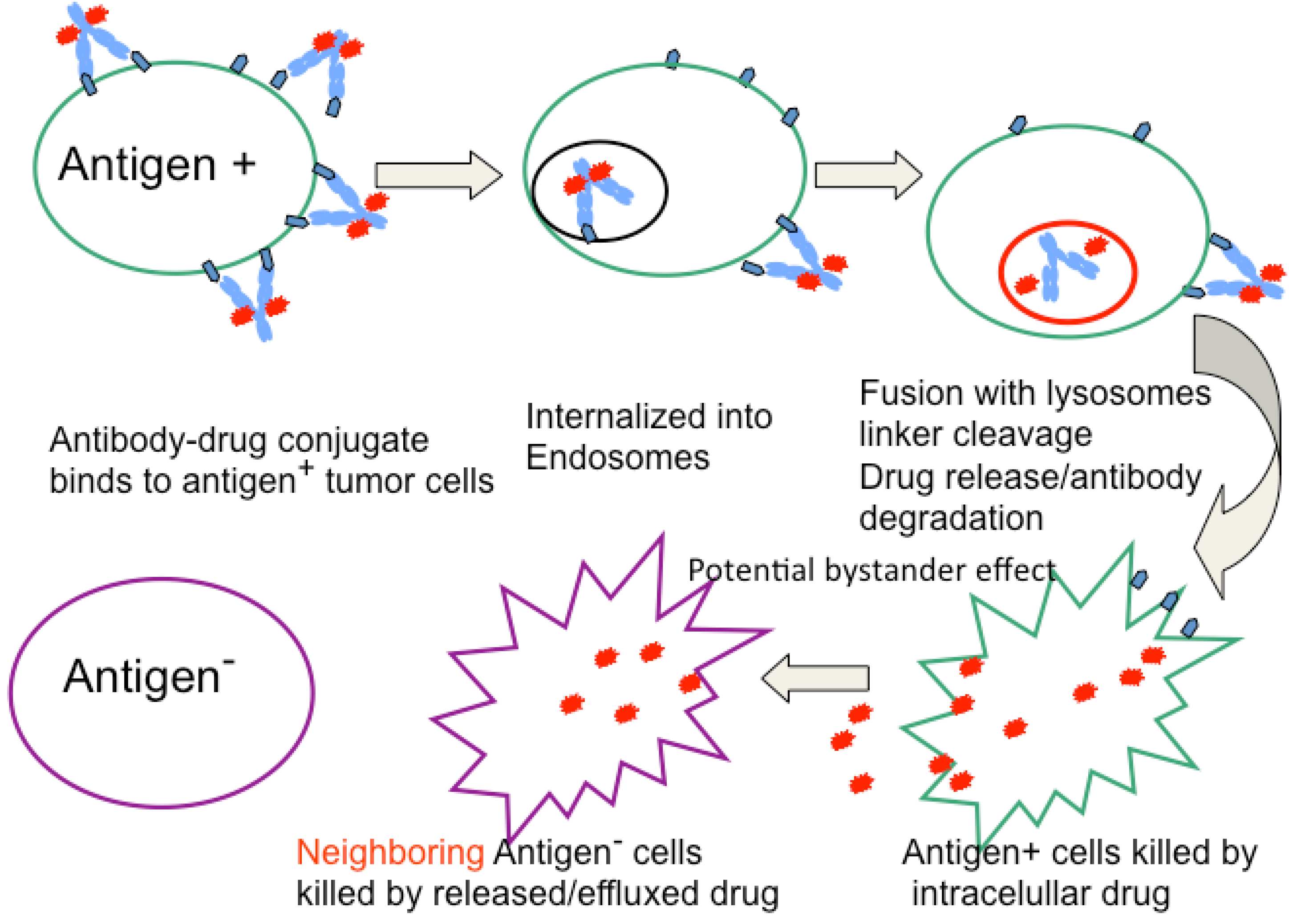{kind=link}
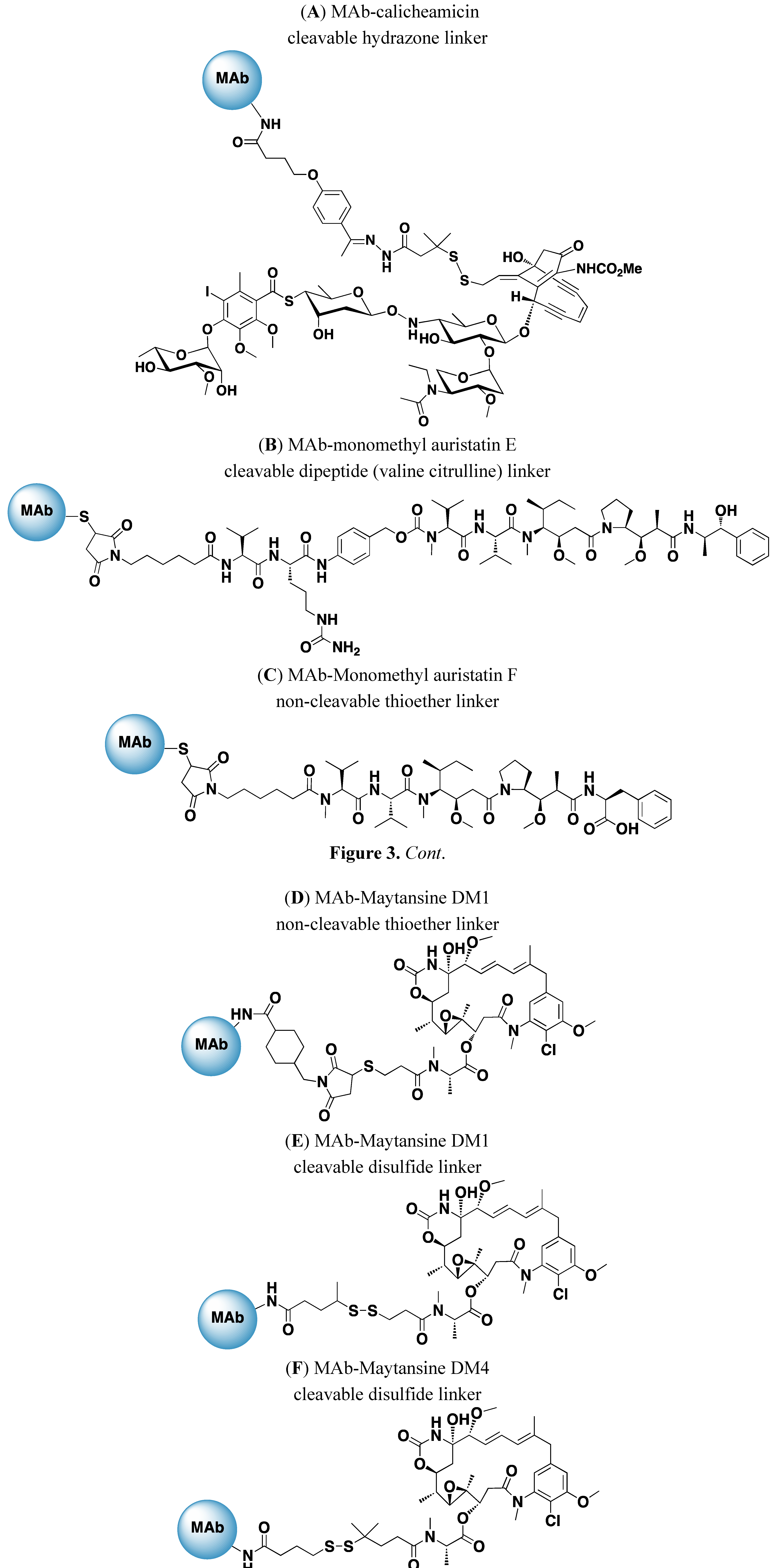{kind=link}
| ADC Designations | Target Antigen | Antibody | Linker | Drug Class | Stage b | Tumor Indication(s) | Developer |
|---|---|---|---|---|---|---|---|
| Brentuximab vedotin | CD30 | Ch IgG1 | Valine-citrulline | Auristatin MMAE | Approved | HL/ALCL | Seattle Genetics |
| Inotuzumab ozogamicin | CD22 | Hz IgG4 | Hydrazone | Calicheamicin | Phase III | NHL | Pfizer |
| Gemtuzumab ozogamicin | CD33 | Hz IgG4 | Hydrazone | Calicheamicin | Phase II | Relapsed AML | Pfizer |
| SAR3419 | CD19 | Hz IgG1 | SPDB | Maytansine DM4 | Phase II | NHL | sanofi |
| BT062 | CD138 | Ch IgG4 | SPDB | Maytansine DM4 | Phase II | MM | Biotest |
| RG7593/DCDT2980S | CD22 | Hz IgG1 | Valine-citrulline | Auristatin MMAE | Phase I | NHL | Genetech/Roche |
| RG-7596 | CD79b | Hz IgG1 | Valine-citrulline | Auristatin MMAE | Phase I | NHL | Genentech/Roche |
| Milatuzumab-doxorubicin | CD74 | HzIgG1 | Hydrazone | Doxorubicin | Phase I | MM | Immunomedics |
| Trastuzumab-emtansine | HER2 | Hz IgG1 | SMCC | Maytansine DM1 | Phase III | Breast Cancer | Genentech/Roche |
| Glembatumomab vedotin | GPNMB | Hu IgG2 | Valine-citrulline | Auristatin MMAE | Phase II | Breast Cancer, Melanoma | Celldex Therapeutics |
| Anti-PSMA ADC | PSMA | Hu IgG1 | Valine-citrulline | Auristatin MMAE | Phase II | Prostate Cancer | Progenics |
| Lorvotuzumab mertansine | CD56 | Hz IgG1 | SPP | Maytansine DM1 | Phase I/II | Solid tumors, MM | Immunogen |
| AGS-5ME | SLC44A4 | Hu IgG2 | Valine-citrulline | Auristatin MMAE | Phase I | Pancreatic, Prostate Cancer | Astellas |
| SAR566658 | CA6 | Hu IgG1 | SPDB | Maytansine DM4 | Phase I | Solid Tumors | Sanofi |
| BAY 79-4620 | CA-IX | Hu IgG1 | Valine-citrulline | Auristatin MMAE | Phase I | Solid Tumors | Bayer |
| BAY 94-9343 | Mesothelin | Hu IgG1 | SPDB | Maytansine DM4 | Phase I | Solid Tumors | Bayer |
| SGN-75 | CD70 | Hz IgG1 | Maleimidocaproyl | Auristatin MMAF | Phase I | RCC, NHL | Seattle Genetics |
| Labestuzumab-SN-38 | CD66e/CEACAM5 | Hz IgG1 | Phenylalanine-lysine | CPT-11 SN38 | Phase I | CRC | Immunomedics |
| ASG-22ME | Nectin-4 | Hu IgG1 | Valine-citrulline | Auristatin MMAE | Phase I | Solid Tumors | Astellas |
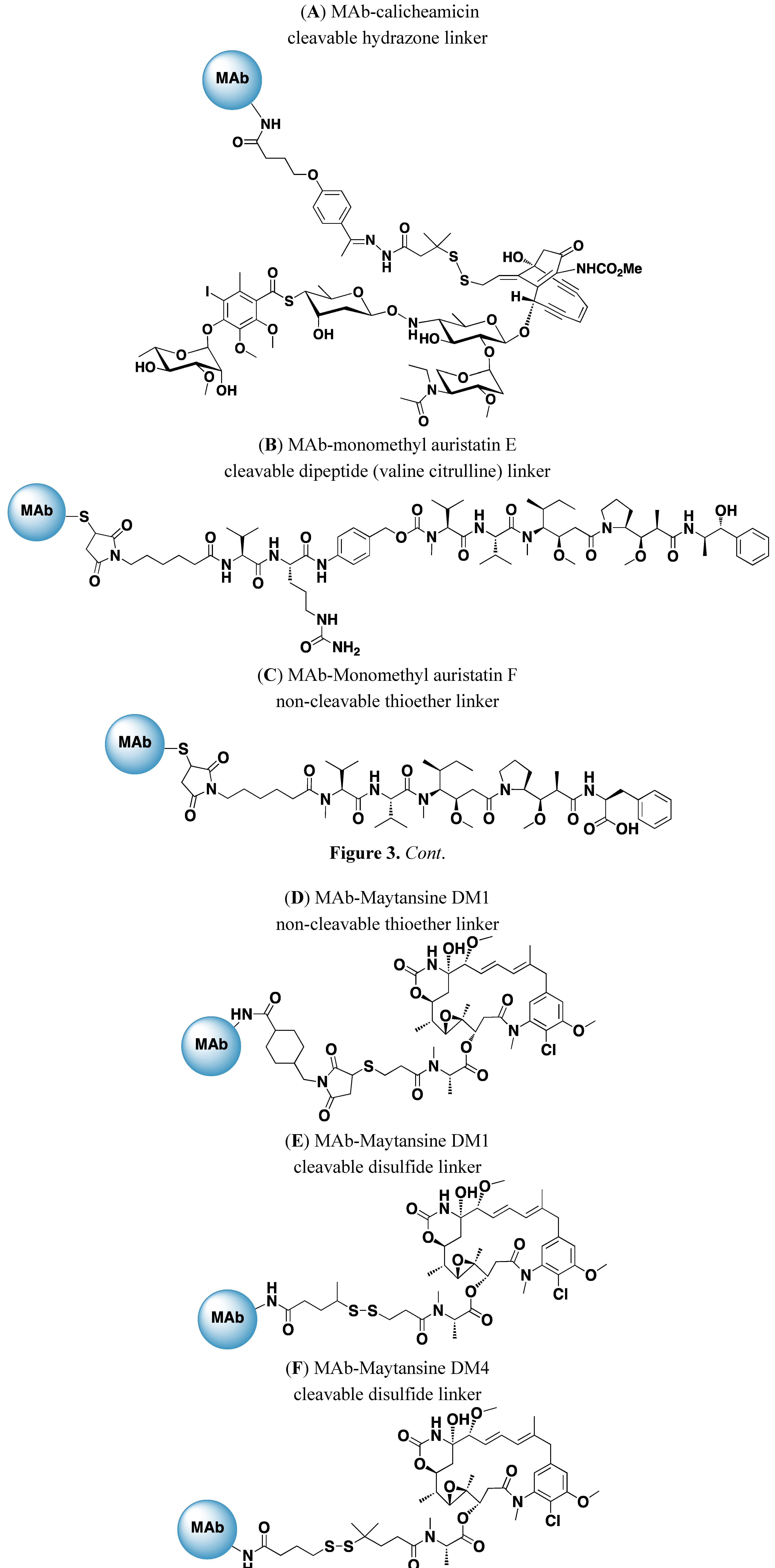
4. Drugs and Linkers Used in ADCs
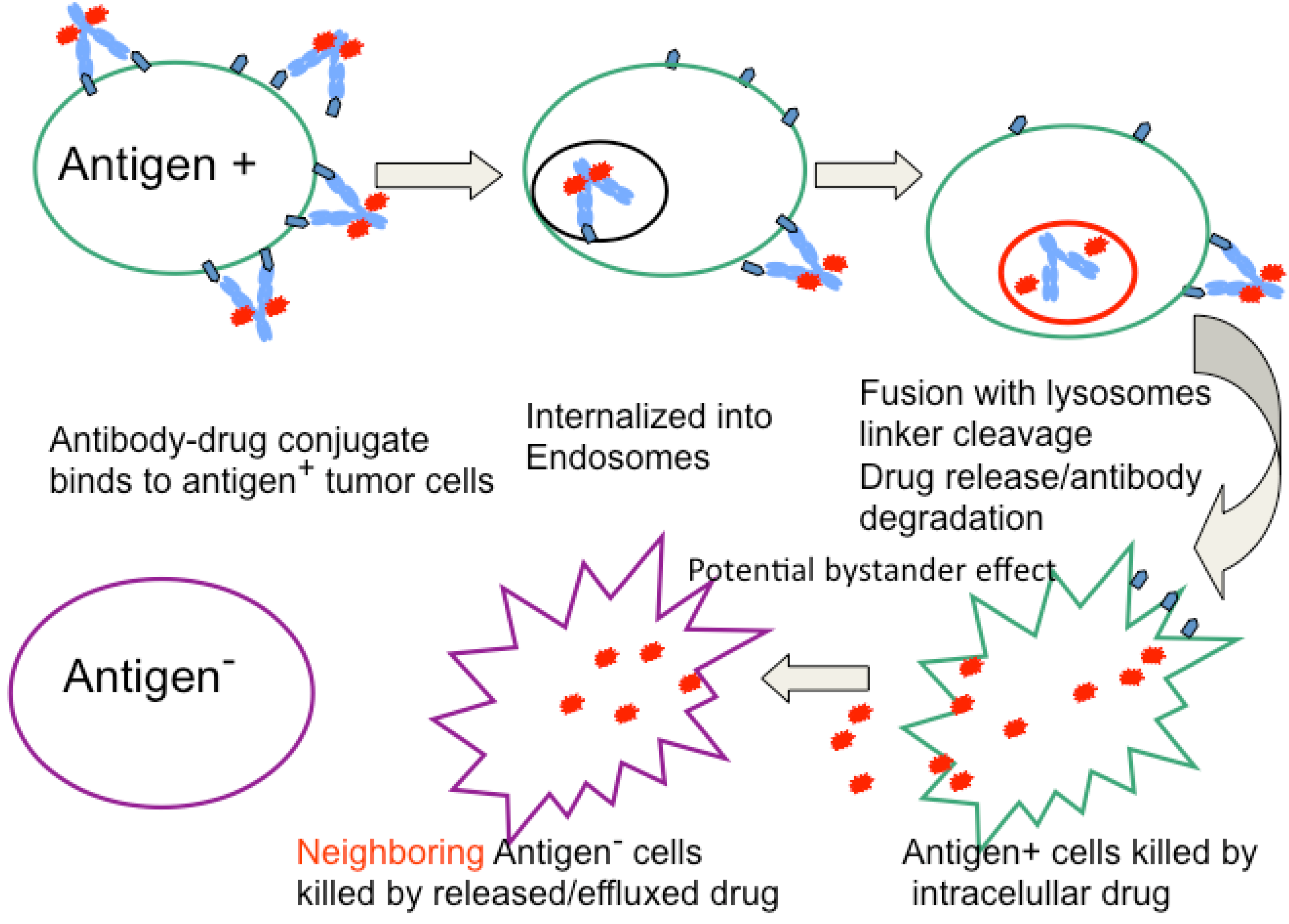
5. Optimization of ADCs
