Fluobodies against Bioactive Natural Products and their Application in Fluorescence-Linked Immunosorbent Assay


Abstract
:1. Introduction
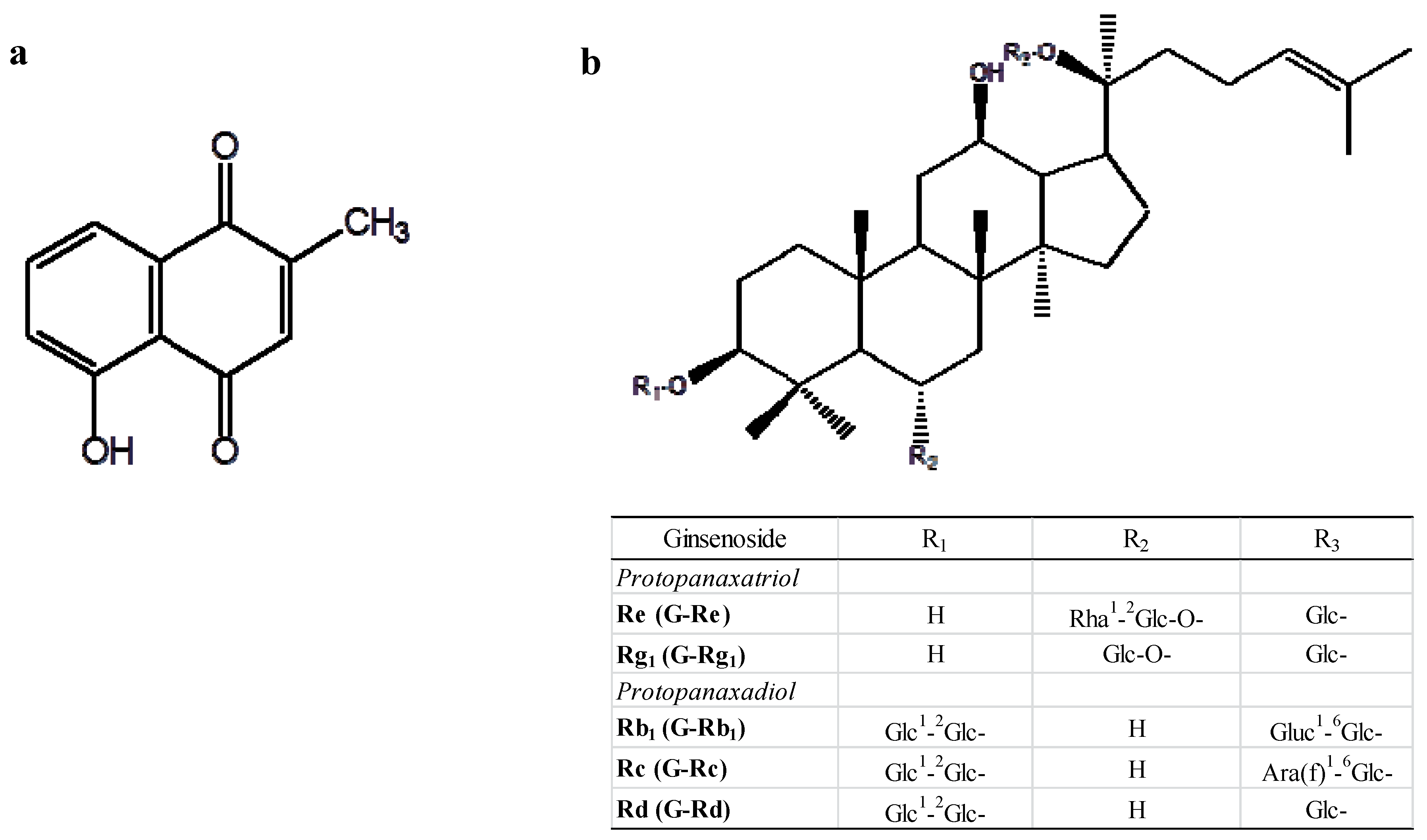
2. Results and Discussion
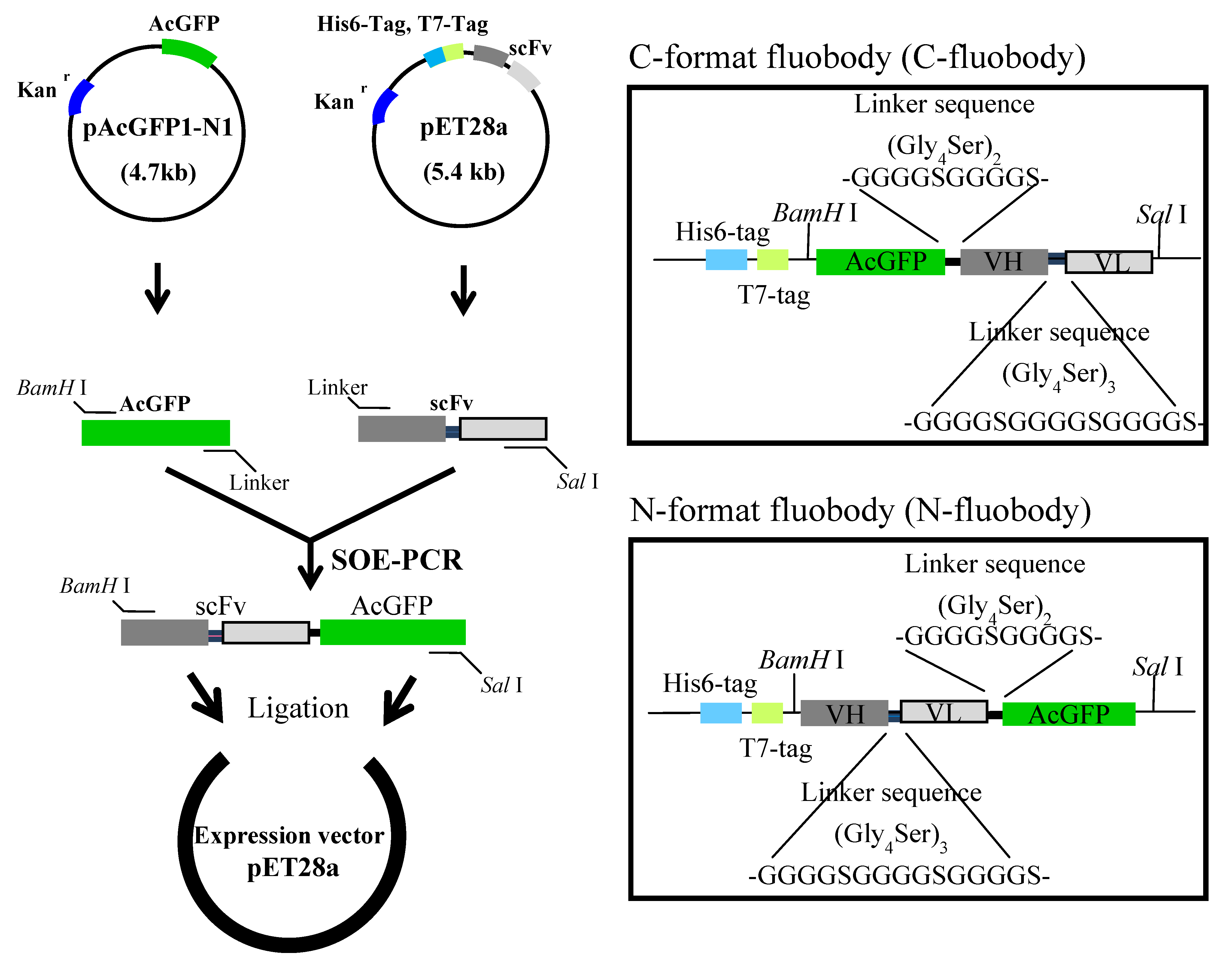
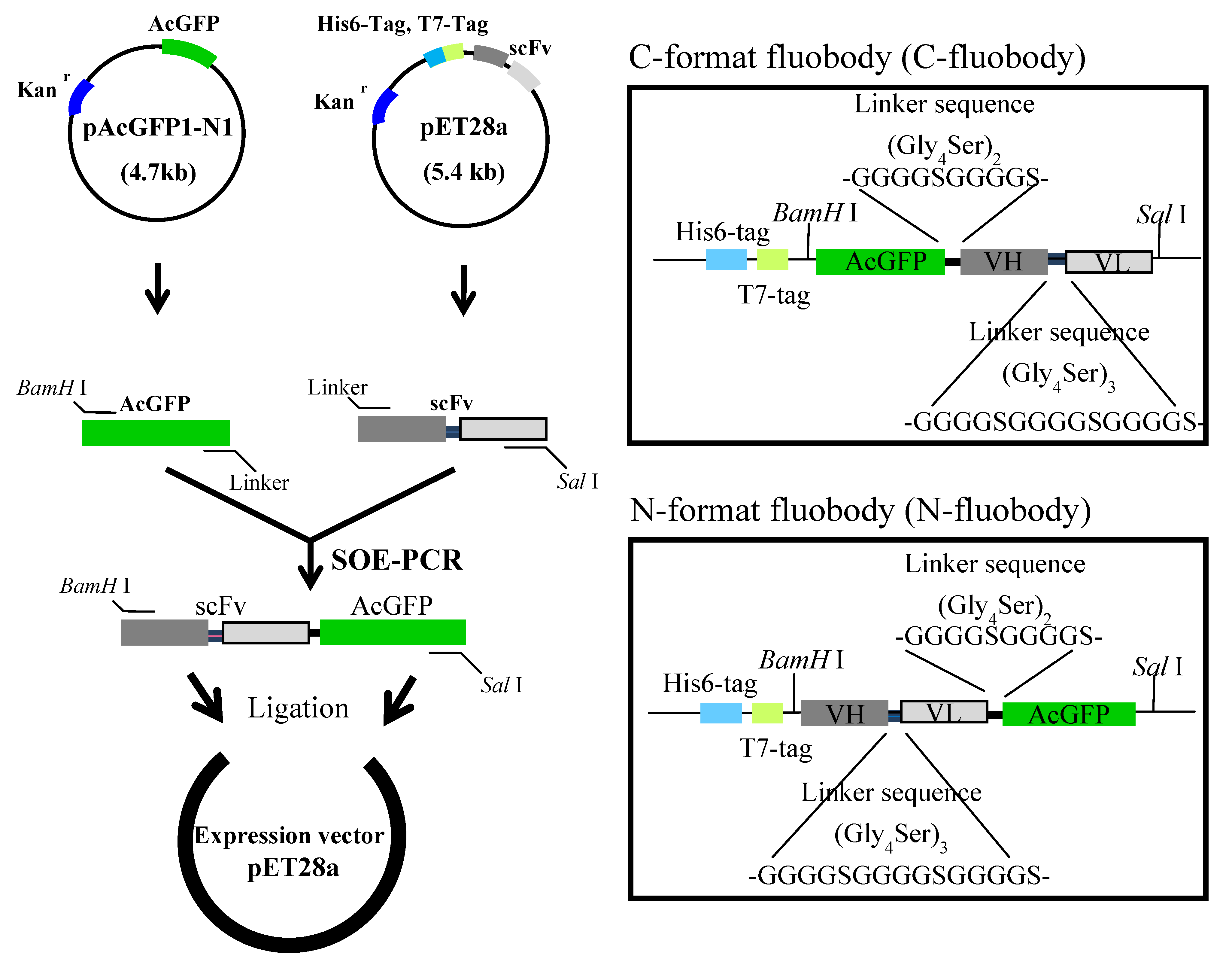
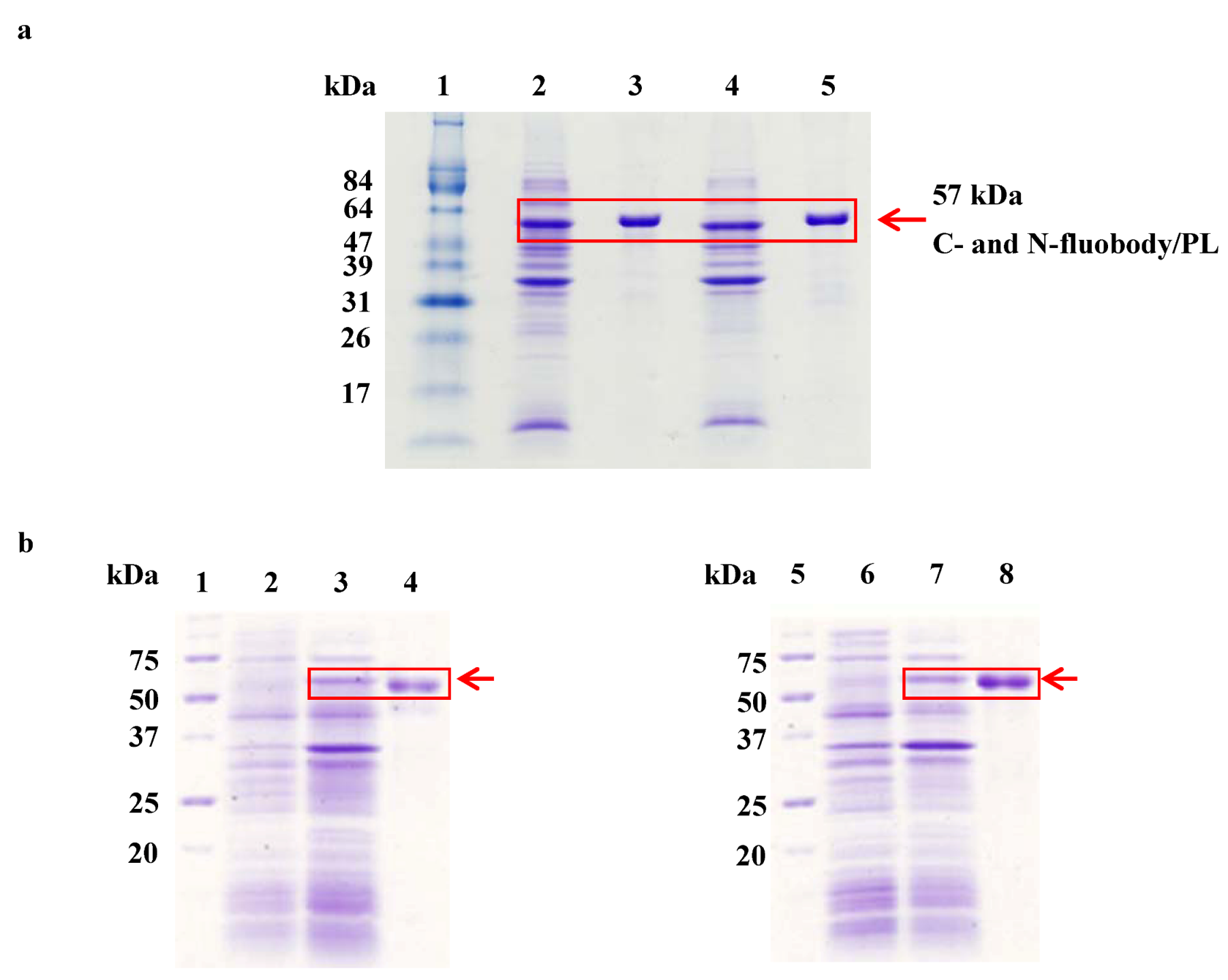
2.1. Construction and Expression of the Fluobodies
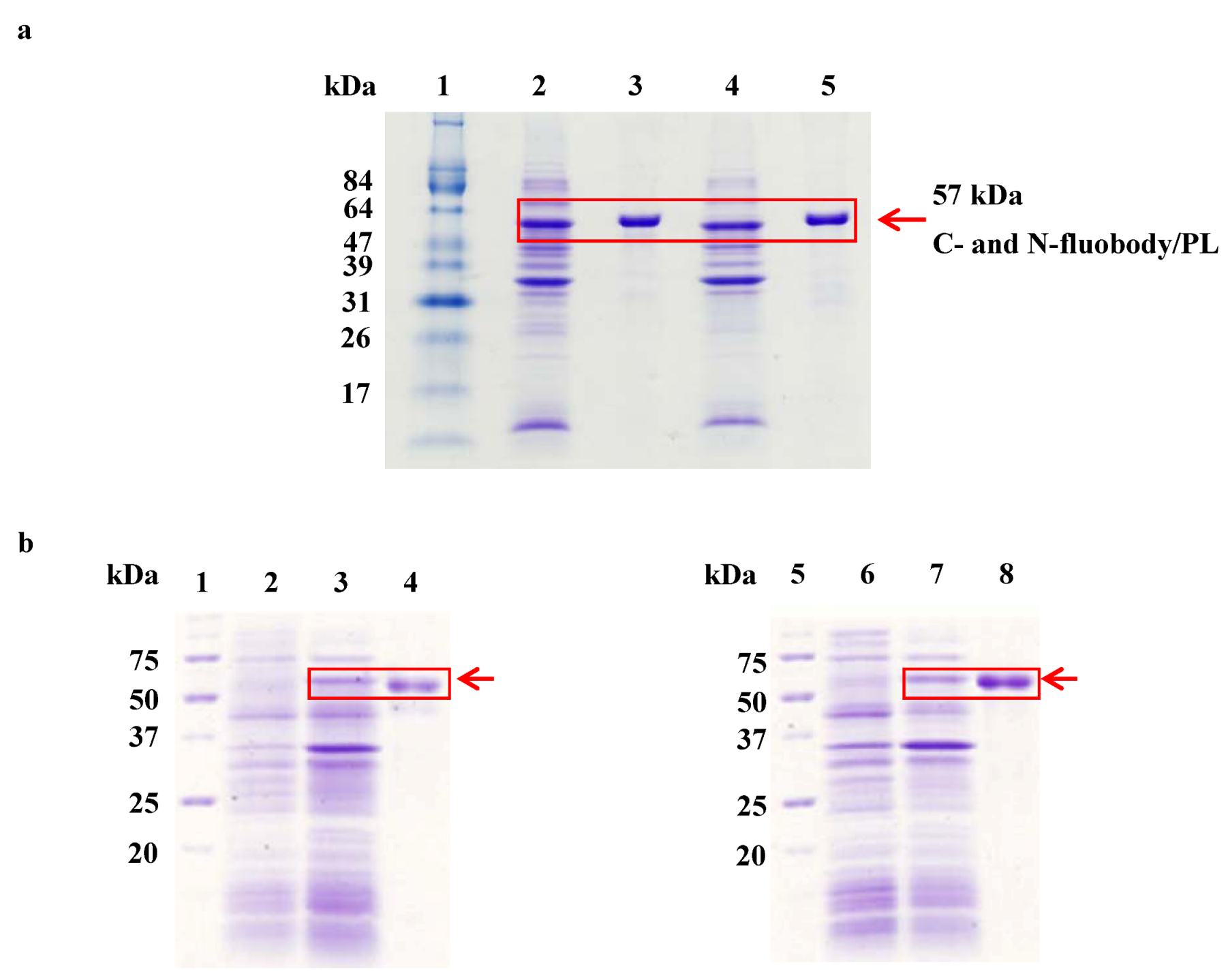
2.2. Purification and Refolding of the Fluobodies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C-format | N-format | |
|---|---|---|
| Fluobody/PL | 29.0 mg | 41.4 mg |
| Fluobody/G-Re | 32.9 mg | 52.6 mg |
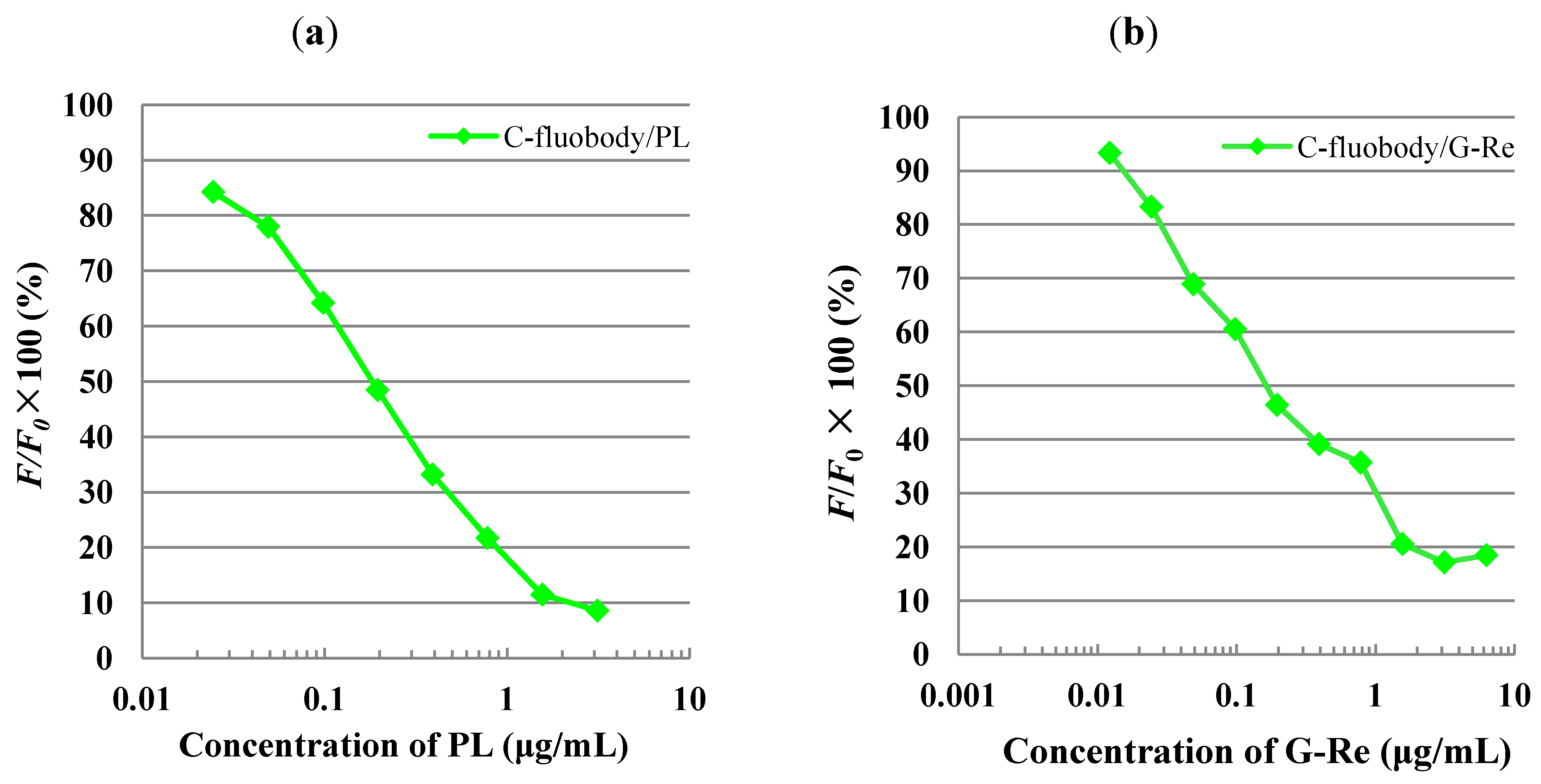
2.3. Measurement of Fluorescence Intensity

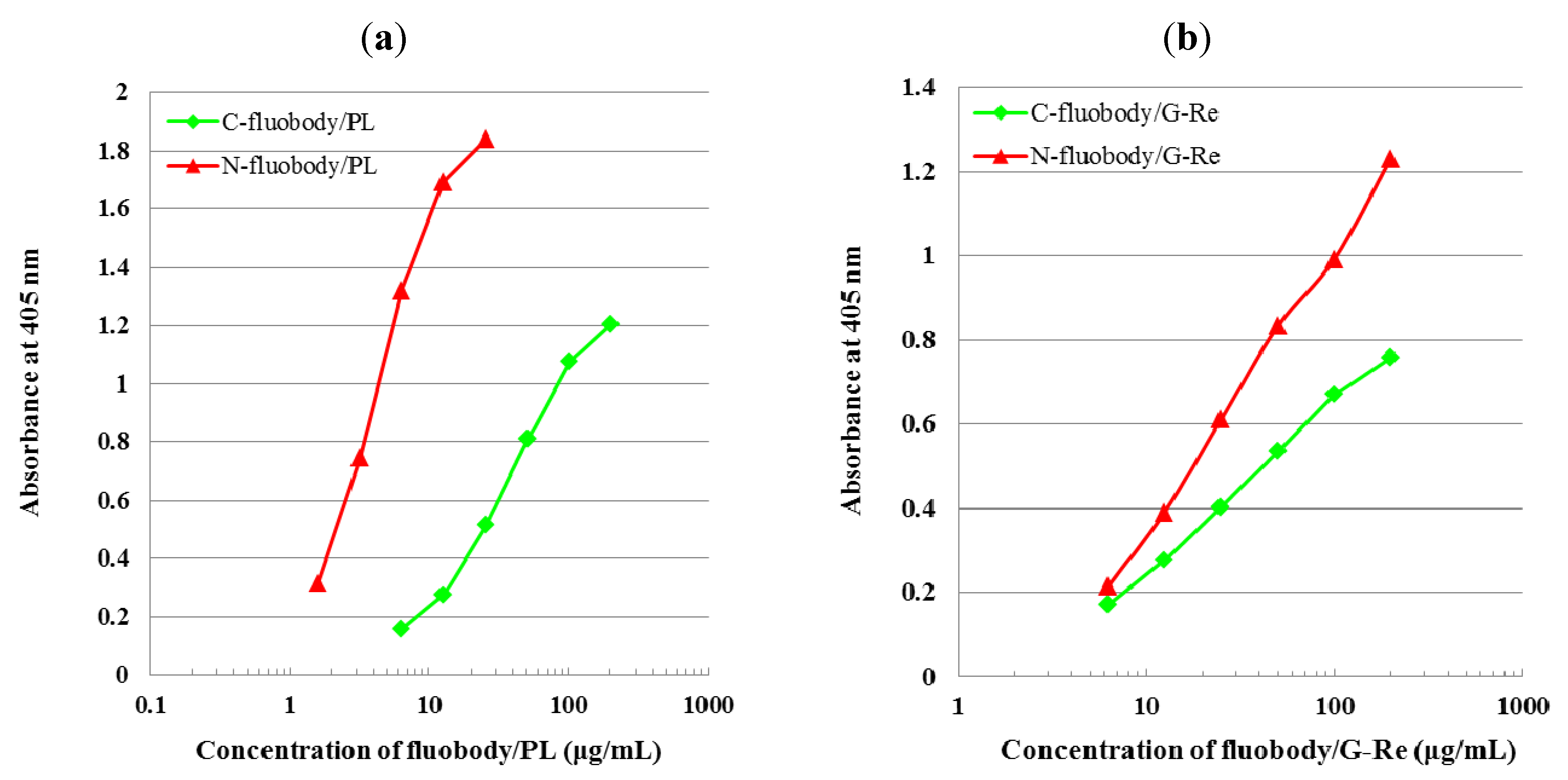
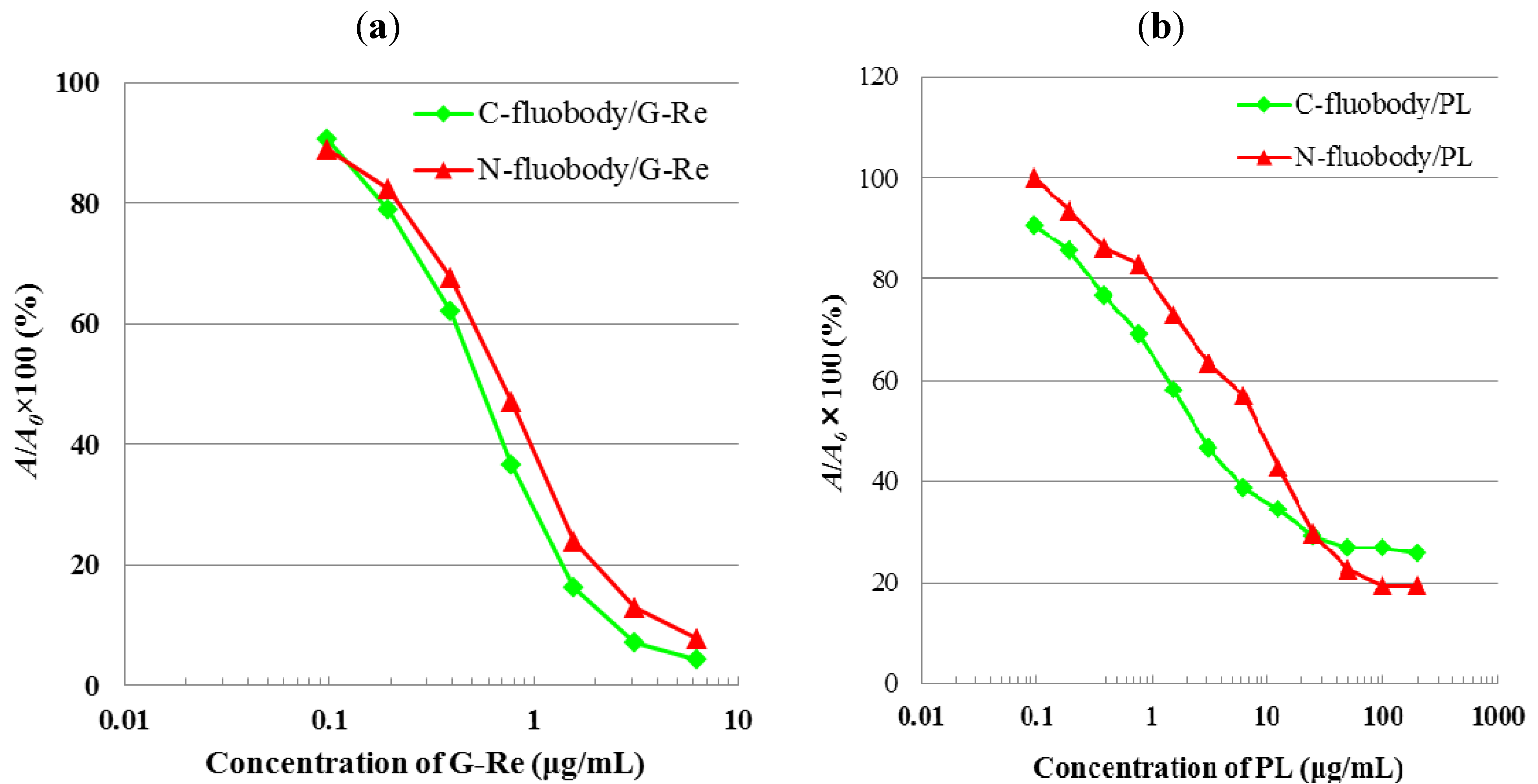
2.4. Indirect ELISA and Indirect Competitive ELISA (icELISA)


2.5. Characterization of Fluobodies/PL and Fluobodies/G-Re
| Compounds | CRs (%) | |
|---|---|---|
| C-fluobody/PL | N-fluobody/PL | |
| Plumbagin (2-methyl-5-hydroxy-1,4-naphthoquinone) | 100 | 100 |
| Menadion (2-methyl-1,4-naphthoquinone) | 11 | 4.4 |
| Juglone (5-hydroxy-1,4-naphthoquinone) | 20 | 22 |
| Lawsone (2-hydroxy-1,4-naphthoquinone) | <0.005 | <0.005 |
| 1,4-Naphthoquinone | 2.5 | 0.9 |
| 1,2-Naphthoquinone | 23 | 83 |
| Shikonin | 0.4 | 0.1 |
| Sennoside A | <0.005 | <0.005 |
| Sennoside B | <0.005 | <0.005 |
| 1-Naphthol | <0.005 | <0.005 |
| 2-Naphthol | <0.005 | <0.005 |
| C-fluobody/G-Re | N-fluobody/G-Re | |
| Ginsenoside Re (G-Re) | 100 | 100 |
| Ginsenoside Rg1 (G-Rg1) | 72 | 79 |
| Ginsenoside Rd (G-Rd) | 15 | 13 |
| Ginsenoside Rb1 (G-Rb1) | 0.4 | 0.4 |
| Ginsenoside Rc (G-Rc) | 0.4 | 0.4 |
| Glycyrrhizin | <0.005 | <0.005 |
| Saikosaponin A | <0.005 | <0.005 |
| Digitonin | <0.005 | <0.005 |
| Swertiamarin | <0.005 | <0.005 |
| Sennoside A | <0.005 | <0.005 |
| Sennoside B | <0.005 | <0.005 |
| Deoxycholic acid | <0.005 | <0.005 |
2.6. Indirect FLISA and Indirect Competitive FLISA (icFLISA)
3. Experimental Section
3.1. Chemicals and Immunochemicals
3.2. Preparation of PL-Ova and GRe-HSA Conjugates for Coated Antigen
3.3. Construction of Expression Vector for Fluobody
3.4. Expression and Purification of Recombinant Fluobodies
3.5. Refolding of Recombinant Fluobodies
