The Origin of Major Ions of Groundwater in a Loess Aquifer



1
Key Laboratory of Shale Gas and Geoengineering, Institute of Geology and Geophysics, Chinese Academy of Sciences, Beijing 100029, China
2
College of Earth and Planetary Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
3
College of Geoscience and Surveying Engineering, China University of Mining & Technology, Beijing 100083, China
*
Author to whom correspondence should be addressed.
Water 2019, 11(12), 2464; https://doi.org/10.3390/w11122464
Submission received: 26 October 2019
/
Revised: 21 November 2019
/
Accepted: 22 November 2019
/
Published: 23 November 2019
(This article belongs to the Section Hydrology)
Abstract
:When groundwater represents most of the world’s reserves of unfrozen freshwaters, water quality is commonly a limiting factor in quantifying usable fresh groundwater storage, especially in arid and semi-arid areas. Tracing the origin of major ions is important to the understanding of hydrochemical evolution and water–rock interaction. Strontium (Sr) and calcium (Ca) are geochemically similar in terms of ionic radius and ionic charge and can substitute for each other in mineral lattices such as in carbonates and silicates. This allows the use of Sr as a proxy for Ca during water–rock interaction. Geochemical mass balance and carbon and strontium isotope techniques were employed to study the origin of each major ion (such as Na, Ca, Mg, Cl, and SO4) in a loess aquifer (there is 10% to 20% of carbonate in the loess). Geochemical mass balance between atmospheric deposition and groundwater shows that the Cl (average concentration of 5.5 mg/L) and SO4 (average concentration of 6.8 mg/L) in groundwater originated from atmospheric deposition. The dissolution of loess using acetic acid was used to analyze the 87Sr/86Sr ratio of carbonate (mainly deposited in continental environments). Groundwater 87Sr/86Sr ratios (0.710677 to 0.712319) are consistent with 87Sr/86Sr ratios in carbonate (0.710329 to 0.711085) but are significantly lower than the whole-rock (0.715136 to 0.717155) and residue (0.719091 to 0.720438), suggesting that Ca and Mg mainly originated from the dissolution of carbonate in the loess aquifer. However, Na originated from the dissolution of albite, suggesting saturation controls the ability of dissolution. There are cation exchanges between Ca + Mg and Na, resulting in Na concentration increases and Ca + Mg concentration decreases in groundwater. This study is important to the understand of the origin of the major ions in groundwater and the geochemical processes in silicate-carbonate aquifers.
1. Introduction
Groundwater represents about 96% of the world’s reserves of unfrozen freshwaters [1]. Groundwater is of fundamental significance for human development to meet the rapidly expanding urban, industrial, and agricultural water requirements, especially in arid and semi-arid areas [2], and more than two billion people in the world depend on groundwater for their daily supply [3]. Meanwhile, water quality is a limiting factor for usable fresh groundwater utilization [4,5]. Poor water quality can be caused by a natural condition (such as strong aridity or many soluble salts in an aquifer) and by human activities (such as fertilizer utilization and drainage of waste waters). The impact of human activities on water quality were only recognized in the past half-century before which time, pristine conditions reflecting natural processes controlled the hydrochemistry [6]. The impact of diffuse pollution from intensive agriculture, waste disposal, and point source pollution from urban and industrial sources, relied on geochemistry to solve questions of origin and attenuation. In semi-arid regions facing water scarcity, geochemical approaches have been vital in the assessment of renewability [6,7] and characterizing water–rock interaction.
The Cl, SO4, HCO3/CO3, Na, K, Ca, and Mg are the major ions in groundwaters [4,8], which reflect the geochemical evolution and water–rock interaction. Their sources vary from different sites and become more complex when they are affected by human intervention. Strontium (Sr) and calcium (Ca) are geochemically similar in terms of ionic radius and ionic charge and can substitute for each other in mineral lattices such as in carbonates and some silicates [9]. Therefore, Sr has been used as a proxy for Ca in many studies [10,11,12]. In most cases, carbonate rocks have higher contents of strontium but lower 87Sr/86Sr as compared to strontium derived from silicate rocks with lower contents of strontium and higher 87Sr/86Sr [12,13]. Marine carbonate commonly has a 87Sr/86Sr value that is less than 0.709 [12]. The difference in 87Sr/86Sr makes it possible to identify the water–rock interaction from carbonate or silicate. For example, based on the 87Sr/86Sr, the chemical composition of the Wujiang River (Guizhou Province, SW China) waters are characterized mainly by limestone weathering, while those of the adjacent Qingshuijiang River waters are characterized mainly by dolomite and silicate weathering [14]. The pore waters extracted from the matrix from the Cretaceous Chalk of southern England were intermediate between the calcite matrix (acetic acid soluble, low 87Sr/86Sr ratios) and clay component (high 87Sr/86Sr ratios), suggesting groundwater is strongly influenced by the weathering or exchange with clays [15].
Most studies over the world have focused on carbonates deposited in marine environments in aquifers and the 87Sr/86Sr ratios for the marine carbonates have been well constructed [12,15]. However, for carbonates deposited in continental environments, the 87Sr/86Sr ratios [16,17,18] and δ13C values vary largely [19,20,21] because of changes in deposition environments. This study aims to determine the origin of the major ions in the Xifeng loess aquifer, in NW China, mainly using major ion chemistry and C isotopes along with Sr isotopes to study the natural geochemical processes for groundwater. The calcites in the loess were deposited in continental environments (i.e., loessification) [22,23]. The aquifer was chosen for several reasons which include: (1) there are typically 10% to 20% carbonate in loess [22,24], presenting a relatively complex matrix of aquifer; (2) the hydrogeological condition is relatively simple and the groundwater cycle has been well recognized [21,25]; and (3) groundwaters are all old water [21], presenting a natural groundwater quality level with little human interference. The chemical (temperature, EC, pH, TDS, Cl, SO4, NO3, HCO3, F, Br, Na, K, Ca, and Mg) and isotopic composition (δ2H-H2O, δ18O-H2O, δ15N-NO3, δ18O-NO3, δ13C-DIC, 3H, and 14C-DIC) of groundwater and isotopic composition (δ13C and δ18O) of carbonate of loess have been reported in our previous study [21]. In this study, we further analyzed Sr concentration and 87Sr/86Sr in groundwater samples, and 87Sr/86Sr in loess as whole-rock and conducted dissolution experiment using acetic acid to determine the 87Sr/86Sr ratios of carbonate in loess.
2. Study Area and Sampling
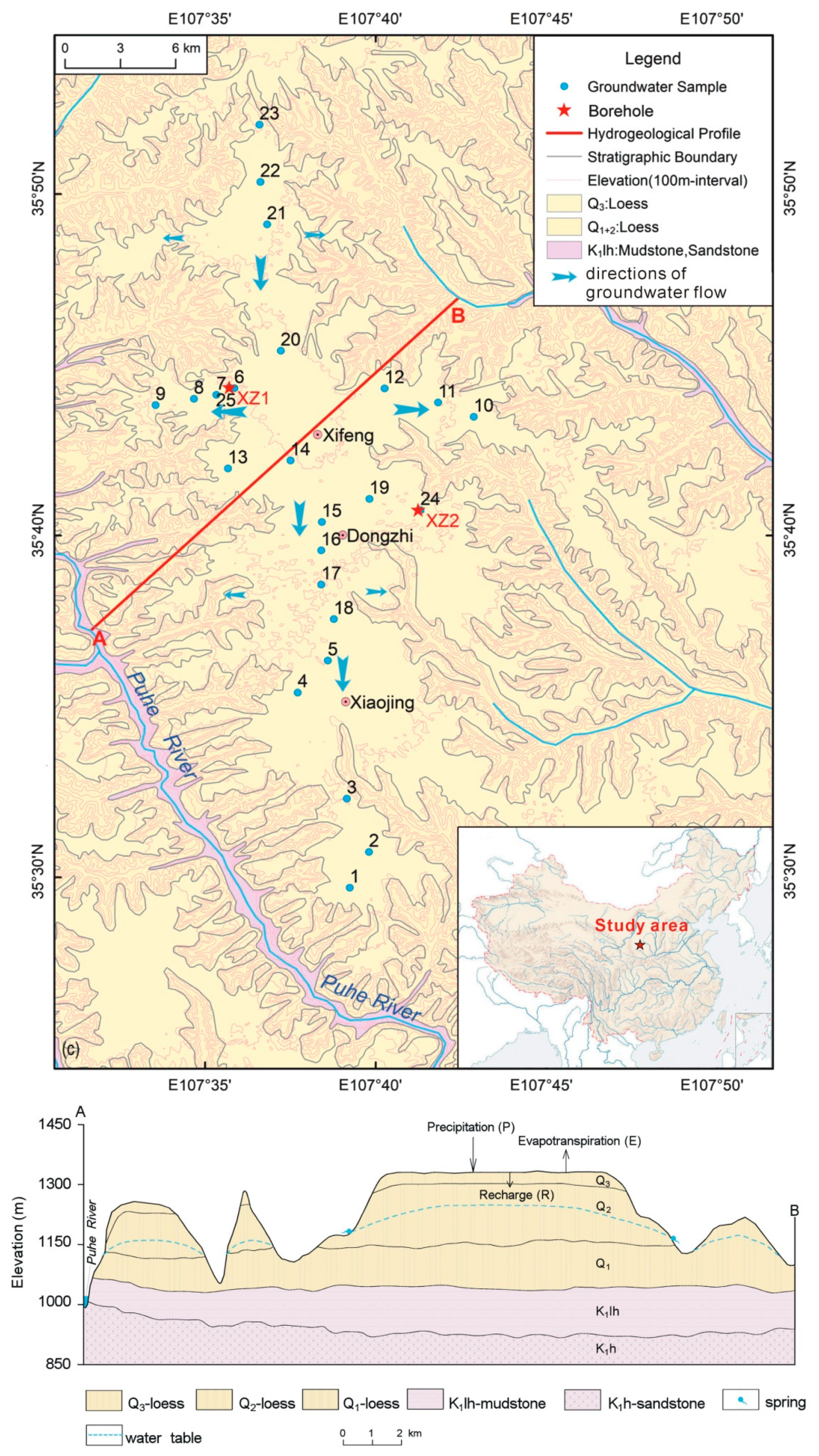
The Xifeng loess tableland is located in the central part of the Loess Plateau of China (LPC) and is the largest tableland in the LPC with a loess depth of 150 to 200 m [25]. The horizontal Cretaceous mudstone (K1lh) is covered by Quaternary loess (Figure 1) composed of lower Pleistocene (Q1), middle Pleistocene (Q2), and upper Pleistocene (Q3) deposits. The Q1 Loess, with a thickness of 40 to 50 m, commonly crops out at the bottom of the upper and middle reaches of gullies. The hard and compacted Q1 Loess has low permeability and is usually considered as an aquitard [25]. The Q2 Loess has a thickness of 120 to 170 m and commonly crops out on the sides of valleys and at the heads and cliffs of gullies. The Q2 Loess is unconsolidated and has relatively large porosity and is considered to be a good aquifer. The Q3 Loess has a thickness of 10 to 15 m and is distributed above the Q2 Loess as the topsoil in the area (Figure 1).