References
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef]
- Jakobovits, A. The long-awaited magic bullets: Therapeutic human monoclonal antibodies from transgenic mice. Expert Opin. Investig. Drugs 1998, 7, 607–614. [Google Scholar] [CrossRef]
- Lonberg, N. Human antibodies from transgenic animals. Nat. Biotechnol. 2005, 23, 1117–1125. [Google Scholar] [CrossRef]
- Reichert, J.M.; Dhimolea, E. The future of antibodies as cancer drugs. Drug Discov. Today 2012, 17, 954–963. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M. Radioactive antibodies: A historical review of selective targeting and treatment of cancer. Hosp. Pract. (Minneap) 2010, 38, 82–93. [Google Scholar] [CrossRef]
- Steiner, M.; Neri, D. Antibody-radionuclide conjugates for cancer therapy: Historical considerations and new trends. Clin. Cancer Res. 2011, 17, 6406–6416. [Google Scholar] [CrossRef]
- Weldon, J.E.; Pastan, I. A guide to taming a toxin—Recombinant immunotoxins constructed from pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011, 278, 4683–4700. [Google Scholar] [CrossRef]
- Lorberboum-Galski, H. Human toxin-based recombinant immunotoxins/chimeric proteins as a drug delivery system for targeted treatment of human diseases. Expert Opin. Drug Deliv. 2011, 8, 605–621. [Google Scholar] [CrossRef]
- Choudhary, S.; Mathew, M.; Verma, R.S. Therapeutic potential of anticancer immunotoxins. Drug Discov. Today 2011, 16, 495–503. [Google Scholar] [CrossRef]
- Trail, P.A.; Willner, D.; Lasch, S.J.; Henderson, A.J.; Hofstead, S.; Casazza, A.M.; Firestone, R.A.; Hellstrom, I.; Hellstrom, K.E. Cure of xenografted human carcinomas by BR96-doxorubicin immunoconjugates. Science 1993, 261, 212–215. [Google Scholar]
- Hinman, L.M.; Hamann, P.R.; Wallace, R.; Menendez, A.T.; Durr, F.E.; Upeslacis, J. Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: A novel and potent family of antitumor antibiotics. Cancer Res. 1993, 53, 3336–3342. [Google Scholar]
- Liu, C.; Chari, R.V. The development of antibody delivery systems to target cancer with highly potent maytansinoids. Expert Opin. Investig. Drugs 1997, 6, 169–172. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Trail, P.A.; Bianchi, A.B. Monoclonal antibody drug conjugates in the treatment of cancer. Curr. Opin. Immunol. 1999, 11, 584–588. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Walker, M.A. Receptor-mediated and enzyme-dependent targeting of cytotoxic anticancer drugs. Pharmacol. Ther. 1999, 83, 67–123. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar]