3.6. Measurement of Fluorescence Intensity
3.7. Indirect ELISA and icELISA
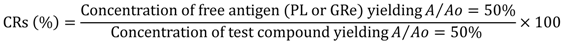
3.8. Indirect FLISA and icFLISA
4. Conclusions
Acknowledgements
References
- Yalow, R.S.; Berson, S.A. Immunoassay of endogenous plasma insulin in man. J. Clin. Invest. 1960, 39, 1157–1175. [Google Scholar] [CrossRef]
- Lê, H.Q.A.; Sauriat-Dorizon, H.; Korri-Youssoufi, H. Investigation of SPR and electrochemical detection of antigen with polypyrrole functionalized by biotinylated single-chain antibody: A review. Anal. Chim. Acta 2010, 674, 1–8. [Google Scholar] [CrossRef]
- Cullen, D.C.; Brown, R.G.W.; Lowe, C.R. Detection of immuno-complex formation via surface plasmon resonance on gold-coated diffraction gratings. Biosensors 1987, 3, 211–225. [Google Scholar] [CrossRef]
- Dangl, J.L.; Herzenberg, L.A. Selection of hybridomas and hybridoma variants using the fluorescence activated cell sorter. J. Immunol. Methods 1982, 52, 1–14. [Google Scholar] [CrossRef]
- Lanier, L.L.; Warner, N.L. Paraformaldehyde fixation of hematopoietic cells for quantitative flow cytometry (FACS) analysis. J. Immunol. Methods 1981, 47, 25–30. [Google Scholar] [CrossRef]
- Putalun, W.; Fukuda, N.; Tanaka, H.; Shoyama, Y. A one-step immunochromatographic assay for detecting ginsenosides Rb1 and Rg1. Anal. Bioanal. Chem. 2004, 378, 1338–1341. [Google Scholar] [CrossRef]
- Putalun, W.; Morinaga, O.; Tanaka, H.; Shoyama, Y. Development of a one-step immunochromatographic strip test for the detection of sennosides A and B. Phytochem. Anal. 2004, 15, 112–116. [Google Scholar] [CrossRef]
- Carnegie, P.R.; Pacheco, G. Immunochromatography: A combination of chromatography and immunodiffusion on a micro-scale. Proc. Soc. Exp. Biol. Med. 1964, 117, 137–141. [Google Scholar]
- Shan, S.; Tanaka, H.; Shoyama, Y. Enzyme-linked immunosorbent assay for glycyrrhizin using anti-glycyrrhizin monoclonal antibody and an eastern blotting technique for glucuronides of glycyrrhetic acid. Anal. Chem. 2001, 73, 5784–5790. [Google Scholar] [CrossRef]
- Morinaga, O.; Zhu, S.; Tanaka, H.; Shoyama, Y. Visual detection of saikosaponins by on-membrane immunoassay and estimation of traditional Chinese medicines containing Bupleuri radix. Biochem. Biophys. Res. Commun. 2006, 346, 687–692. [Google Scholar] [CrossRef]