The depth of the water table is in the range of 40–75 m with an increasing trend from the center of the plain to its surrounding areas. The discharge of groundwater is mainly in the form of suspension gravity springs in gullies (Figure 1). The average annual precipitation is 546 mm/year, approximately 68% of which falls between June and September during the Asian monsoon. The average annual temperature is 9.2 °C according to data reported from 1956 to 2010. The main crops are winter wheat and maize, with one crop each per year.
According to our recent study [21], diffuse recharge through the thick unsaturated zone is the only recharge source to the aquifer. The recent infiltrated recharging water, since the 1950s, is still retained in the upper 10 m of the unsaturated zone with a recharge rate in the range of 37–41 mm/year. Therefore, the groundwater is all old water (tritium free) with the corrected 14C ages in the range of 220–19,220 years.
Twenty-six shallow groundwater samples, with well depths in the range of 55–180 m, were sampled in 2018 (Figure 1). Meanwhile, loess samples were collected from two deep profiles (XZ1 with a depth of 55 m and XZ2 with a depth of 45 m) by using an engineering geological survey drill. There were no fluids added during drilling. The bulk samples of ~2000 g were collected, at 0.25 m intervals at 0 to 20 m and 0.50 m intervals below 20 m (280 samples in total). The samples were immediately sealed in polyethylene bags after collection.
3. Experiment and Analyses
In this study, we further analyzed the following: (1) Sr concentration (n = 26) and 87Sr/86Sr (n = 14) in groundwater samples and (2) 87Sr/86Sr (n = 10) in loess as whole-rock and conducted a dissolution experiment using acetic acid (HAc) with different concentrations (0.5 M and 1 M) to determine the 87Sr/86Sr ratios of carbonate in loess.
The 0.5 M HAc can only dissolve calcite (CaCO3) and magnesite (MgCO3), and 1 M HAc can dissolve dolomite (CaMg(CO3)2) [17,18,26]. The HCl was not used because it can dissolve some silicates [17,26,27]. Four loess samples (unsaturated zone sample, S1 and S2; saturated zone sample, S3 and S4) were chosen to conduct the dissolution experiment. First, mineral and geochemical compositions were analyzed. Then, 400 mL of 0.5-M HAc was added into 40 g of dried loess. The samples were agitated intermittently for 4 h, and the supernatant solution was centrifuged and filtered through 0.45 μm filters for Sr, major cation, and 87Sr/86Sr analyses. The residual loess was dried and 150 mL of 1-M HAc was added into 15 g of dried residual loess. The samples were also agitated intermittently for 4 h, and the supernatant solution was centrifuged and filtered through 0.45 μm filters for Sr, major cation, and 87Sr/86Sr analyses. The residual loess after dissolution by 1 M of HAc was dried for mineral composition and 87Sr/86Sr analyses.
The mineral composition was analyzed using an X-ray diffraction system (XRD) (X’Pert Pro, Malvern Panalytical Ltd., Malvern, UK). The major elements were analyzed using an X-ray fluorescence spectrometer (XRF) (Axios-mAX, Malvern Panalytical Ltd., Malvern, UK). Major cation (Na, K, Ca, and Mg) was analyzed using an inductively coupled plasma optical emission spectrometry (ICP-OES) (5300DV, Perkin Elmer, Waltham, MA, USA) and Sr was analyzed using an inductively coupled plasma mass spectrometry (ICP-MS) (NexION 300D, Perkin Elmer, Waltham, MA, USA). The 87Sr/86Sr ratio was measured using a multi-collector thermal ionization mass spectrometry (TIMS) (Finnigan MAT 261, Thermo Scientific, Waltham, MA, USA), which gave a ratio of 0.710235 ± 0.000026 for the NIST NBS 987 standard. All the analyses were conducted at the Beijing Research Institute of Uranium Geology.
4. Results
4.1. Geochemical Characteristics of Loess
Loess is a kind of aeolian deposit and has been deposited in succession since 2.4 Ma [22]. Under different deposition environments (such as temperature and precipitation), particle sizes and geochemical compositions for loess with different ages change but with limited variations [19,22]. The loess samples in this study contain a significant proportion of silt-sized particles (2 to 50 µm), accounting for 70% to 84%, and less clay (<2 µm and <12%) and sand (>50 µm and ~12%). Loess is composed of quartz, potassium feldspar, plagioclase, clay, and carbonate (calcite and dolomite) (Table 1). The illite-smectite, illite, kaolinite, and chlorite are the main components for clay (Table 2). The loess samples are enriched in carbonate ranging from 9.1% to 22.1% (Table 1). The CaCO3 in loess mainly originated from secondary deposit (accounting for 90%) that occurs during the loessification processes [17,22,23,24,28]. Secondary carbonates originate from the vertical, horizontal, or in situ resettlement of carbonates during pedogenesis in the soil-sedimentary environment [28]. Table 3 shows the element composition for the four loess samples.
The carbon isotopic composition (δ13C) for marine carbonate is 0.6‰ ± 1.6‰ [29] and it typically ranges from −10‰ to −2‰ for continental carbonate [20] with an average value of −4.9‰ ± 2.8‰ [29]. The δ13C values for carbonate in loess in the study area range from −8.3‰ to −3.3‰ (Table 4), showing the continental origin of the carbonate. The values are consistent with that in the Luochuan tableland ranging from −9‰ to −4‰ [23].
The strontium isotopic composition (87Sr/86Sr) for the whole loess ranges from 0.715136 to 0.717155 (Table 4). Because the whole loess includes silicate, evaporite (if there was), and carbonate minerals, the values are the mixed results for the two kinds of minerals.
4.2. Hydrogeochemical Characteristics of Groundwater
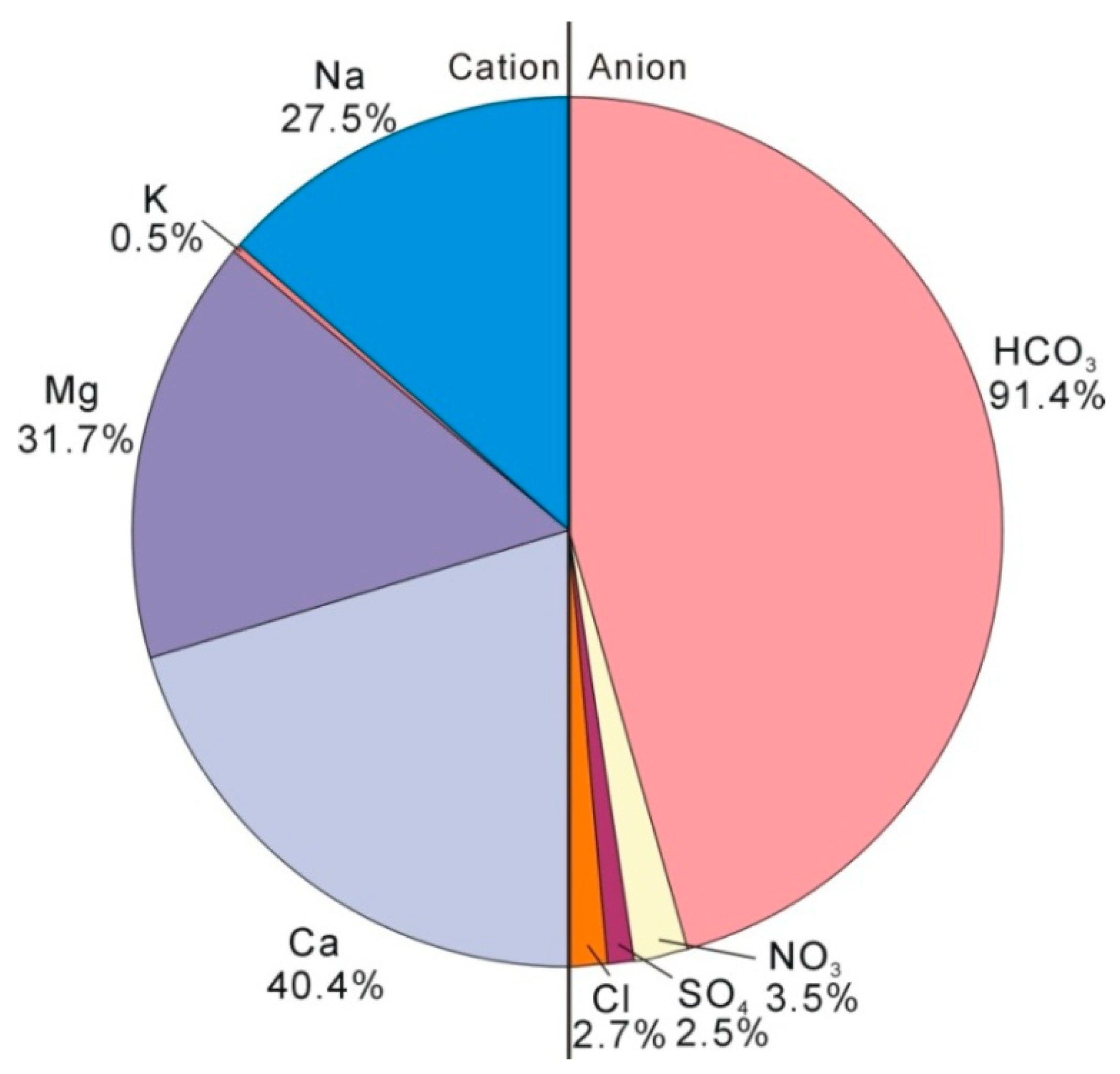
The groundwater is fresh water with TDS ranging from 225 to 316 mg/L with an average value of 288 mg/L (Table 5). There is no tritium detected and the corrected 14C age ranges from 220 to 19,220 years [21], suggesting that groundwaters are all old water with little influence from human activities. The Cl and SO4 concentrations are rather low, with an average value of 5.5 mg/L and 6.8 mg/L, respectively. The main anion is HCO3 (91.4%) and the main cations are Ca (40.4%), Mg (31.7%), and Na (27.5%) (Figure 2). The saturation indices (SI) calculated by using Phreeqc software [30] for calcite (SIcal) and dolomite (SIdol) are all larger than zero, suggesting groundwaters are all over-saturated with respect to calcite and dolomite. The SI values for fluorite, gypsum, magnesite, and strontianite are less than zero, suggesting that groundwaters are all under-saturated with respect to these minerals (Table 5).