- Trail, P.A.; King, H.D.; Dubowchik, G.M. Monoclonal antibody drug immunoconjugates for targeted treatment of cancer. Cancer Immunol. Immunother. 2003, 52, 328–337. [Google Scholar]
- Teicher, B.A. Antibody-drug conjugate targets. Curr. Cancer Drug Targets 2009, 9, 982–1004. [Google Scholar] [CrossRef]
- Blanc, V.; Bousseau, A.; Caron, A.; Carrez, C.; Lutz, R.J.; Lambert, J.M. SAR3419: An anti-CD19-maytansinoid immunoconjugate for the treatment of B-cell malignancies. Clin. Cancer Res. 2011, 17, 6448–6458. [Google Scholar] [CrossRef]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blattler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar]
- Petrul, H.M.; Schatz, C.A.; Kopitz, C.C.; Adnane, L.; McCabe, T.J.; Trail, P.; Ha, S.; Chang, Y.S.; Voznesensky, A.; Ranges, G.; et al. Therapeutic mechanism and efficacy of the antibody-drug conjugate BAY 79-4620 targeting human carbonic anhydrase 9. Mol. Cancer Ther. 2012, 11, 340–349. [Google Scholar] [CrossRef]
- Wahl, A.F.; Klussman, K.; Thompson, J.D.; Chen, J.H.; Francisco, L.V.; Risdon, G.; Chace, D.F.; Siegall, C.B.; Francisco, J.A. The anti-CD30 monoclonal antibody SGN-30 promotes growth arrest and DNA fragmentation in vitro and affects antitumor activity in models of Hodgkin's disease. Cancer Res. 2002, 62, 3736–3742. [Google Scholar]
- Barginear, M.F.; John, V.; Budman, D.R. Trastuzumab-DM1: A clinical update of the novel antibody-drug conjugate for HER2-overexpressing breast cancer. Mol. Med. 2012, 18, 1473–1479. [Google Scholar] [CrossRef]
- Ogura, M.; Hatake, K.; Ando, K.; Tobinai, K.; Tokushige, K.; Ono, C.; Ishibashi, T.; Vandendries, E. Phase I study of anti-CD22 immunoconjugate inotuzumab ozogamicin plus rituximab in relapsed/refractory B-cell non-Hodgkin lymphoma. Cancer Sci. 2012, 103, 933–938. [Google Scholar] [CrossRef]
- Ricart, A.D. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef]
- Younes, A.; Kim, S.; Romaguera, J.; Copeland, A.; Farial Sde, C.; Kwak, L.W.; Fayad, L.; Hagemeister, F.; Fanale, M.; Neelapu, S.; et al. Phase I multidose-escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous infusion every 3 weeks to patients with relapsed/refractory B-cell lymphoma. J. Clin. Oncol. 2012, 30, 2776–2782. [Google Scholar]