- Tanaka, H.; Fukuda, N.; Shoyama, Y. Eastern blotting and immunoaffinity concentration using monoclonal antibody for ginseng saponins in the field of traditional Chinese medicines. J. Agric. Food Chem. 2007, 55, 3783–3787. [Google Scholar] [CrossRef]
- Shoyama, Y. Pharmacognosical study during 40 years. Yakugaku Zasshi 2007, 127, 1593–1620. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Winter, G.; Milstein, C. Man-made antibodies. Nature 1991, 349, 293–299. [Google Scholar] [CrossRef]
- Karawajew, L.; Micheel, B.; Behrsing, O.; Gaestel, M. Bispecific antibody-producing hybrid hybridomas selected by a fluorescence activated cell sorter. J. Immunol. Methods 1987, 96, 265–270. [Google Scholar] [CrossRef]
- Sakamoto, S.; Putalun, W.; Tsuchihashi, R.; Morimoto, S.; Kinjo, J.; Tanaka, H. Development of an enzyme-linked immunosorbent assay (ELISA) using highly-specific monoclonal antibodies against plumbagin. Anal. Chim. Acta 2008, 607, 100–105. [Google Scholar] [CrossRef]
- Morinaga, O.; Tanaka, H.; Shoyama, Y. Enzyme-linked immunosorbent assay for the determination of total ginsenosides in ginseng. Anal. Lett. 2006, 39, 287–296. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Malonne, H.; Duez, P.; Vanhaelen-Fastre, R.; Vanhaelen, M.; Fontaine, J. Cytotoxic constituents from Plumbago zeylanica. Fitoterapia 2004, 75, 500–504. [Google Scholar] [CrossRef]
- Gangopadhyay, M.; Dewanjee, S.; Bhattacharyya, S.; Bhattacharya, S. Effect of different strains of Agrobacterium rhizogenes and nature of explants on Plumbago indica hairy root culture with special emphasis on root biomass and plumbagin production. Nat. Prod. Commun. 2010, 5, 1913–1916. [Google Scholar]
- Serrilli, A.M.; Sanfilippo, V.; Ballero, M.; Sanna, C.; Poli, F.; Scartezzini, P.; Serafini, M.; Bianco, A. Polar and antioxidant fraction of Plumbago europaea L., a spontaneous plant of Sardinia. Nat. Prod. Res. 2010, 24, 633–639. [Google Scholar] [CrossRef]
- Sandur, S.K.; Ichikawa, H.; Sethi, G.; Ahn, K.S.; Aggarwal, B.B. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-κB activation and NF-κB-regulated gene products through modulation of p65 and IκBα kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J. Biol. Chem. 2006, 281, 17023–17033. [Google Scholar] [CrossRef]