4.3. Characteristics of Carbonate in Loess
Because only CaCO3 and MgCO3 can be dissolved in the 0.5 M acetic acid (HAc) [17,26], the CaCO3 and MgCO3 content in loess can be calculated. The results show that during the dissolution processes using 0.5 M of HAc, there is plenty of Ca dissolved, with the concentration ranging from 5260 mg/L to 7445 mg/L, however, the Mg concentration is relatively limited (127 to 161 mg/L). Here, we have not considered the limited desorption process on the clay surface.
The content of CaCO3 in loess, calculated using the HAc dissolution experiment, ranges from 13.15% to 18.61% (Table 6), and overlaps with the results (9.1% to 22.1%) from the XRD analyses (Table 1). The MgCO3 content in loess ranges from 0.44% to 0.56% (Table 6), much lower than CaCO3. The Sr concentration ranges from 5.1 mg/L to 8.3 mg/L, and 87Sr/86Sr ranges from 0.710455 to 0.711085.
The dolomite (CaMg(CO3)2) can be dissolved in 1 M of HAc [26]. The Mg concentration ranged from 72 mg/L to 219 mg/L. The dolomite content in the whole loess ranges from 0.47% to 1.43% (Table 6). The 87Sr/86Sr ratios in the solution of 1 M of HAc range from 0.710329 to 0.710969, close to the 87Sr/86Sr ratios in the 0.5 M of HAc solution (0.710455 to 0.711085), suggesting little silicate dissolution for 1 M of HAc dissolution.
5. Discussion
5.1. Atmospheric Deposition
Chloride is one of the most conservative tracers in the water cycle. It seldom substitutes into minerals and only precipitates at very high concentrations [31]. It does not significantly participate in oxidation and reduction reactions [32]. Chloride is commonly used to estimate groundwater recharge based on the mass balance between Cl from atmospheric deposition and Cl in soil water [33,34], or between Cl from atmospheric deposition and Cl in groundwater [35,36]. Chloride in groundwater, in the study area, originated from atmospheric deposition based on that Br/Cl values (0.0034 to 0.0061) for groundwater are equal to or exceed the marine-derived precipitation of 0.0034 [37,38]. This is a reasonable assumption in well-drained soil [33,39,40]. Feth [41] also reported that where groundwater contains less than 10 mg/L chloride, atmospheric sources are probably the major source (the average Cl concentration in groundwater is 5.5 mg/L). Therefore, we can use Cl to estimate the atmospheric deposition amount for a specific ion.
On the basis of the chemical observation of precipitation in the adjacent Jiwozi station, from 2000 to 2015 [42], which is about 220 km from the study area, the weighted average concentration of Cl, SO4, Na, Ca, and Mg are 1.13 mg/L, 8.63 mg/L, 1.59 mg/L, 6.08 mg/L, and 0.67 mg/L, respectively. The atmospheric deposition amount can be calculated using the enrichment factor based on the Cl concentration between precipitation and groundwater. For example, when the groundwater Cl concentration is 5.7 mg/L, the enrichment factor between groundwater and precipitation would be 5 (i.e., 5.7 mg/L of groundwater divided by 1.13 mg/L of precipitation), and then, the atmospheric deposition amount for SO4 is expected to be 5 × 8.63 mg/L = 43.2 mg/L. If the SO4 concentration in groundwater is larger than 43.2 mg/L it suggests that there are other SO4 sources, whereas a concentration less than 43.2 mg/L suggests that the atmospheric deposition can explain the existence of SO4 in groundwater.
The results show that enrichment factor in the study area ranges from 3.5 to 9.6 and the estimated atmospheric deposition for SO4 ranges from 30.2 mg/L (3.5 × 8.63 mg/L) to 82.8 mg/L (9.6 × 8.63 mg/L), while SO4 in groundwater is only 3.2–13.1 mg/L, suggesting groundwater SO4 originated from atmospheric deposition. The estimated values are very high, mainly caused by the high concentration in modern precipitation affected by human activities, such as SO2 emission [43,44]. Because there are no sulfur minerals (such as gypsum and pyrite) in the loess, and loess is well-drained soil, the origin from an atmospheric deposition for groundwater SO4 is, therefore, reasonable. However, for the Na, Ca, and Mg, the estimated atmospheric deposition is significantly lower than those in groundwater, suggesting there are other sources for them.
5.2. The Origin of Na, Ca, and Mg