- Gerber, H.P.; Senter, P.D.; Grewal, I.S. Antibody drug-conjugates targeting the tumor vasculature: Current and future developments. MAbs 2009, 1, 247–253. [Google Scholar] [CrossRef]
- Baccala, A.; Sercia, L.; Li, J.; Heston, W.; Zhou, M. Expression of prostate-specific membrane antigen in tumor-associated neovasculature of renal neoplasms. Urology 2007, 70, 385–390. [Google Scholar] [CrossRef]
- Haffner, M.C.; Kronberger, I.E.; Ross, J.S.; Sheehan, C.E.; Zitt, M.; Muhlmann, G.; Ofner, D.; Zelger, B.; Ensinger, C.; Yang, X.J.; et al. Prostate-specific membrane antigen expression in the neovasculature of gastric and colorectal cancers. Hum. Pathol. 2009, 40, 1754–1761. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; Demarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140ra86. [Google Scholar]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Lutz, R.J.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Vallet, S.; Pozzi, S.; Santo, L.; et al. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin. Cancer Res. 2009, 15, 4028–4037. [Google Scholar] [CrossRef]
- DeFrancesco, L. Seattle genetics rare cancer drug sails through accelerated approval. Nat. Biotechnol. 2011, 29, 851–852. [Google Scholar] [CrossRef]
- Jiang, X.R.; Song, A.; Bergelson, S.; Arroll, T.; Parekh, B.; May, K.; Chung, S.; Strouse, R.; Mire-Sluis, A.; Schenerman, M. Advances in the assessment and control of the effector functions of therapeutic antibodies. Nat. Rev. Drug Discov. 2011, 10, 101–111. [Google Scholar]
- Junttila, T.T.; Li, G.; Parsons, K.; Phillips, G.L.; Sliwkowski, M.X. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res. Treat. 2011, 128, 347–356. [Google Scholar]
- Kovtun, Y.V.; Goldmacher, V.S. Cell killing by antibody-drug conjugates. Cancer Lett. 2007, 255, 232–240. [Google Scholar] [CrossRef]
- Okeley, N.M.; Miyamoto, J.B.; Zhang, X.; Sanderson, R.J.; Benjamin, D.R.; Sievers, E.L.; Senter, P.D.; Alley, S.C. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin. Cancer Res. 2010, 16, 888–897. [Google Scholar] [CrossRef]
- Chari, R.V. Targeted cancer therapy: Conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef]
- Doronina, S.O.; Bovee, T.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.E.; Morris-Tilden, C.A.; Senter, P.D. Novel peptide linkers for highly potent antibody-auristatin conjugate. Bioconjug. Chem. 2008, 19, 1960–1963. [Google Scholar] [CrossRef]