- Itogawa, M.; Takeya, K.; Furukawa, H. Cardiotonic action of plumbagin on guinea-pig papillary muscle. Planta Med. 1991, 57, 317–319. [Google Scholar] [CrossRef]
- Paiva, de S.R.; Figueiredo, M.R.; Aragão, T.V.; Kaplan, M.A.C. Antimicrobial activity in vitro of plumbagin isolated from Plumbago species. Mem. Inst. Oswaldo. Cruz. 2003, 98, 959–961. [Google Scholar] [CrossRef]
- Bhargava, S.K. Effects of plumbagin on reproductive function of male dog. Indian J. Exp. Biol. 1984, 22, 153–156. [Google Scholar]
- Srinivasan, L.; Mathew, N.; Muthuswamy, K. In vitro antifilarial activity of glutathione S-transferase inhibitors. Parasitol. Res. 2009, 105, 1179–1182. [Google Scholar] [CrossRef]
- Bermejo-Bescós, P.; Martín-Aragón, S.; Jiménez-Aliaga, K.L.; Ortega, A.; Molina, M.T.; Buxaderas, E.; Orellana, G.; Csákÿ, A.G. In vitro antiamyloidogenic properties of 1,4-naphthoquinones. Biochem. Biophys. Res. Commun. 2010, 400, 169–174. [Google Scholar] [CrossRef]
- Stensen, W.; Jensen, E. High-performance liquid chromatrographic separations of naphthoquinones and their derivatives. J. Chromatogr. A 1994, 659, 87–93. [Google Scholar] [CrossRef]
- Babula, P.; Mikelova, R.; Adam, V.; Kizek, R.; Havel, L.; Sladky, Z. Using of liquid chromatography coupled with diode array detector for determination of naphthoquinones in plants and for investigation of influence of pH of cultivation medium on content of plumbagin in Dionaea muscipula. J. Chromatogr. B 2006, 842, 28–35. [Google Scholar] [CrossRef]
- Hsieh, Y.J.; Lin, L.C.; Tsai, T.H. Determination and identification of plumbagin from the roots of Plumbago zeylanica L. by liquid chromatography with tandem mass spectrometry. J. Chromatogr. A 2005, 1083, 141–145. [Google Scholar] [CrossRef]
- Lee, T.K.; Johnke, R.M.; Allison, R.R.; O’Brien, K.F.; Dobbs, L.J. Radioprotective potential of ginseng. Mutagenesis 2005, 20, 237–243. [Google Scholar] [CrossRef]
- Kiefer, D.; Pantuso, T. Panax ginseng. Am. Fam. Physician 2003, 68, 1539–1542. [Google Scholar]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Brenner, C.; Larochette, N.; Prévost, M.; Alzari, P.M.; Kroemer, G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med. 1999, 189, 381–394. [Google Scholar] [CrossRef]