When precipitation infiltrates through the unsaturated zone to recharge groundwater, soil CO2 is dissolved in soil water, making soil water weakly acid. The weakly acid soil water could further dissolve minerals to produce Na, Ca, and Mg [8,37].
The Na concentration in groundwater ranges from 18 mg/L to 84 mg/L with an average value of 35 mg/L, larger than the concentration in 0.5 M of HAc dissolution (11.7 to 12.7 mg/L). This suggests that Na originated from the dissolution of albite.
2NaAlSi3O8 + 11H2O + 2CO2→Al2Si2O5(OH)4 + 2Na+ + 2HCO3− + 4H4SiO4
Because there is carbonate in loess, CO2 would also dissolve carbonate.
CaCO3 + H2O + CO2→Ca2+ + 2HCO3−
MgCO3 + H2O + CO2→Mg2+ + 2HCO3−
CaMg(CO3)2 + 2H2O + 2CO2→Ca2+ + Mg2+ + 4HCO3−
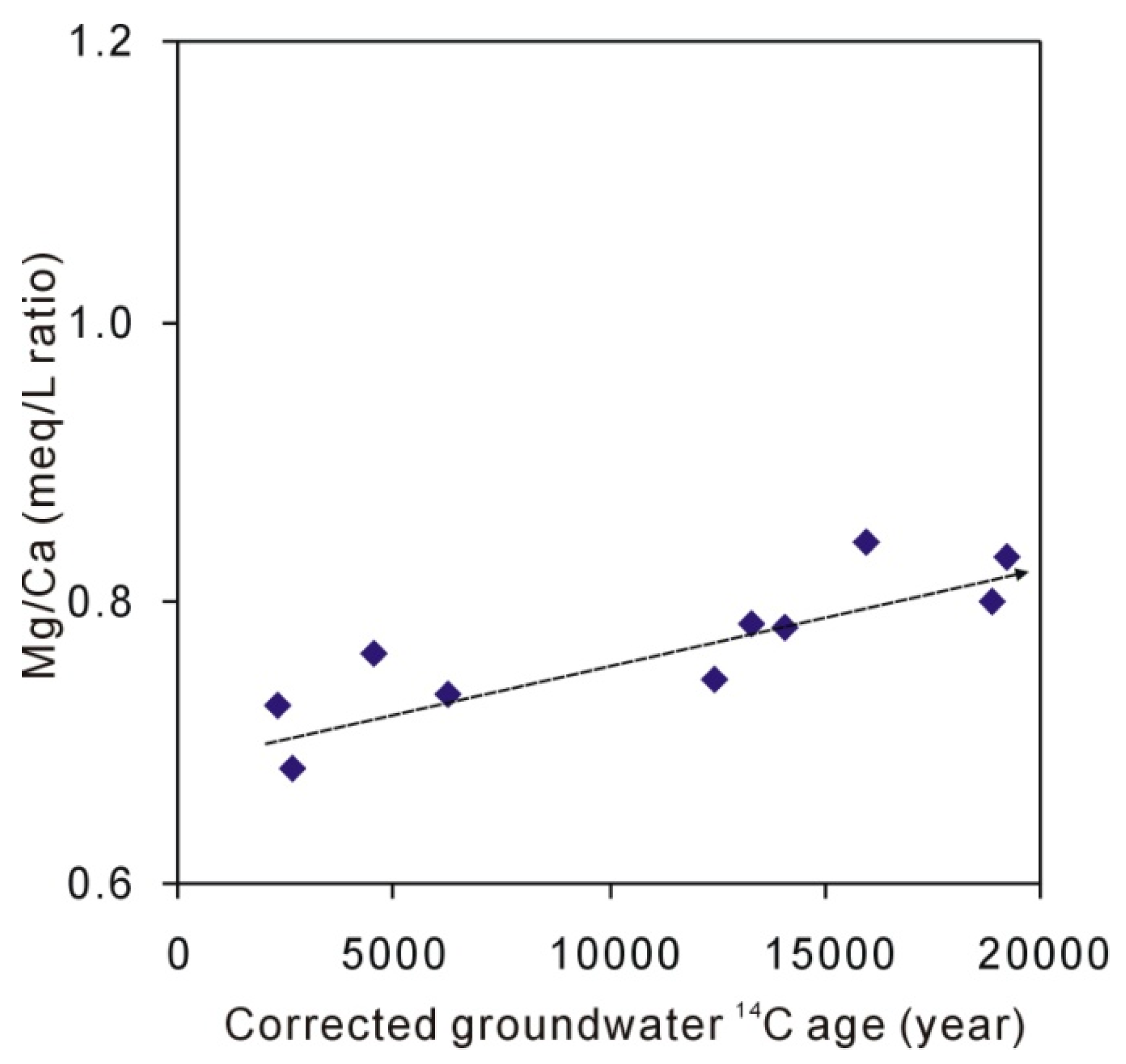
Recharge through an unsaturated zone and an aquifer containing a sufficient amount of carbonate will result in almost immediate calcite saturation by dissolution. The plot of Ca/Na and Mg/Ca (Figure 3a) shows that the increase of Ca and Mg is synchronous, suggesting that the above geochemical reactions occur, Equations (2)–(4). Meanwhile, the dissolution of carbonates always results in an increase in Ca/Na or Mg/Na and the dissolution of silicates results in a decrease in Ca/Na or Mg/Na [45,46]. The endmember for dissolution of silicates is commonly defined as a milligram equivalent ratio of <2 (moral ratio of <1) for Ca/Na and of <1 for Mg/Na [46] and the dissolution of carbonate (including dolomite) always results in high ratios of Ca/Na and Mg/Na. The results reflect a mixed influence of silicate and carbonate rock weathering (Figure 3a). When Sr concentrations in silicates are relatively lower than that in carbonates [12], the dissolution of silicates results in a low concentration of Sr in groundwater (Figure 3b). Figure 4 shows that the Mg/Ca ratios (milligram equivalent ratio) range from 0.68 to 1.04 and increase with the groundwater age. When groundwater is over-saturated with respect to calcite, the subsequent dissolution of carbonate along the flow path originates from incongruent dissolution involving concomitant precipitation and the dissolution of carbonate minerals [12,47]. This suggests that the incongruent dissolution may be involved as follows:
Ca2+ + CaMg(CO3)2→2CaCO3 + Mg2+
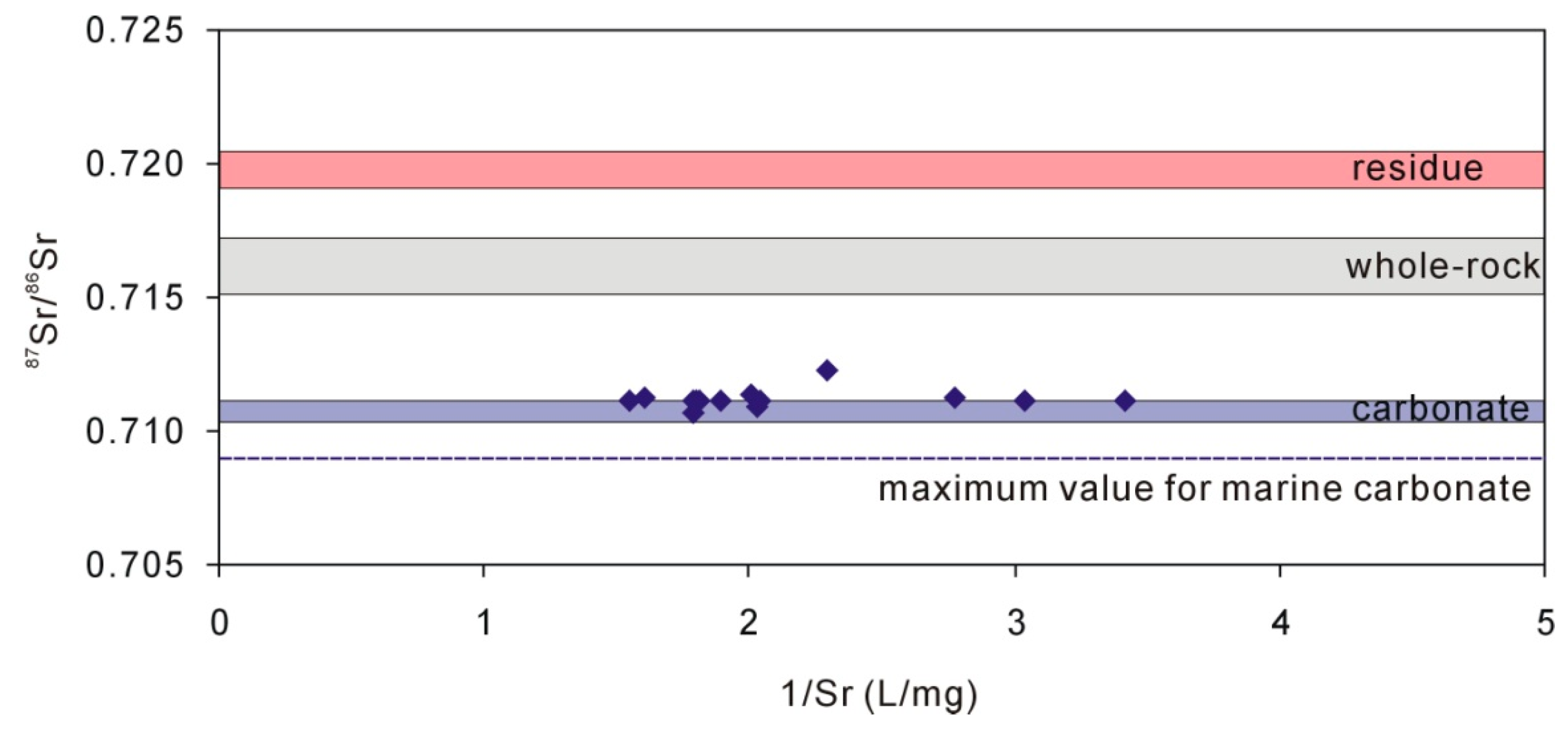
The groundwater 87Sr/86Sr ratios ranged from 0.710677 to 0.712319 (n = 14) with an average of 0.711207. The 87Sr/86Sr ratios for carbonate extracted from loess by 0.5 M HAc and 1 M HAc ranged from 0.710329 to 0.711085. The coincidence of 87Sr/86Sr ratios for carbonate and groundwater (Figure 5) suggests that the groundwater Ca + Mg mainly originated from the dissolution of carbonate rather than the dissolution of calcium-containing plagioclase because further dissolution of calcium-containing plagioclase would increase the 87Sr/86Sr ratios [27]. Because there is very little Sr in albite, about 20 to 170 times less than oligoclase, labradorite, and anorthite [48], the dissolution of albite would not change the 87Sr/86Sr ratios of groundwater (Figure 5), which remain constant. The groundwater 87Sr/86Sr ratios suggest that the dissolution of calcium-containing plagioclase is little.
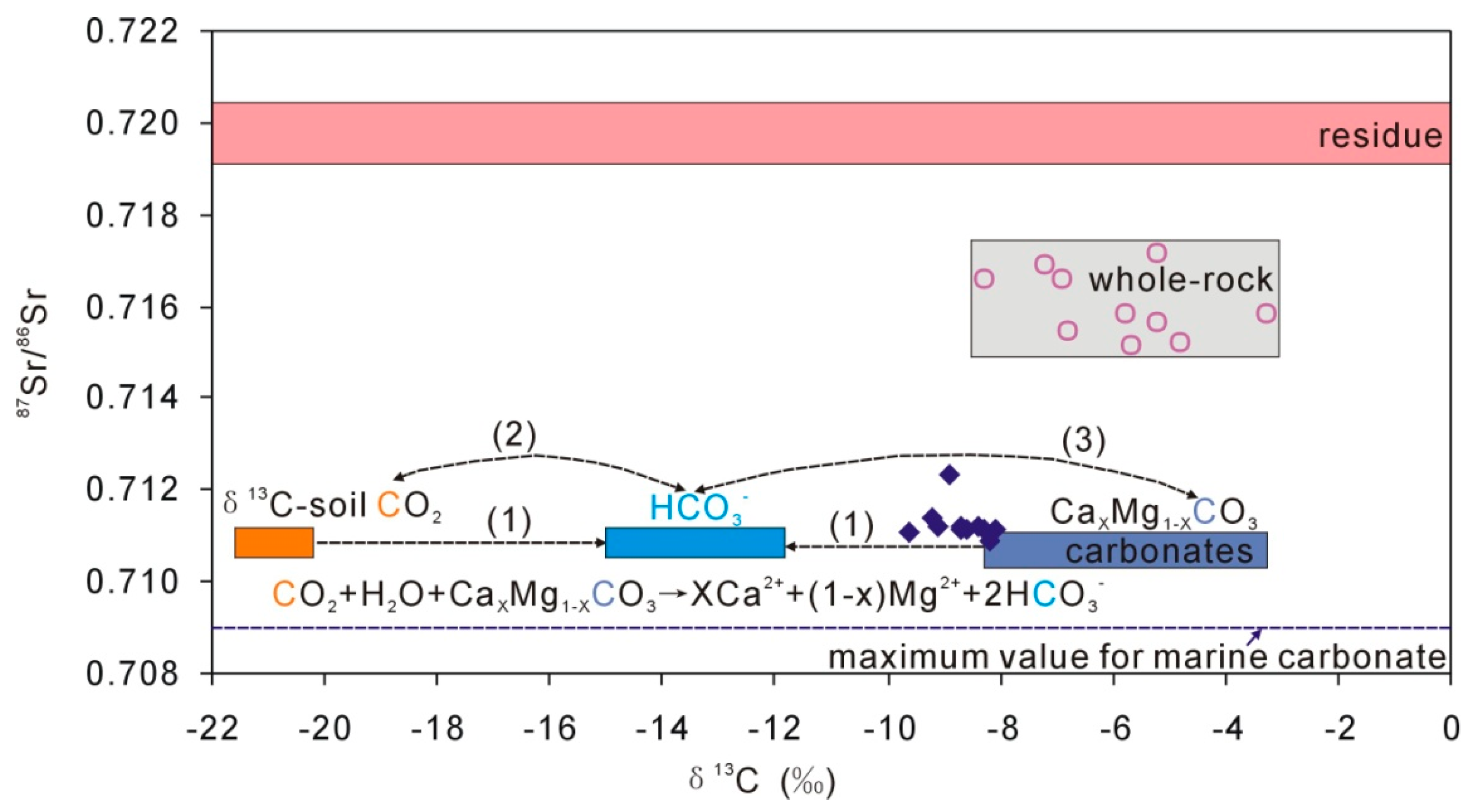
In the Loess Plateau, there were both C3 and C4 plants during the past 2.4 Ma [19], and there was 10% to 20% C4 vegetation in the Xifeng Loess tableland, resulting in δ13C of soil CO2 of −21.6‰ to −20.2‰ [39]. Meanwhile, the δ13C values of loess carbonate range from −8.3‰ to −3.3‰ (Table 4). The dissolution of carbonates by soil CO2 would directly result in δ13C values of groundwater ranging from −15.0‰ to −11.8‰ through carbonates dissolution, Equations (2)–(4), indicated as (1) in Figure 6. However, the later carbon exchange between groundwater DIC (mainly HCO3) and soil CO2 (as (2) in Figure 6), and between groundwater DIC and carbonates in loess (as (3) in Figure 6) would change the groundwater δ13C values (−9.6‰ to −8.1‰). In addition, there is no carbon production during the dissolution of albite except for soil CO2, Equation (1), i.e., the carbon in HCO3 is all from soil CO2. These complicated processes make the quantitative interpretation of groundwater δ13C more difficult.
