1. Introduction
Onsite wastewater treatment systems (OWTS) rely on biogeochemical processes and ecological interactions that take place in the soil treatment area (STA; also known as drainfield or leachfield) to renovate wastewater, many of which are driven by microorganisms. To maximize septic tank effluent (STE) treatment potential, we need to improve our understanding of the composition and structure of the microbial community that develops in STAs in response to STE inputs. However, information on the structure and composition of the bacterial community of STAs is scant and is often limited to the soil at the infiltrative surface (the biomat) [
1,
2]. Although the biomat is considered an important zone because of its high biological activity and potential for transformation of nutrients and organic contaminants [
3], other treatment processes are likely to occur deeper in the soil profile.
Much of the data available on the microbial community of STAs is based on culture-dependent methods, which capture a very small fraction of the microbial diversity in soil, limiting its usefulness for understanding community structure and composition. Recent studies have used clone libraries to examine the microbial communities of STAs, shedding new light on their structure and phylogenetic diversity [
1,
4]. They suggest that these communities are very complex, and respond differently to differences in availability of terminal electron acceptors and antibiotics. These studies also raise questions about the sources of different microorganisms, the prevalence of particular strains, and the functions they carry out.
The relative contribution of soil and STE to the composition of the microbial community of STAs is an open question. In general, the soil microbial community is extremely diverse, with up to 10
9 microorganisms—representing thousands of different species—occupying one gram of soil [
5]. STAs are usually built using soil from the B and C horizons, which tends to be low in carbon substrates and nutrients. A few studies have examined the structure of the soil microbial community below the top few centimeters of the soil profile using modern molecular methods. Fierer
et al. [
6] observed lower microbial biomass at 2 m than at the surface of two Mollisols and a decrease in microbial diversity with depth. Furthermore, they reported that Gram-positive bacteria and actinomycetes were proportionally more abundant with depth, whereas Gram-negative bacteria, fungi and protozoa were less abundant at greater depths. Similarly, Zhou
et al. [
7] examined two sandy soils from the surface (0.05 m) and vadose (1.57 m) zones and found microbial diversity to decrease with depth. These studies suggest that the depth at which an STA is built may be an important determinant of the composition of the resulting bacterial community. STE is much less phylogenetically diverse than soil [
1], more closely resembling that of the human gut which, at the phylum level, is one of the least diverse ecosystems on Earth [
8]. Beyond the differences in diversity between soil and STE, there are clear differences in properties between these two sources of microorganisms to the STA in terms physical structure, pH, redox potential and availability of C, nutrients and electron acceptors, suggesting a low probability of establishment of STE bacteria in soil. However, the gradients of moisture, C, and nutrient resources and electron acceptors established with depth in STAs, as well as the continuous influx of microorganisms from STE inputs, may provide niches suitable for organisms from both sources.
Soil type is also likely to play an important role in determining the structure and composition of STA microbial communities. For example, soil texture can control the size, structure and function of soil microbial communities. A number of studies examining communities associated with different particle size fractions of the same surface soil [
9,
10] have found higher diversity to be associated with smaller particle size fractions. Sessitsch
et al. [
9] hypothesized that this is due to a higher availability of nutrients in soil with a high proportion of clay-sized particles. Knops
et al. [
11] found a positive correlation between clay content and nutrient retention in soils. Furthermore, the high spatial isolation in soils with a high proportion of clay-sized particles is thought to encourage microbial diversity by limiting competition and enhancing the probability of colonization by new species without allowing them to achieve dominance [
7]. Tiedje
et al. [
12] hypothesize that soils with high spatial isolation also have a higher heterogeneity of carbon resources, leading to greater niche variation. The spatial isolation and substrate heterogeneity found in soils may be reduced in STAs by periodic inputs of wastewater, which would be expected to result in greater hydrologic connection and a shift to more restrictive environmental conditions.