- Luo, J.Z.; Luo, L. Ginseng on hyperglycemia: Effects and mechanisms. Evid. Based Complement Altern. Med. 2009, 6, 423–427. [Google Scholar] [CrossRef]
- Xie, J.T.; Mchendale, S.; Yuan, C.S. Ginseng and diabetes. Am. J. Chin. Med. 2005, 33, 397–404. [Google Scholar] [CrossRef]
- Liu, L.; Huang, J.; Hu, X.; Li, K.; Sun, C. Simultaneous determination of ginsenoside (G-Re, G-Rg1, G-Rg2, G-F1, G-Rh1) and protopanaxatriol in human plasma and urine by LC-MS/MS and its application in a pharmacokinetics study of G-Re in volunteers. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011, 879, 2011–2017. [Google Scholar] [CrossRef]
- Li, L.; Luo, G.A.; Liang, Q.L.; Hu, P.; Wang, Y.M. Rapid qualitative and quantitative analyses of Asian ginseng in adulterated American ginseng preparations by UPLC/Q-TOF-MS. J. Pharm. Biomed. Anal. 2010, 52, 66–72. [Google Scholar] [CrossRef]
- Kanazawa, H.; Nagata, Y.; Matsushima, Y.; Tomoda, M.; Takai, N. Simultaneous determination of ginsenosides and saikosaponins by high-performance liquid chromatography. J. Chromatogr. 1990, 507, 327–332. [Google Scholar] [CrossRef]
- Fukuda, N.; Tanaka, H.; Shoyama, Y. Applications of ELISA, western blotting and immunoaffinity concentration for survey of ginsenosides in crude drugs of Panax species and traditional Chinese herbal medicines. Analyst 2000, 125, 1425–1429. [Google Scholar] [CrossRef]
- Deng, G.F.; Wang, D.L.; Meng, M.X.; Hu, F.; Yao, T.W. Simultaneous determination of notoginsenoside R1, ginsenoside Rg1, Re, Rb1 and icariin in rat plasma by ultra-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2113–2122. [Google Scholar] [CrossRef]
- Sakamoto, S.; Taura, F.; Putalun, W.; Pongkitwitoon, B.; Tsuchihashi, R.; Morimoto, S.; Kinjo, J.; Shoyama, Y.; Tanaka, H. Construction and expression of specificity-improved single-chain variable fragments against the bioactive naphthoquinone, plumbagin. Biol. Pharm. Bull. 2009, 32, 434–439. [Google Scholar] [CrossRef]
- Pongkitwitoon, B.; Sakamoto, S.; Morinaga, O.; Juengwatanatrakul, T.; Shoyama, Y.; Tanaka, H.; Morimoto, S. Single-chain variable fragment antibody against ginsenoside Re as an effective tool for the determination of ginsenosides in various ginsengs. J. Nat. Med. 2011, 65, 24–30. [Google Scholar] [CrossRef]