Although the dissolution rate of albite is significantly less than that of calcium-containing plagioclase [49], the groundwater Na is mainly from the dissolution of albite, and the Ca+Mg is from the dissolution of carbonate. This is mainly attributed to the saturation of calcite and dolomite, which has limited further dissolution of carbonate and calcium-containing plagioclase [37]. In addition, the times for groundwater residence are relatively long (220 to 19,220 year) and there is sufficient time for albite to dissolve.
Loess contains materials like clay minerals [22] and organic matter [50], which can sorb chemicals. The cation exchange capacity (CEC) of a soil in meq/kg is used to assess the adsorbents in soils and aquifers [4]. An empirical formula which relates the CEC to the percentages of clay (<2 μm) and organic carbon (OC) at near neutral pH given by Breeuwsma et al. [51] is used to calculate the CEC of loess in the study area:
CEC (meq/kg) = 500 clay + 1500 OC
In the study area, the average percentage of clay is 8.3% [21] and the OC is 0.35% [50]. Therefore, the average CEC is expected to be 47 meq/kg. The average water content in loess is 20% for the unsaturated zone and 23% for the saturated zone [21], and therefore the CEC for pore water is expected to be 204 to 235 meq/L. Groundwater contains less than 6.2 meq/L cations in the study area, more than 30 times less than the amount of cations located on the exchanger. The principle factor influencing the absorption affinity of cations is valence. The following are the ions in decreasing adsorption affinity: Ca2+ = Mg2+ > Na+ [4,8]. Therefore, in this neutral soil, the cation exchange can be involved:
2NaX + Ca2+→CaX2 + 2Na+
2NaX + Mg2+→MgX2 + 2Na+
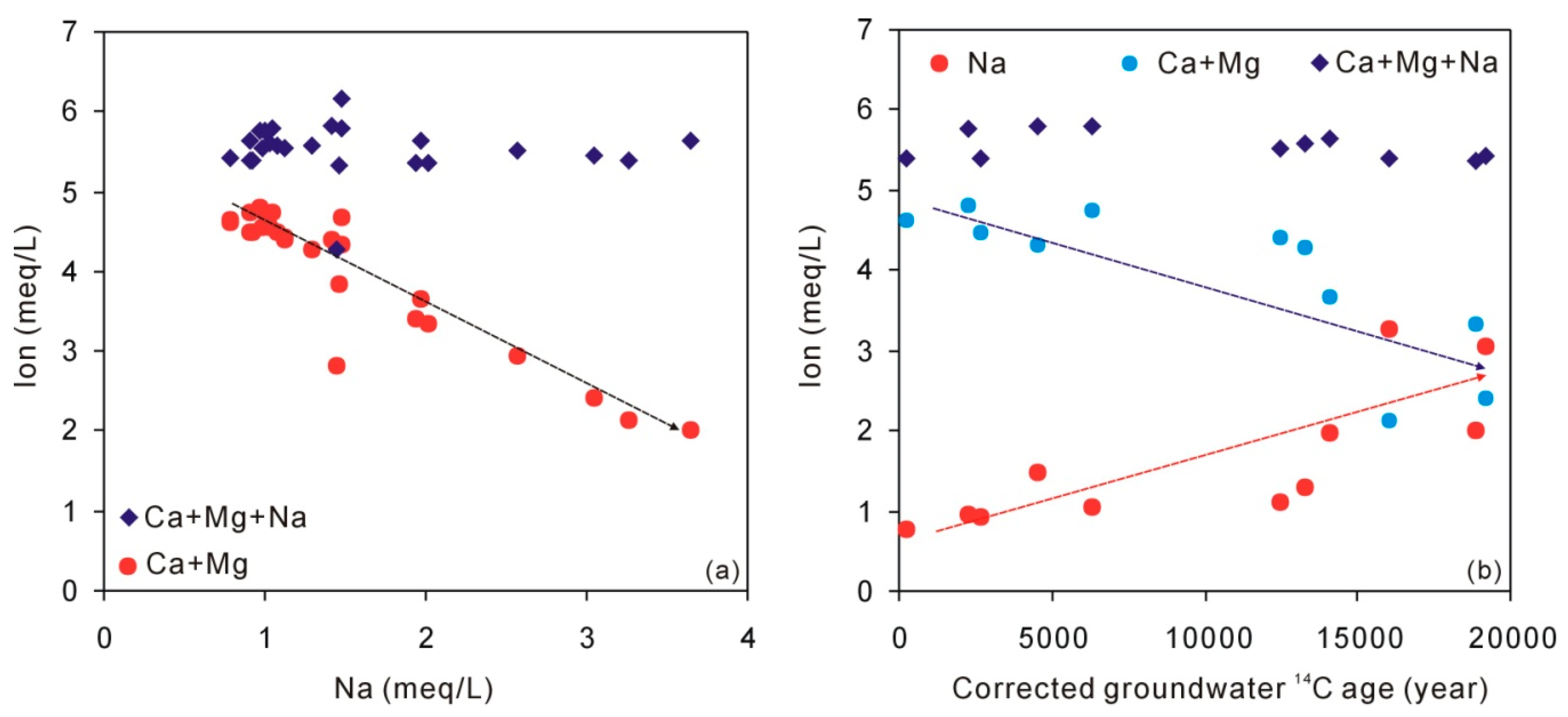
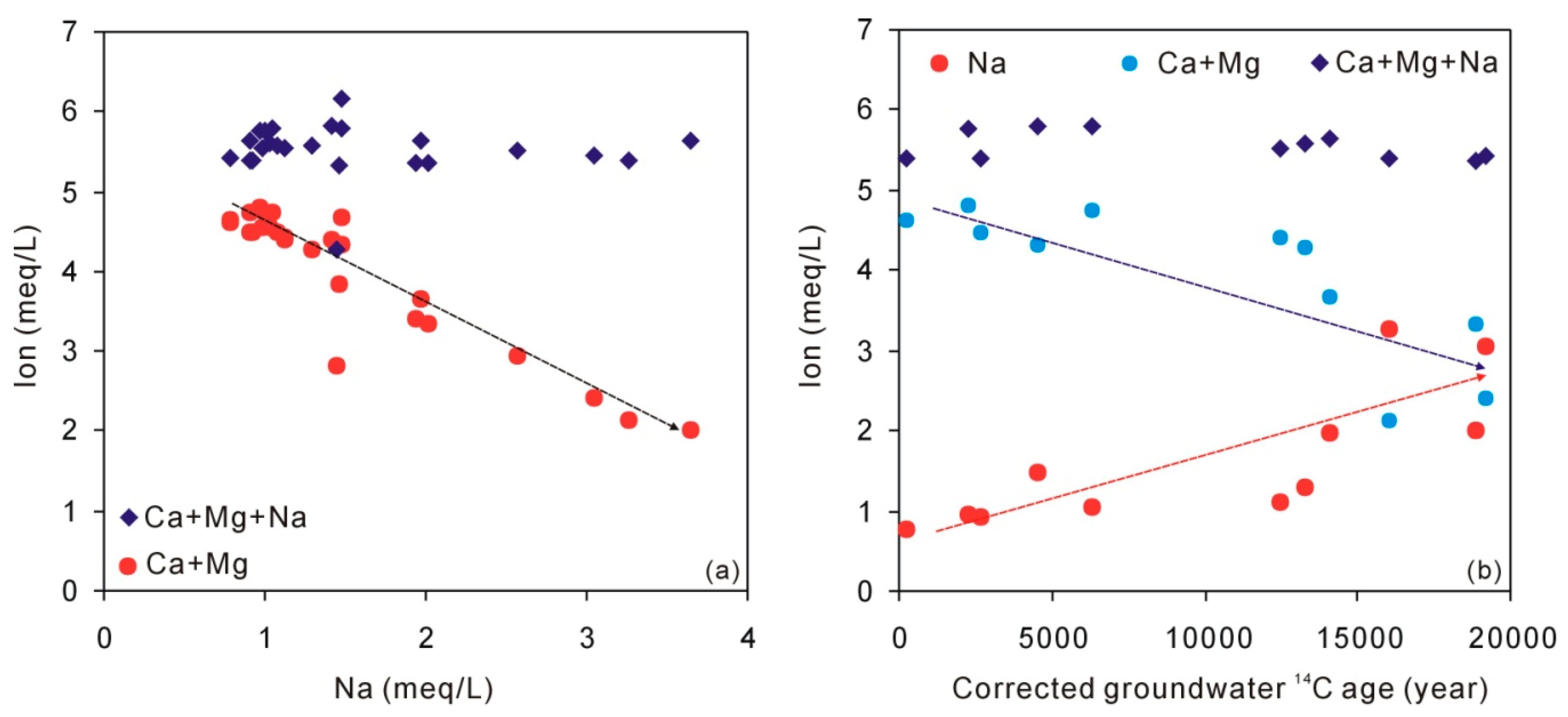
During cation exchange, the total cations (as meq/L) would not change. Figure 7 shows that when Na concentrations increase with time, the Ca+Mg concentrations decrease. Meanwhile, the total concentrations (Ca + Mg + Na) have not changed, suggesting that cation exchange between Ca + Mg and Na has been involved in the loess aquifer and contributes additional Na in groundwater.