An understanding of the factors that control the composition and structure of the bacterial community of STAs is important for developing our ability to design systems that renovate water quality in a predictable manner and adjusting system conditions to promote desirable processes. To this end, we conducted a mesocosm-scale experiment to (i) evaluate the structure and composition of the bacterial communities of STAs as a function of soil texture and depth below the infiltrative surface; and (ii) to determine the relative contribution of STE and native soil to these bacterial communities. Mesocosms received STE at a rate of 2.4 cm/day. Community composition and structure were assessed before and after development of a stable biomat by TRFLP analysis of PCR-amplified 16S rDNA fragments extracted from the soil. The presence of a biomat is regarded as an indicator that the system has achieved maturity with respect to capacity for STE renewal [
3]. Three soil types—sand, sandy loam, and clay—were evaluated, with analysis conducted for soils at depths of 0–4, 4–14, 14–24, and 24–34 cm below the infiltrative surface. These data, in conjunction with TRFLP data for STE and native soil unexposed to STE, were also used to examine the relative contribution of native soil and STE to the resulting STA bacterial communities.
2. Results and Discussion
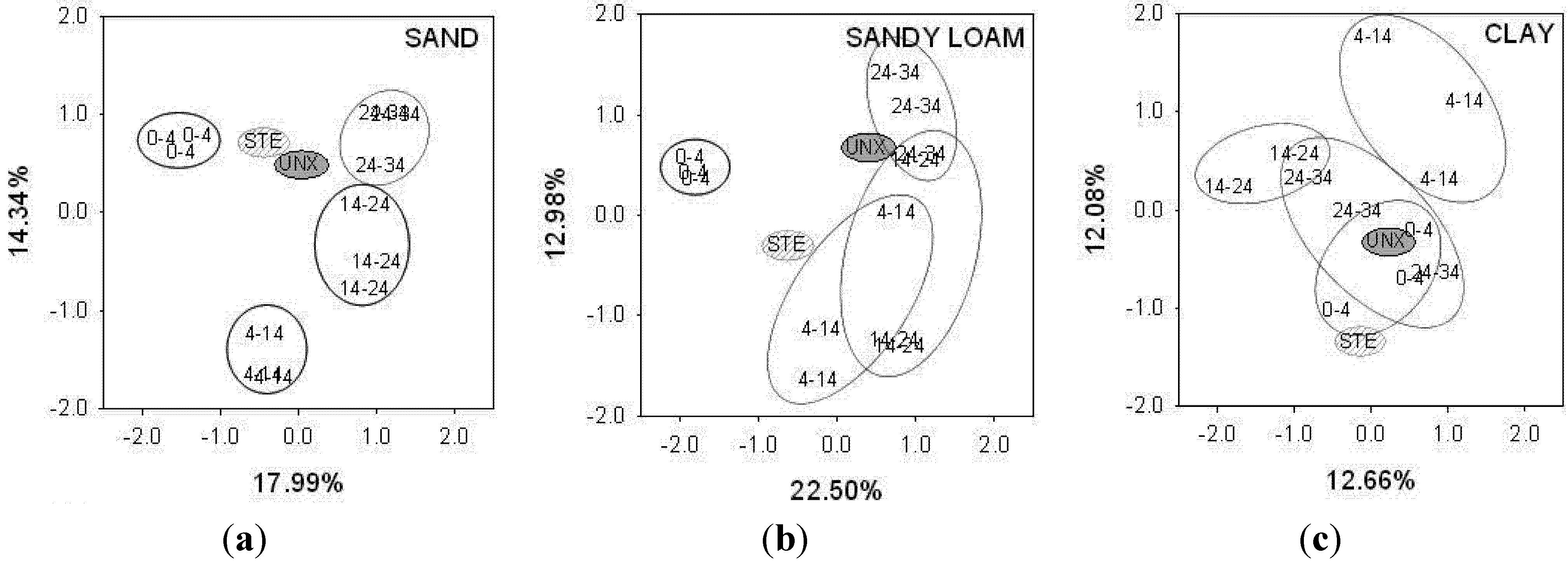
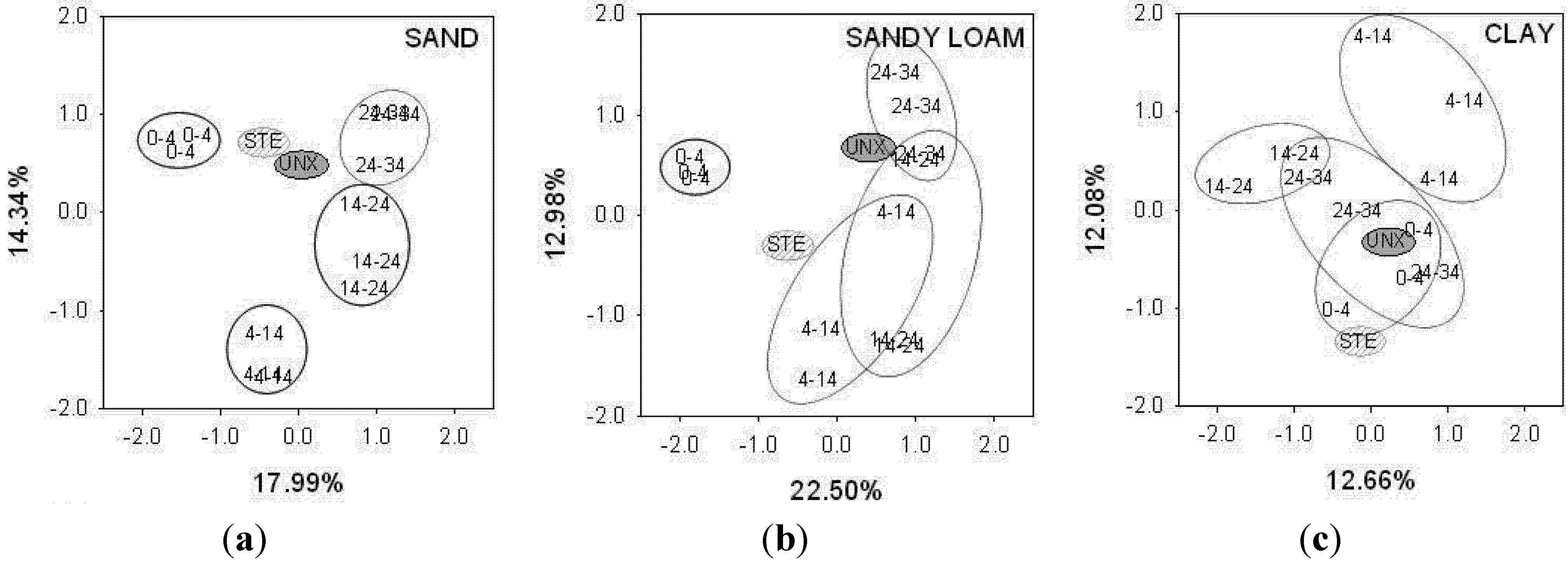
Principal component analysis (PCA) based on the presence or absence of terminal restriction fragments (TRFs) shows that the bacterial communities of unexposed sand (UNX) and septic tank effluent (STE) differs from sand exposed to STE (EXP) (
Figure 1). In addition, PCA separated EXP sand from different depths. In mesocosms with sandy loam the bacterial communities in EXP soil were distinct from STE and UNX sandy loam. Sandy loam communities from 4–14, 14–24, and 24–34 cm overlapped, whereas communities from 0–4 cm were distinct from those of other depths (
Figure 1). Clay soil bacterial communities had the least separation, with the UNX soil community overlapping that of EXP soil from 0–4 and 24–34 cm, with overlap also apparent between 24–34 cm and 14–24 cm. By contrast, the bacterial communities at 4–14 cm were distinct from all other EXP soils, as well as UNX soil and STE.
Figure 1.
Principal component analysis of (a) sand; (b) sandy loam and (c) clay showing the degree of similarity among bacterial communities from septic tank effluent (STE), unexposed soil (UNX), and soil receiving STE from depths of 0–4, 4–14, 14–24, and 24–34 cm. Values in bold indicate the proportion of the variability accounted for by principal component 1 (horizontal axis) and 2 (vertical axis).
Figure 1.