- Sakamoto, S.; Taura, F.; Tsuchihashi, R.; Putalun, W.; Kinjo, J.; Tanaka, H.; Morimoto, S. Expression, purification, and characterization of anti-plumbagin single-chain variable fragment antibody in Sf9 insect cell. Hybridoma (Larchmt) 2010, 29, 481–488. [Google Scholar] [CrossRef]
- Sakamoto, S.; Pongkitwitoon, B.; Nakamura, S.; Maenaka, K.; Tanaka, H.; Morimoto, S. Efficient silkworm expression of single-chain variable fragment antibody against ginsenoside Re using Bombyx mori nucleopolyhedrovirus bacmid DNA system and its application in enzyme-linked immunosorbent assay for quality control of total ginsenosides. J. Biochem. 2010, 148, 335–340. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate Techniques; Academic Press: San Diego, CA, USA, 1996; pp. 297–364. [Google Scholar]
- Goldstein, G.; Slizys, I.S.; Chase, M.W. Studies on fluorescent antibody staining. I. Non-specific fluorescence with fluorescein-coupled sheep anti-rabbit globulins. J. Exp. Med. 1961, 114, 89–110. [Google Scholar] [CrossRef]
- Sommerville, R.G. The production of fluorescent antibody reagents for virus diagnosis in the albino mouse. II. Coupling of mouse immune globulin with fluorescein isothiocyanate (FITC). Arch. Gesamte. Virusforsch. 1967, 20, 452–458. [Google Scholar] [CrossRef]
- Souriu, C.; Hudson, P.J. Recombinant antibodies for cancer diagnosis and therapy. Expert Opin. Biol. Ther. 2001, 1, 845–855. [Google Scholar] [CrossRef]
- Casey, J.L.; Coley, A.M.; Tilley, L.M.; Foley, M. Green fluorescent antibodies: novel in vitro tools. Protein Eng. 2000, 13, 445–452. [Google Scholar] [CrossRef]
- Peipp, M.; Saul, D.; Barbin, K.; Bruenke, J.; Zunino, S.J.; Niederweis, M.; Fey, G.H. Efficient eukaryotic expression of fluorescent scFv fusion proteins directed against CD antigens for FACS applications. J. Immunol. Methods 2004, 285, 265–280. [Google Scholar] [CrossRef]
- Cao, M.; Cao, P.; Yan, H.J.; Ren, F.; Lu, W.G.; Hu, Y.L.; Zhang, S.Q. Construction and characterization of an enhanced GFP-tagged anti-BAFF scFv antibody. Appl. Microbiol. Biotechnol. 2008, 79, 423–431. [Google Scholar] [CrossRef]
- Olichon, A.; Surrey, T. Selection of genetically encoded fluorescent single domain antibodies engineered for efficient expression in Escherichia coli. J. Biol. Chem. 2007, 282, 36314–36320. [Google Scholar] [CrossRef]