6. Conclusions
The analyses of 87Sr/86Sr of carbonate in the matrix help to determine the origin of groundwater Ca and Mg in a complex aquifer (silicate and carbonate both exist). On the basis of the comparison of 87Sr/86Sr concentrations between groundwater and carbonate, groundwater Ca and Mg mainly originated from the dissolution of carbonate, but Na originated from the dissolution of albite. Meanwhile, there are cation exchanges between Ca + Mg and Na, resulting in Na concentration increases and Ca + Mg concentration decreases in groundwater. Cl and SO4 originated from atmospheric deposition. This study is important to the understand of the origin of the major ions in groundwater and the geochemical processes in silicate-carbonate aquifers.
Author Contributions
Conceptualization, T.H.; methodology, B.M. and T.H.; investigation, T.H. and B.M.; writing, T.H.
Funding
The work is supported by the National Natural Science Foundation of China, grant numbers 41672254 and 41877207, and the Youth Innovation Promotion Association CAS, grant number 2018087.
Acknowledgments
The authors thank Zhenbin Li for his help in field sampling and Yiman Li for her help in conducting the experiment. The authors also wish to express their appreciation to the editor and three anonymous reviewers whose detailed comments were very helpful in improving the clarity and focus of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shiklomanov, I.A.; Rodda, J.C. World Water Resources at the Beginning of the 21st Century; Cambridge University Press: Cambridge, UK, 2003; pp. 1–18. [Google Scholar]
- De Vries, J.J.; Simmers, I. Groundwater recharge: An overview of processes and challenges. Hydrogeol. J. 2002, 10, 5–17. [Google Scholar] [CrossRef]
- Kemper, K.E. Groundwater from development to management. Hydrogeol. J. 2004, 12, 3–5. [Google Scholar] [CrossRef]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution, 2nd ed.; A. A. Balkema Publishers: Amsterdam, The Netherlands, 2005; pp. 1–634. [Google Scholar]
- Edmunds, W.M. Limits to the availability of groundwater in Africa. Environ. Res. Lett. 2012, 7, 021003. [Google Scholar] [CrossRef]
- Edmunds, W.M. Geochemistry’s vital contribution to solving water resource problems. Appl. Geochem. 2009, 24, 1058–1073. [Google Scholar] [CrossRef]
- Huang, T.; Pang, Z.; Li, J.; Xiang, Y.; Zhao, Z. Mapping groundwater renewability using age data in the Baiyang alluvial fan, NW China. Hydrogeol. J. 2017, 25, 743–755. [Google Scholar] [CrossRef]
- Shen, Z.; Zhu, Y.; Zhong, Y. Hydrogeochemistry; Geological Publishing House: Beijing, China, 1993; pp. 1–189. [Google Scholar]
- Marcus, Y.; Kertes, A.S. Ion Exchange and Solvent Extraction of Metal Complexes; Wiley Interscience: New York, NY, USA, 1969; pp. 1–1037. [Google Scholar]
- Salifu, M.; Aiglsperger, T.; Hällström, L.; Martinsson, O.; Billström, K.; Ingri, J.; Dold, B.; Alakangas, L. Strontium (87Sr/86Sr) isotopes: A tracer for geochemical processes in mineralogically-complex mine wastes. Appl. Geochem. 2018, 99, 42–54. [Google Scholar] [CrossRef]
- Tipper, E.T.; Bickle, M.J.; Galy, A.; West, A.J.; Pomiès, C.; Chapman, H.J. The short term climatic sensitivity of carbonate and silicate weathering fluxes: Insight from seasonal variations in river chemistry. Geochim. Cosmochim. Acta 2006, 70, 2737–2754. [Google Scholar] [CrossRef]
- McNutt, R.H. Strontium Isotopes. In Environmental Tracers in Subsurface Hydrology; Cook, P.G., Herczeg, A.L., Eds.; Kluwer Academic Publishers: New York, NY, USA, 2000; pp. 233–260. [Google Scholar]
- Palmer, M.R.; Edmond, J.M. Controls over the strontium isotope composition of river water. Geochim. Cosmochim. Acta 1992, 56, 2099–2111. [Google Scholar] [CrossRef]
- Han, G.; Liu, C.Q. Water geochemistry controlled by carbonate dissolution: A study of the river waters draining karst-dominated terrain, Guizhou Province, China. Chem. Geol. 2004, 204, 1–21. [Google Scholar] [CrossRef]
- Shand, P.; Darbyshire, D.P.F.; Love, A.J.; Edmunds, W.M. Sr isotopes in natural waters: Applications to source characterisation and water–rock interaction in contrasting landscapes. Appl. Geochem. 2009, 24, 574–586. [Google Scholar] [CrossRef]
- Bickle, M.J.; Harris, N.B.W.; Bunbury, J.M.; Chapman, H.J.; Fairchild, I.J.; Ahmad, T. Controls on the 87Sr/86Sr Ratio of Carbonates in the Garhwal Himalaya, Headwaters of the Ganges. J. Geol. 2001, 109, 737–753. [Google Scholar] [CrossRef]
- Yang, J.; Chen, J.; An, Z.; Shield, G.; Tao, X.; Zhu, H.; Ji, J.; Chen, Y. Variations in 87Sr/86Sr ratios of calcites in Chinese loess: A proxy for chemical weathering associated with the East Asian summer monsoon. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2000, 157, 151–159. [Google Scholar] [CrossRef]
- Chen, J.; Li, G.; Yang, J.; Rao, W.; Lu, H.; Balsam, W.; Sun, Y.; Ji, J. Nd and Sr isotopic characteristics of Chinese deserts: Implications for the provenances of Asian dust. Geochim. Cosmochim. Acta 2007, 71, 3904–3914. [Google Scholar] [CrossRef]
- Yang, S.; Ding, Z.; Li, Y.; Wang, X.; Jiang, W.; Huang, X. Warming-induced northwestward migration of the East Asian monsoon rain belt from the last glacial maximum to the mid-Holocene. Proc. Natl. Acad. Sci. USA 2015, 112, 13178–13183. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, J. Geochemistry of Stable Isotopes; Science Press: Beijing, China, 2000; pp. 193–217. [Google Scholar]
- Huang, T.; Ma, B.; Pang, Z.; Li, Z.; Li, Z.; Long, Y. How does precipitation recharge groundwater in loess aquifers? Evidence from multiple environmental tracers. J. Hydrol. 2019. in revision. [Google Scholar]
- Liu, T.S. Loess and the Environment; Science Press: Beijing, China, 1985; pp. 1–481. [Google Scholar]
- Geng, A.; Wen, Q. Some geochemical characteristics of carbonates in Luochuan loess, Shaanxi Province. Geochimica 1988, 17, 267–275. [Google Scholar]
- Ning, Y.; Liu, W.; An, Z. Variation of soil Δδ13C values in Xifeng loess–paleosol sequence and its paleoenvironmental implication. Chin. Sci. Bull. 2006, 51, 1350–1354. [Google Scholar] [CrossRef]
- Qu, H. Assessment of Groundwater Resources in the Arid and Semiarid Land of China; Science Press: Beijing, China, 1991; pp. 1–457. [Google Scholar]
- Sheng, X.; Yang, J.; Li, C.; Chen, J.; Tao, X. A method for separation of calcite and dolomite in loess and sedimentary rocks. Rock Miner. Anal. 2000, 19, 264–267. [Google Scholar]
- Yokoo, Y.; Nakano, T.; Nishikawa, M.; Quan, H. Mineralogical variation of Sr–Nd isotopic and elemental compositions in loess and desert sand from the central Loess Plateau in China as a provenance tracer of wet and dry deposition in the northwestern Pacific. Chem. Geol. 2004, 204, 45–62. [Google Scholar] [CrossRef]
- Barta, G. Secondary carbonates in loess-paleosoil sequences: A general review. Cent. Eur. J. Geosci. 2011, 3, 129–146. [Google Scholar] [CrossRef]
- Keith, M.L.; Weber, J.N. Carbon and oxygen isotopic composition of selected limestones and fossils. Geochim. Cosmochim. Acta 1964, 18, 1787–1816. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQC (Version 2)—A Compute Program for Speciation, Batch–Reaction, One–Dimensional Transport, and Inverse Geochemical Calculations; USGS: Reston, VA, USA, 1999; pp. 1–312.
- Herczeg, A.L.; Edmunds, W.M. Inorganic ions as tracers. In Environmental Tracers in Subsurface Hydrology; Cook, P.G., Herczeg, A.L., Eds.; Kluwer Academic Publishers: New York, NY, USA, 2000; pp. 31–78. [Google Scholar]
- Hem, J.D. Study and Interpretation of the Chemical Characteristics of Natural Water; Water Supply Paper, no. 2254; US Geological Survey: Reston, VA, USA, 1985; pp. 1–264.