- Dubowchik, G.M.; Radia, S.; Mastalerz, H.; Walker, M.A.; Firestone, R.A.; Dalton King, H.; Hofstead, S.J.; Willner, D.; Lasch, S.J.; Trail, P.A. Doxorubicin immunoconjugates containing bivalent, lysosomally-cleavable dipeptide linkages. Bioorg. Med. Chem. Lett. 2002, 12, 1529–1532. [Google Scholar]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C.; et al. Synthesis and evaluation of hydrophilic linkers for antibody-maytansinoid conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar]
- Smyth, M.J.; Pietersz, G.A.; McKenzie, I.F. The mode of action of methotrexate-monoclonal antibody conjugates. Immunol. Cell Biol. 1987, 65, 189–200. [Google Scholar] [CrossRef]
- Ghose, T.; Ferrone, S.; Blair, A.H.; Kralovec, Y.; Temponi, M.; Singh, M.; Mammen, M. Regression of human melanoma xenografts in nude mice injected with methotrexate linked to monoclonal antibody 225.28 to human high molecular weight-melanoma associated antigen. Cancer Immunol. Immunother. 1991, 34, 90–96. [Google Scholar] [CrossRef]
- Elias, D.J.; Kline, L.E.; Robbins, B.A.; Johnson, H.C., Jr.; Pekny, K.; Benz, M.; Robb, J.A.; Walker, L.E.; Kosty, M.; Dillman, R.O. Monoclonal antibody KS1/4-methotrexate immunoconjugate studies in non-small cell lung carcinoma. Am. J. Respir. Crit. Care Med. 1994, 150, 1114–1122. [Google Scholar]
- Schrappe, M.; Bumol, T.F.; Apelgren, L.D.; Briggs, S.L.; Koppel, G.A.; Markowitz, D.D.; Mueller, B.M.; Reisfeld, R.A. Long-term growth suppression of human glioma xenografts by chemoimmunoconjugates of 4-desacetylvinblastine-3-carboxyhydrazide and monoclonal antibody 9.2.27. Cancer Res. 1992, 52, 3838–3844. [Google Scholar]
- Petersen, B.H.; DeHerdt, S.V.; Schneck, D.W.; Bumol, T.F. The human immune response to KS1/4-desacetylvinblastine (ly256787) and KS1/4-desacetylvinblastine hydrazide (LY203728) in single and multiple dose clinical studies. Cancer Res. 1991, 51, 2286–2290. [Google Scholar]
- Yang, H.M.; Reisfeld, R.A. Doxorubicin conjugated with a monoclonal antibody directed to a human melanoma-associated proteoglycan suppresses the growth of established tumor xenografts in nude mice. Proc. Natl. Acad. Sci. USA 1988, 85, 1189–1193. [Google Scholar] [CrossRef]
- Shih, L.B.; Goldenberg, D.M.; Xuan, H.; Lu, H.W.; Mattes, M.J.; Hall, T.C. Internalization of an intact doxorubicin immunoconjugate. Cancer Immunol. Immunother. 1994, 38, 92–98. [Google Scholar] [CrossRef]