Principal component analysis of (a) sand; (b) sandy loam and (c) clay showing the degree of similarity among bacterial communities from septic tank effluent (STE), unexposed soil (UNX), and soil receiving STE from depths of 0–4, 4–14, 14–24, and 24–34 cm. Values in bold indicate the proportion of the variability accounted for by principal component 1 (horizontal axis) and 2 (vertical axis).
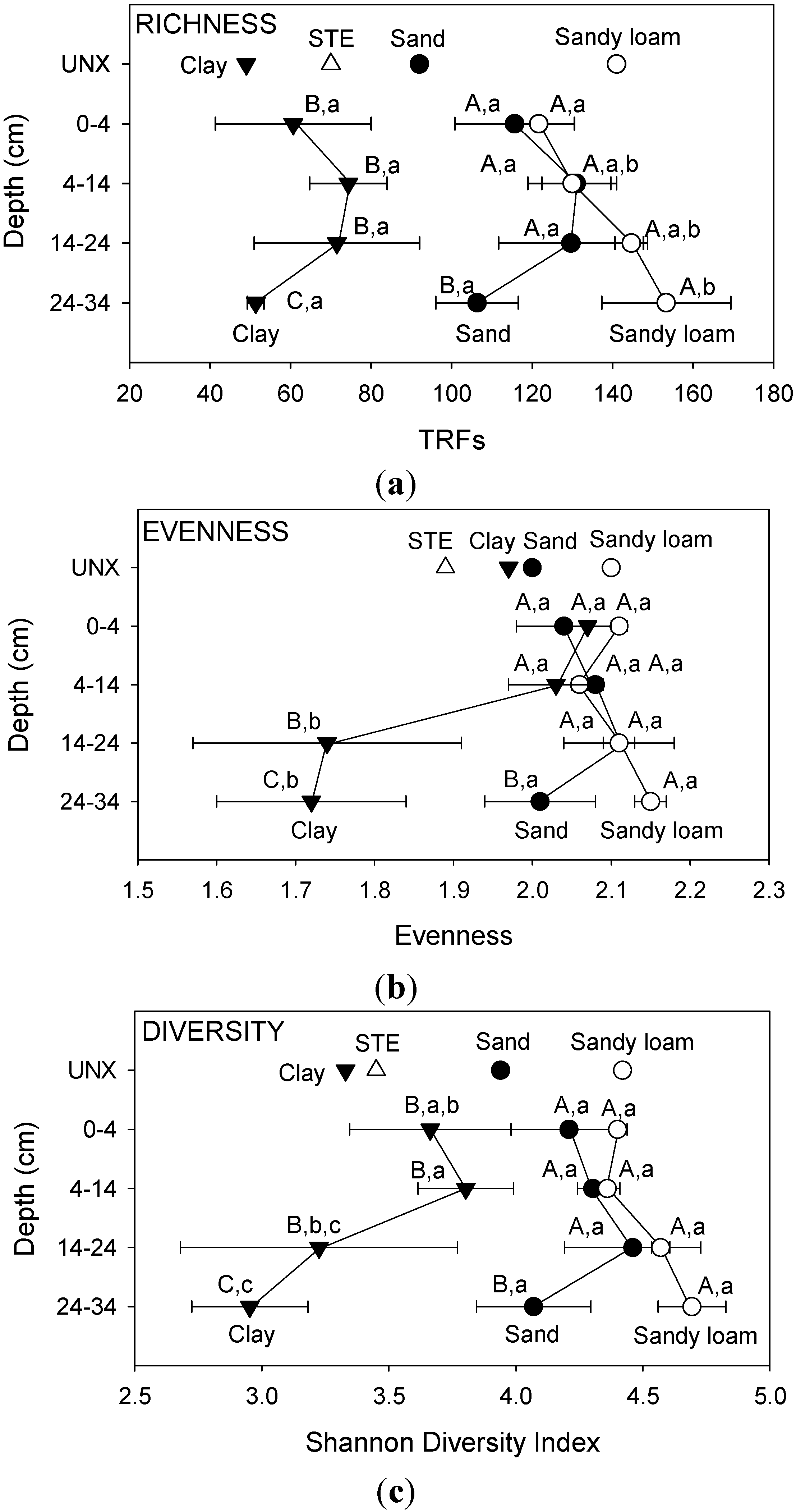
Mean values of community richness (all depths) were significantly different among EXP soil types, following the order: sandy loam (137) > sand (121) > clay (64) (
Table 1). This pattern was identical to that observed for UNX soil types. Richness values ranged from 49.0 TRFs for UNX clay to 153.3 TRFs for EXP sandy loam at 24–34 cm (
Figure 2). In addition, the sum of unique TRFs from all depths for EXP soil was significantly different among soil types, following the order: sandy loam (550) > sand (483) > clay (258) (data not shown). Comparisons among depths within a soil type showed a significant difference in TRF richness only between 0–4 and 24–34 cm in sandy loam. Comparisons among EXP soil types at particular depths showed that clay was significantly less rich than either sand or sandy loam at all depths. In addition, sand was significantly less rich than sandy loam at 24–34 cm (
Figure 2).