- Allison, G.B.; Hughes, M.W. The use of environmental chloride and tritium to estimate total recharge to an unconfined aquifer. Aust. J. Soil Res. 1978, 16, 181–195. [Google Scholar] [CrossRef]
- Edmunds, W.M.; Walton, N.R.G. A geochemical and isotopic approach to recharge evaluation in semi-arid zones—Past and present. In Arid–Zone Hydrology: Investigations with Isotope Techniques, Proceeding of Advisory Group Meeting; Fontes, J.C., Ed.; IAEA: Vienna, Austria, 1980; pp. 47–68. [Google Scholar]
- Eriksson, E.; Khunakasem, V. Chloride concentration in groundwater, recharge rate and rate of deposition of chloride in the Israel Coastal Plain. J. Hydrol. 1969, 7, 178–197. [Google Scholar] [CrossRef]
- Wood, W.W.; Sanford, W.E. Chemical and isotopic methods for quantifying groundwater recharge in a regional, semiarid environment. Ground Water 1995, 33, 458–468. [Google Scholar] [CrossRef]
- Mazor, E. Chemical and Isotopic Groundwater Hydrology, 3rd ed.; Dekker: New York, NY, USA, 2004; pp. 1–453. [Google Scholar]
- Edmunds, W.M.; Ma, J.; Aeschbach–Hertig, W.; Kipfer, R.; Darbyshire, D.P.F. Groundwater recharge history and hydrogeochemical evolution in the Minqin Basin, North West China. Appl. Geochem. 2006, 21, 2148–2170. [Google Scholar] [CrossRef]
- Huang, T.; Pang, Z.; Liu, J.; Ma, J.; Gates, J. Groundwater recharge mechanism in an integrated tableland of the Loess Plateau, northern China: Insights from environmental tracers. Hydrogeol. J. 2017, 25, 2049–2065. [Google Scholar] [CrossRef]
- Huang, T.; Pang, Z. Estimating groundwater recharge following land-use change using chloride mass balance of soil profiles: A case study at Guyuan and Xifeng in the Loess Plateau of China. Hydrogeol. J. 2011, 19, 177–186. [Google Scholar] [CrossRef]
- Feth, J.H. Chloride in Natural Continental Water: A Review; Water Supply Paper, no. 2176; US Geological Survey: Reston, VA, USA, 1981; pp. 1–30.
- The Acid Deposition Monitoring Network in East Asia (EANET). Available online: http://www.eanet.asia (accessed on 12 October 2018).
- Wang, T.; Wang, P.; Theys, N.; Tong, D.; Hendrick, F.; Zhang, Q.; van Roozendael, M. Spatial and temporal changes in SO2 regimes over China in the recent decade and the driving mechanism. Atmos. Chem. Phys. 2018, 18, 18063–18078. [Google Scholar] [CrossRef]
- Huang, T.; Fan, Y.; Long, Y.; Pang, Z. Quantitative calculation for the contribution of acid rain to carbonate weathering. J. Hydrol. 2019, 568, 360–371. [Google Scholar] [CrossRef]
- Gaillardet, J.; Dupré, B.; Louvat, P.; Allègre, C.J. Global silicate weathering and CO2 consumption rates deduced from the chemistry of large rivers. Chem. Geol. 1999, 159, 3–30. [Google Scholar] [CrossRef]
- Petelet-Giraud, E.; Luck, J.M.; Othman, D.B.; Joseph, C.; Négrel, P. Chemical and isotopic fingerprinting of small ungauged watershed: How far the hydrological functioning can be understood? Comptes Rendus Geosci. 2016, 348, 379–386. [Google Scholar] [CrossRef]
- Bishop, P.K.; Smalley, P.C.; Emery, D.; Dickson, J.A.D. Strontium isotopes as indicators of the dissolving phase in a carbonate aquifer: Implications for 14C dating of groundwaters. J. Hydrol. 1994, 154, 301–321. [Google Scholar] [CrossRef]
- Giletti, B.J.; Casserly, J.E.D. Strontium diffusion kinetics in plagioclase feldspars. Geochim. Cosmochim. Acta 1994, 58, 3785–3793. [Google Scholar] [CrossRef]
- Welch, W.S.A.; Ullman, J. Feldspar dissolution in acidic and organic solutions: Compositional and pH dependence of dissolution rate. Geochim. Cosmochim. Acta 1996, 60, 2939–2948. [Google Scholar] [CrossRef]
- Rao, Z.; Zhang, X.; Xue, S.; Xu, Y.; Liu, X. Primary organic carbon isotopic study result of Xifeng loess/red clay profile. Quat. Sci. 2012, 32, 825–827. [Google Scholar]
- Breeuwsma, A.; Wösten, J.H.M.; Vleeshouwer, J.J.; van Slobbe, A.M.; Bouma, J. Derivation of land qualities to assess environmental problems from soil surveys. Soil Sci. Soc. Am. J. 1986, 50, 186–190. [Google Scholar] [CrossRef]
Figure 1.
The hydrogeological condition of the study area and sampling location modified from [21].
Figure 1.
The hydrogeological condition of the study area and sampling location modified from [21].
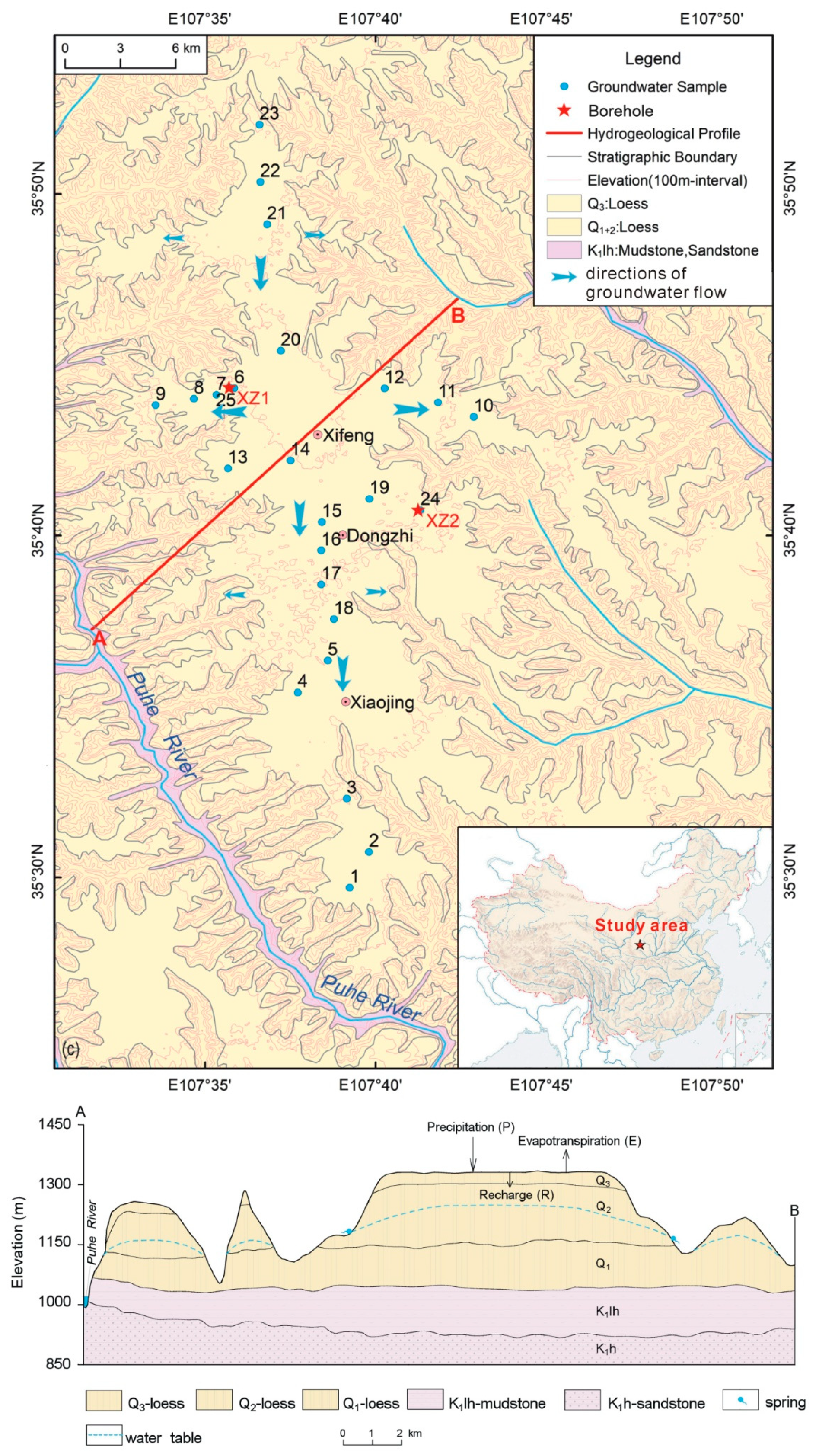
Figure 2.
The ions composition (% in meq/L) for groundwater.