- Trail, P.A.; Willner, D.; Lasch, S.J.; Henderson, A.J.; Greenfield, R.S.; King, D.; Zoeckler, M.E.; Braslawsky, G.R. Antigen-specific activity of carcinoma-reactive BR64-doxorubicin conjugates evaluated in vitro and in human tumor xenograft models. Cancer Res. 1992, 52, 5693–5700. [Google Scholar]
- King, H.D.; Yurgaitis, D.; Willner, D.; Firestone, R.A.; Yang, M.B.; Lasch, S.J.; Hellstrom, K.E.; Trail, P.A. Monoclonal antibody conjugates of doxorubicin prepared with branched linkers: A novel method for increasing the potency of doxorubicin immunoconjugates. Bioconjug. Chem. 1999, 10, 279–288. [Google Scholar] [CrossRef]
- King, H.D.; Dubowchik, G.M.; Mastalerz, H.; Willner, D.; Hofstead, S.J.; Firestone, R.A.; Lasch, S.J.; Trail, P.A. Monoclonal antibody conjugates of doxorubicin prepared with branched peptide linkers: Inhibition of aggregation by methoxytriethyleneglycol chains. J. Med. Chem. 2002, 45, 4336–4343. [Google Scholar]
- Shih, L.B.; Goldenberg, D.M.; Xuan, H.; Lu, H.; Sharkey, R.M.; Hall, T.C. Anthracycline immunoconjugates prepared by a site-specific linkage via an amino-dextran intermediate carrier. Cancer Res. 1991, 51, 4192–4198. [Google Scholar]
- Saleh, M.N.; LoBuglio, A.F.; Trail, P.A. Immunoconjugate Therapy of Solid Tumors: Studies with BR96-Doxorubicin. In Monoclonal Antibody-Based Therapy of Cancer, 1th ed.; Grossbard, M.L., Ed.; Marcel Dekker, Inc: New York, NY, USA, 1998; Volume 15, pp. 397–416. [Google Scholar]
- Terrett, J.; Gangwar, S.; Rao-Naik, C.; Pan, C.; Guerlavais, V.; Huber, M.; Chong, C.; Green, L.; Cardarelli, P.; King, D.; et al. Single, low dose treatment of lymphoma and renal cancer xenografts with human anti-CD70 antibody-toxin conjugates, results in long term cures. In Proceedings of the 98th Annual Meeting of the American Association for Cancer Research, Los Angeles, CA, USA, 14–18 April 2007.
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Lowenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef]
- Jurcic, J.G. What happened to anti-CD33 therapy for acute myeloid leukemia? Curr. Hematol. Malig. Rep. 2012, 7, 65–73. [Google Scholar] [CrossRef]
- DiJoseph, J.F.; Dougher, M.M.; Evans, D.Y.; Zhou, B.B.; Damle, N.K. Preclinical anti-tumor activity of antibody-targeted chemotherapy with CMC-544 (inotuzumab ozogamicin), a CD22-specific immunoconjugate of calicheamicin, compared with non-targeted combination chemotherapy with CVP or CHOP. Cancer Chemother. Pharmacol. 2011, 67, 741–749. [Google Scholar] [CrossRef]