Table 1.
Mean (±S.D.) values of richness, diversity and evenness indices for the bacterial communities of septic tank effluent (STE) and sand, sandy loam, and clay soils before (UNX) and after (EXP) exposure to STE. Values for a particular index followed by the same letter are not significantly different.
Table 1.
Mean (±S.D.) values of richness, diversity and evenness indices for the bacterial communities of septic tank effluent (STE) and sand, sandy loam, and clay soils before (UNX) and after (EXP) exposure to STE. Values for a particular index followed by the same letter are not significantly different.
| Index | STE | Sand | Sandy loam | Clay |
|---|
| UNX | EXP | UNX | EXP | UNX | EXP |
|---|
| Richness | 70 | 91 | 121 ± 16 a | 141 | 137 ± 15 b | 49 | 64 ± 15 c |
| Diversity | 3.5 | 3.9 | 4.3 ± 0.2 a | 4.4 | 4.5 ± 0.1 b | 3.3 | 3.4 ± 0.5 c |
| Evenness | 1.9 | 2.0 | 2.1 ± 0.1 a | 2.1 | 2.1 ± 0.0 a | 2.0 | 1.9 ± 0.2 b |
Mean values of diversity indices among soil types (all depths) were significantly different, following the order: sandy loam (4.5) > sand (4.3) > clay (3.4) (
Table 1), the same pattern observed for UNX soil types. Diversity index values ranged from a low of 2.95 for EXP clay at 24–34 cm to 4.69 in EXP sandy loam at 24–34 cm (
Figure 2). Comparisons within an individual soil type showed that there were no significant differences among soil from different depths for sand and sandy loam (
Figure 2). By contrast, in the clay soil there were significant differences in diversity index between 0–4 cm and 24–34 cm, and between 14–24 and 24–34 cm. When soil from particular depths were compared among soil types, sand and sandy loam communities were significantly more diverse than clay for all depths, and sandy loam was also significantly more diverse than sand at 24–34 cm.
Figure 2.
(a) Bacterial community richness; (b) evenness and (c) diversity index values based on the number of TRFs in septic tank effluent (STE) and sand, sandy loam, and clay soils before (UNX) or after (EXP) exposure to STE at different depths. Bars represent one standard deviation. Values within a soil type followed by the same lowercase letter are not significantly different; values within the same depth followed by the same uppercase letter are not significantly different.
Figure 2.
(a) Bacterial community richness; (b) evenness and (c) diversity index values based on the number of TRFs in septic tank effluent (STE) and sand, sandy loam, and clay soils before (UNX) or after (EXP) exposure to STE at different depths. Bars represent one standard deviation. Values within a soil type followed by the same lowercase letter are not significantly different; values within the same depth followed by the same uppercase letter are not significantly different.