- Kim, I.S.; Shim, J.H.; Suh, Y.T.; Yau, K.Y.F.; Hall, J.C.; Trevors, J.T.; Lee, H. Green fluorescent protein-labeled recombinant antibody for detecting the picloram herbicide. Biosci. Biotechnol. Biochem. 2002, 66, 1148–1151. [Google Scholar] [CrossRef]
- Oelschlaeger, P.; Srikant-Iyer, S.; Lange, S.; Schmitt, J.; Schmid, R.D. Fluorophor-linked immunosorbent assay: a time- and cost-saving method for the characterization of antibody fragments using a fusion protein of a single-chain antibody fragment and enhanced green fluorescent protein. Anal. Biochem. 2002, 309, 27–34. [Google Scholar]
- Sakamoto, S.; Taura, F.; Pongkitwitoon, B.; Putalun, W.; Tsuchihashi, R.; Kinjo, J.; Tanaka, H.; Morimoto, S. Development of sensitivity-improved fluorescence-linked immunosorbent assay using a fluorescent single-domain antibody against the bioactive naphthoquinone, plumbagin. Anal. Bioanal. Chem. 2010, 396, 2955–2963. [Google Scholar] [CrossRef]
- Sakamoto, S.; Tanizaki, Y.; Pongkitwitoon, B.; Tanaka, H.; Morimoto, S. A chimera of green fluorescent protein with single chain variable fragment antibody against ginsenosides for fluorescence-linked immunosorbent assay. Protein Expr. Purif. 2011, 77, 124–130. [Google Scholar] [CrossRef]
- Sakamoto, S.; Pongkitwitoon, B.; Sasaki-Tabata, K.; Putalun, W.; Maenaka, K.; Tanaka, H.; Morimoto, S. A fluorescent single domain antibody against plumbagin expressed in silkworm larvae for fluorescence-linked immunosorbent assay (FLISA). Analyst 2011, 136, 2056–2063. [Google Scholar]
- Horton, R.M.; Hunt, H.D.; Ho, S.N.; Pullen, J.K.; Pease, L.R. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 1989, 77, 61–68. [Google Scholar] [CrossRef]
- Umetsu, M.; Tsumoto, K.; Hara, M.; Ashish, K.; Goda, S.; Adschiri, T.; Kumagai, I. How additives influence the refolding of immunoglobulin-folded proteins in a stepwise dialysis system. Spectroscopic evidence for highly efficient refolding of a single-chain Fv fragment. J. Biol. Chem. 2003, 278, 8979–8987. [Google Scholar]
- Ohshima, M.; Inoue, K.; Hayashi, H.; Tsuji, D.; Mizugaki, M.; Itoh, K. Generation of AcGFP fusion with single-chain Fv selected from a phage display library constructed from mice hyperimmunized against 5-methyl 2'-deoxycytidine. Protein Eng. Des. Sel. 2010, 23, 881–888. [Google Scholar] [CrossRef]