- Advani, A.; Coiffier, B.; Czuczman, M.S.; Dreyling, M.; Foran, J.; Gine, E.; Gisselbrecht, C.; Ketterer, N.; Nasta, S.; Rohatiner, A.; et al. Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin's lymphoma: Results of a phase I study. J. Clin. Oncol. 2010, 28, 2085–2093. [Google Scholar]
- Cardarelli, P.; King, D.; Terrett, J.; Gangwar, S.; Cohen, L.; Pan, C.; Rao, C.; Deshpande, S.; Angipuram, R.; Passmore, D.; et al. Efficacy and safety of a human anti-CD70 antibody-MGBA conjugate. In Proceedings of the 99th Annual Meeting of the American Association for Cancer Research, San Diego, CA, USA, 12-16 April 2008; AACR: Philadelphia, PA, USA, 2008; p. Abstract nr 4061. [Google Scholar]
- King, D.; Terrett, J.; Cardarelli, P.; Pan, C.; Rao, C.; Gangwar, S.; Deshpande, S.; Vangipuram, R.; Passmore, D.; Mirjolet, J.; et al. Mechanism of activation of a human anti-CD70 antibody-mgba conjugate and efficacy in a nude rat model of renal carcinoma. In Proceedings of the 99th Annual Meeting of the American Association for Cancer Research, San Diego, CA, USA, 12-16 April 2008; AACR: Philadelphia, PA, USA, 2008; p. Abstract nr 4057. [Google Scholar]
- Beck, A.; Lambert, J.; Sun, M.; Lin, K. Fourth World Antibody-drug Conjugate Summit: February 29-march 1, 2012, Frankfurt, Germany. MAbs 2012, 4, 637–647. [Google Scholar] [CrossRef]
- Chari, R.V.; Martell, B.A.; Gross, J.L.; Cook, S.B.; Shah, S.A.; Blattler, W.A.; McKenzie, S.J.; Goldmacher, V.S. Immunoconjugates containing novel maytansinoids: Promising anticancer drugs. Cancer Res. 1992, 52, 127–131. [Google Scholar]
- Oroudjev, E.; Lopus, M.; Wilson, L.; Audette, C.; Provenzano, C.; Erickson, H.; Kovtun, Y.; Chari, R.; Jordan, M.A. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol. Cancer Ther. 2010, 9, 2700–2713. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Varterasian, M.L.; Almatchy, V.P.; Hannoudi, G.N.; Pettit, G.R.; Al-Katib, A. Successful treatment of human chronic lymphocytic leukemia xenografts with combination biological agents auristatin PE and bryostatin 1. Clin. Cancer Res. 1998, 4, 1337–1343. [Google Scholar]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F.; et al. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar]