Mean values of evenness indices for all depths within EXP soil were significantly higher for sand (2.0) and sandy loam (2.1) than those for clay (1.9) (
Table 1). The bacterial community in EXP clay at 24–34 cm had the lowest evenness (1.72), indicating the presence of dominant populations, whereas EXP sandy loam at the same depth had the highest evenness index (2.15) (
Figure 2). Within a soil type, depth had no significant effect on evenness for sand and sandy loam. By contrast, evenness was significantly affected by depth in clay, with higher values observed at 0–4 cm and 4–14 cm than at 14–24 and 24–34 cm. Comparisons among soil types at a particular depth showed that there were no significant differences at 0–4 and 4–14 cm. On the other hand, evenness indices were significantly higher for sand and sandy loam soil than for clay at 14–24 cm. At 24–34 cm, soil type had a significant effect on evenness index, with values following the order: sandy loam > sand > clay.
Our results show that the effects of STE inputs on bacterial community structure are dependent on soil type. The differences presumably result from differences in the interaction between STE and soil, which can result in conditions that affect richness, diversity and evenness to different extents. How STE interacts with soil depends on soil texture and structure—properties that control pore size distribution, connectivity, and water-filled porosity. These translate into differences in habitable pore space for bacteria, their predators (e.g., protozoa and nematodes) and competitors (e.g., fungi). STE inputs also contain carbon and energy sources, nutrients, and potentially toxic compounds, which will have differential effects on autochthonous bacterial communities by relieving competitive pressures or favoring particular groups of organisms. Finally, repeated inputs of allochthonous bacteria present in STE can alter the composition of the bacterial communities in the STU, as discussed below, and may also alter that of predators and competitors.
Depth had a clear effect on measures of bacterial community structure in sandy loam and clay soil, whereas depth had no apparent effect in sand (
Figure 2). The latter suggests that conditions that control richness, diversity and evenness, such as diversity of habitats and/or availability of resources, are similar at all depths. However, PCA on TRFs suggested that bacterial communities at different depths were distinct from each other. This apparent discrepancy is likely due to differences in the type of data analyzed: whereas PCA analysis is based solely on the presence or absence of individual TRFs, community structure indices also incorporate information about the concentration of TRFs. By contrast to sand, significant changes in richness with depth for sandy loam and in diversity and evenness for clay indicate that habitat and/or resource gradients as a function of depth drive these differences. PCA on individual TRFs in these soils showed considerable overlap in community members between EXP depths and with UNX soil and STE. Differences in spatial differentiation of bacterial communities structure underscore the importance of soil type in shaping the communities that result from the interactions of soil and STE.
TRF profiles for EXP soil were compared to the corresponding UNX soil and STE profiles to calculate the percentage of all TRF peaks that were identical to either of these sources. TRFs that did not appear in either UNX soil or STE profiles were considered unique. The fraction of TRFs contributed by UNX soil was significantly higher than that contributed by STE for sand and sandy loam for all depths, except sand at 0–4 cm, whereas an equal contribution of UNX soil and STE was observed in clay for nearly all depths (
Figure 3). Bacteria from UNX soil made up a higher percentage of the EXP community in sandy loam (22–%42%), followed by sand (16–%18%) and clay (11–%16%). By contrast, the contribution of STE to community composition was similar for all soil types, ranging from 5% to 16%. Together, STE and UNX soil accounted for 18% to 50% of the TRFs found in EXP soils (
Figure 3), pointing to the dominance of unique TRFs in EXP soils. The contribution of unique TRFs to the bacterial community was significantly higher than that for UNX soil or STE for all soil types and depths.
Figure 3.
Mean (n = 3) similarity of TRF profiles from (a) sand; (b) sandy loam and (c) clay loam soils exposed to septic tank effluent (STE) at different depths (EXP) to TRF profiles from STE and unexposed soils (UNX). TRFs that did not match either STE or UNX soil were designated as unique. Bars represent one standard deviation. Values within a soil type and mesocosm depth followed by the same lowercase letter are not significantly different.
Figure 3.
Mean (n = 3) similarity of TRF profiles from (a) sand; (b) sandy loam and (c) clay loam soils exposed to septic tank effluent (STE) at different depths (EXP) to TRF profiles from STE and unexposed soils (UNX). TRFs that did not match either STE or UNX soil were designated as unique. Bars represent one standard deviation. Values within a soil type and mesocosm depth followed by the same lowercase letter are not significantly different.