- Ormo, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal structure of Aequorea victoria green fluorescent protein. Science 1996, 273, 1392–1395. [Google Scholar]
- Friguet, B.; Chaffotte, A.F.; Djavadi-Ohaniance, L.; Goldberg, M.E. Measurements of the true affinity constant in solution of antigen-antibody complexes by enzyme-linked immunosorbent assay. J. Immunol. Methods 1985, 77, 305–319. [Google Scholar] [CrossRef]
- Ishiyama, M.; Shoyama, Y.; Murakami, H.; Shinohara, H. Production of monoclonal antibodies and development of an ELISA for solamargine. Cytotechnology 1996, 18, 153–158. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Weiler, E.; Zenk, M.H. Radioimmunoassay for determination of digoxin and related compounds in Digitalis lanata. Phytochemistry 1976, 15, 1537–1545. [Google Scholar] [CrossRef]
- Orlandi, R.; Gussow, D.H.; Jones, P.T.; Winter, G. Cloning immunoglobulin variable domains for expression by the polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1989, 86, 3833–3837. [Google Scholar] [CrossRef]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature (London) 1990, 348, 552–554. [Google Scholar] [CrossRef]
- Bertani, G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 1952, 62, 293–300. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sakamoto, S.; Pongkitwitoon, B.; Nakahara, H.; Shibata, O.; Shoyama, Y.; Tanaka, H.; Morimoto, S. Fluobodies against Bioactive Natural Products and their Application in Fluorescence-Linked Immunosorbent Assay. Antibodies 2012, 1, 239-258. https://doi.org/10.3390/antib1020239
Sakamoto S, Pongkitwitoon B, Nakahara H, Shibata O, Shoyama Y, Tanaka H, Morimoto S. Fluobodies against Bioactive Natural Products and their Application in Fluorescence-Linked Immunosorbent Assay. Antibodies. 2012; 1(2):239-258. https://doi.org/10.3390/antib1020239
Chicago/Turabian StyleSakamoto, Seiichi, Benyakan Pongkitwitoon, Hiromichi Nakahara, Osamu Shibata, Yukihiro Shoyama, Hiroyuki Tanaka, and Satoshi Morimoto. 2012. "Fluobodies against Bioactive Natural Products and their Application in Fluorescence-Linked Immunosorbent Assay" Antibodies 1, no. 2: 239-258. https://doi.org/10.3390/antib1020239