- Widdison, W.C.; Wilhelm, S.D.; Cavanagh, E.E.; Whiteman, K.R.; Leece, B.A.; Kovtun, Y.; Goldmacher, V.S.; Xie, H.; Steeves, R.M.; Lutz, R.J.; et al. Semisynthetic maytansine analogues for the targeted treatment of cancer. J. Med. Chem. 2006, 49, 4392–4408. [Google Scholar]
- de Claro, R.A.; McGinn, K.; Kwitkowski, V.; Bullock, J.; Khandelwal, A.; Habtemariam, B.; Ouyang, Y.; Saber, H.; Lee, K.; Koti, K.; et al. Food and drug administration approval summary: Brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin. Cancer Res. 2012, 18, 5845–5849. [Google Scholar] [CrossRef]
- Jackson, D.; Gooya, J.; Mao, S.; Kinneer, K.; Xu, L.; Camara, M.; Fazenbaker, C.; Fleming, R.; Swamynathan, S.; Meyer, D.; et al. A human antibody-drug conjugate targeting EphA2 inhibits tumor growth in vivo. Cancer Res. 2008, 68, 9367–9374. [Google Scholar]
- Oflazoglu, E.; Stone, I.J.; Gordon, K.; Wood, C.G.; Repasky, E.A.; Grewal, I.S.; Law, C.L.; Gerber, H.P. Potent anticarcinoma activity of the humanized anti-CD70 antibody h1F6 conjugated to the tubulin inhibitor auristatin via an uncleavable linker. Clin. Cancer Res. 2008, 14, 6171–6180. [Google Scholar]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar]
- Hartley, J.A.; Hochhauser, D. Small molecule drugs - optimizing DNA damaging agent-based therapeutics. Curr. Opin. Pharmacol. 2012, 12, 398–402. [Google Scholar] [CrossRef]
- Junutula, J.R.; Flagella, K.M.; Graham, R.A.; Parsons, K.L.; Ha, E.; Raab, H.; Bhakta, S.; Nguyen, T.; Dugger, D.L.; Li, G.; et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin. Cancer Res. 2010, 16, 4769–4778. [Google Scholar] [CrossRef]
- Sapra, P.; Hooper, A.T.; O'Donnell, C.J.; Gerber, H.P. Investigational antibody drug conjugates for solid tumors. Expert Opin. Investig. Drugs 2011, 20, 1131–1149. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Trail, P.A. Antibody Drug Conjugates as Cancer Therapeutics. Antibodies 2013, 2, 113-129. https://doi.org/10.3390/antib2010113
Trail PA. Antibody Drug Conjugates as Cancer Therapeutics. Antibodies. 2013; 2(1):113-129. https://doi.org/10.3390/antib2010113
Chicago/Turabian StyleTrail, Pamela A. 2013. "Antibody Drug Conjugates as Cancer Therapeutics" Antibodies 2, no. 1: 113-129. https://doi.org/10.3390/antib2010113
APA StyleTrail, P. A. (2013). Antibody Drug Conjugates as Cancer Therapeutics. Antibodies, 2(1), 113-129. https://doi.org/10.3390/antib2010113