The higher contribution of dominant TRFs to the bacterial community of STAs from UNX soil for all soil types underscores the difficulty that allochthonous bacteria have in becoming part of a previously established community. Given the magnitude of differences in physical, chemical and biological properties between STE and soil, it is somewhat surprising that bacteria originating in STE are able to establish themselves as part of the STA community. Numerous studies have reported that allochthonous bacteria inoculated into soil generally do not survive for very long [
13,
14]. However, the development of gradients in physicochemical properties induced by periodic STE inputs, in combination with repeated inoculation of bacteria present in STE, may help to support the survival and growth of bacterial species originating in wastewater.
Dominant TRFs from native soil and STE contributed fewer than half of the total TRFs in the three soil types, indicating that neither dominated the resulting bacterial communities. Furthermore, changes in soil conditions induced by long-term, repeated STE inputs appear to have a significant negative effect on the survival of dominant bacteria from both of these sources, as indicated by the presence of fewer TRFs associated with native soil and STE. Although we did not evaluate bacterial community function, our results suggest that only a fraction of the functions carried out by dominant bacteria in either STE or native soil will be performed by the bacterial community of STAs. Identification of those bacteria that originate in native soil and STE and persist to form part of the STA bacterial community may be useful in terms of predicting functionality of STA microbial communities.
The bacterial communities of STAs of all soil types were constituted primarily by unique TRFs, that is, TRFs that presumably were not among the dominant bacteria in either active soil or STE, and thus were not identified from either source. Because we controlled the sources of bacteria introduced into the mesocosms, it is reasonable to assume that these unique TRFs originated from native soil and/or STE. Their absence from the native and STE libraries indicates that these bacteria were present in sufficiently low numbers that they were not detected in our analysis. The presence of a sizable number of unique TRFs in EXP soil suggests that the conditions established by the long-term addition of STE to soil promote the growth of a sizeable number of bacteria that otherwise are a minor fraction of the community in either source. This effect may result from removal of a limiting factor (e.g., organic substrates, co-factors) and/or establishment of conditions that favor these organisms over others (e.g., anaerobic conditions, toxic compounds). The differences in the relative contribution of unique, native soil and STE TRFs to the leachfield bacterial communities that develop in sand, sandy loam and clay suggest that the functional capacities of these communities may differ as well. As such, analysis of their functional diversity would further contribute to our understanding of differences in system performance.
The mesocosms employed in our study were meant to mimic the most biologically active zone of an STA. Because the broader experiment involved examination of the fate of bacteria and bacteriophage in this zone, mesocosms were designed to allow for sampling of water at discrete depths (
Figure 4). The segmentation of soil within mesocosms represents a deviation from the flow conditions experienced in an STA in the field. In particular, it tends to minimize the opportunities for by-pass flow. This may short-circuit the transport of bacteria in STE to greater soil depths, by-passing the physical, chemical and ecological mechanisms that control the fate of STE bacteria at shallower depths. However, by-pass flow is unlikely to be important under field conditions because (i) the presence of a biomat is expected to limit infiltration, resulting in unsaturated flow under the infiltrative surface; and (ii) soils with significant by-pass flow are unlikely to be used for STAs because they may have percolation rates above acceptable regulatory values. Thus, we believe our mesocosms are likely to provide a fair representation of STAs under field conditions.
There are recognized biases in TRFLP (e.g., DNA extraction, PCR artifacts, incomplete digestion by restriction endonucleases) [
15]. However, this also holds true for most other contemporary community analysis methods. Because TRFLP analysis is a rapid and reproducible way to compare communities in various samples [
16], it allows us to examine STA bacterial community structure across soil types and depth from a molecular perspective.
Figure 4.
(a) Schematic diagram of experimental mesocosms (not to scale); (b) detail of 0–4 cm sand mesocosm showing gravel and connections to STE inputs and soil column; (c) Experimental mesocosms set-up.
Figure 4.
(a) Schematic diagram of experimental mesocosms (not to scale); (b) detail of 0–4 cm sand mesocosm showing gravel and connections to STE inputs and soil column; (c) Experimental mesocosms set-up.



{kind=link}
{kind=link}
{kind=link}
{kind=link}