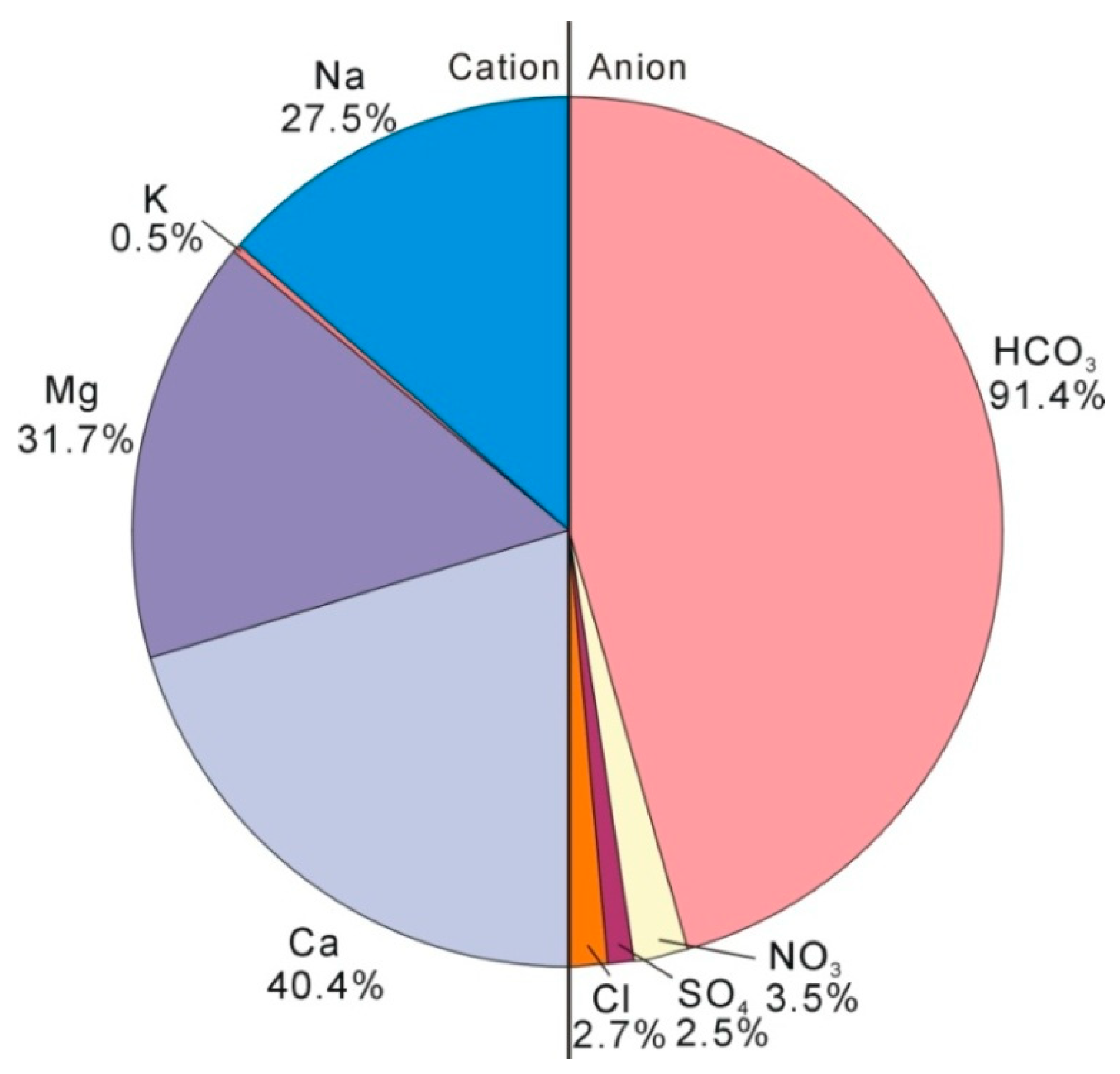
Figure 3.
Plot of groundwater Ca/Na and Mg/Na (a) and plot of Sr concentration and Ca/Na (b).
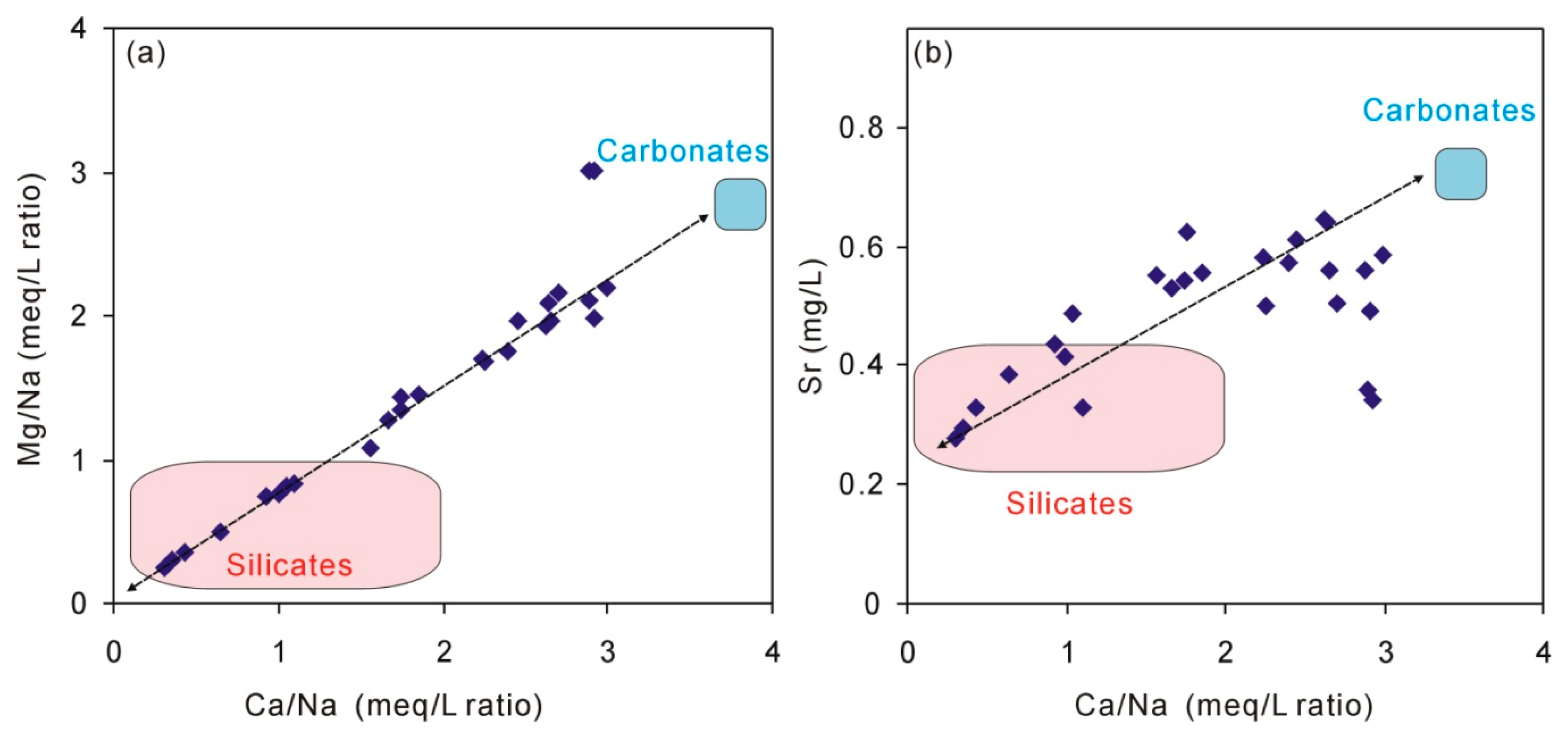
Figure 4.
Plot of corrected groundwater 14C ages and Mg/Ca ratios.
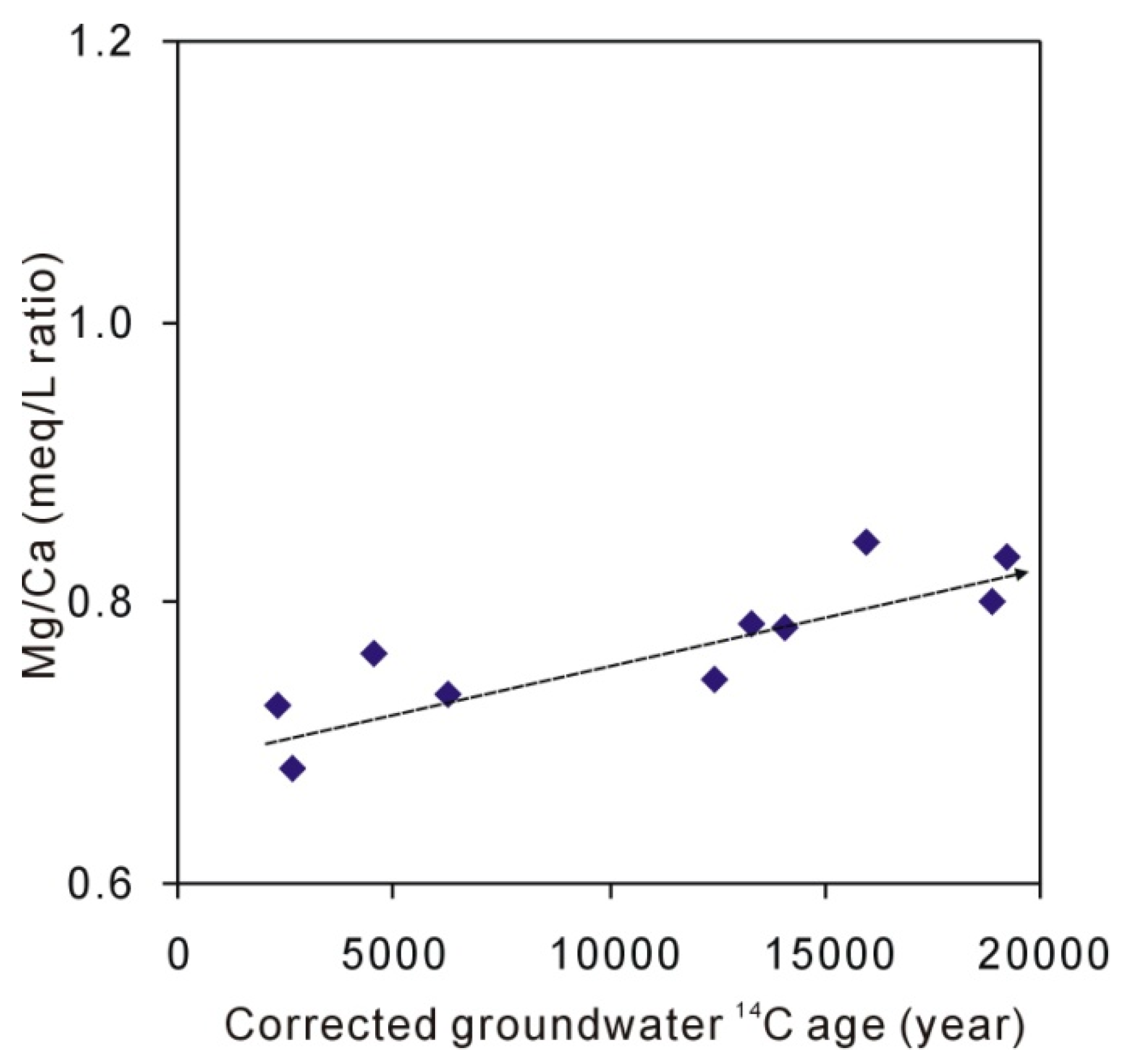
Figure 5.
Groundwater 87Sr/86Sr ratios compared with carbonate minerals, whole-rock (loess), and residue after dissolution of 1-M HAc.
Figure 5.
Groundwater 87Sr/86Sr ratios compared with carbonate minerals, whole-rock (loess), and residue after dissolution of 1-M HAc.
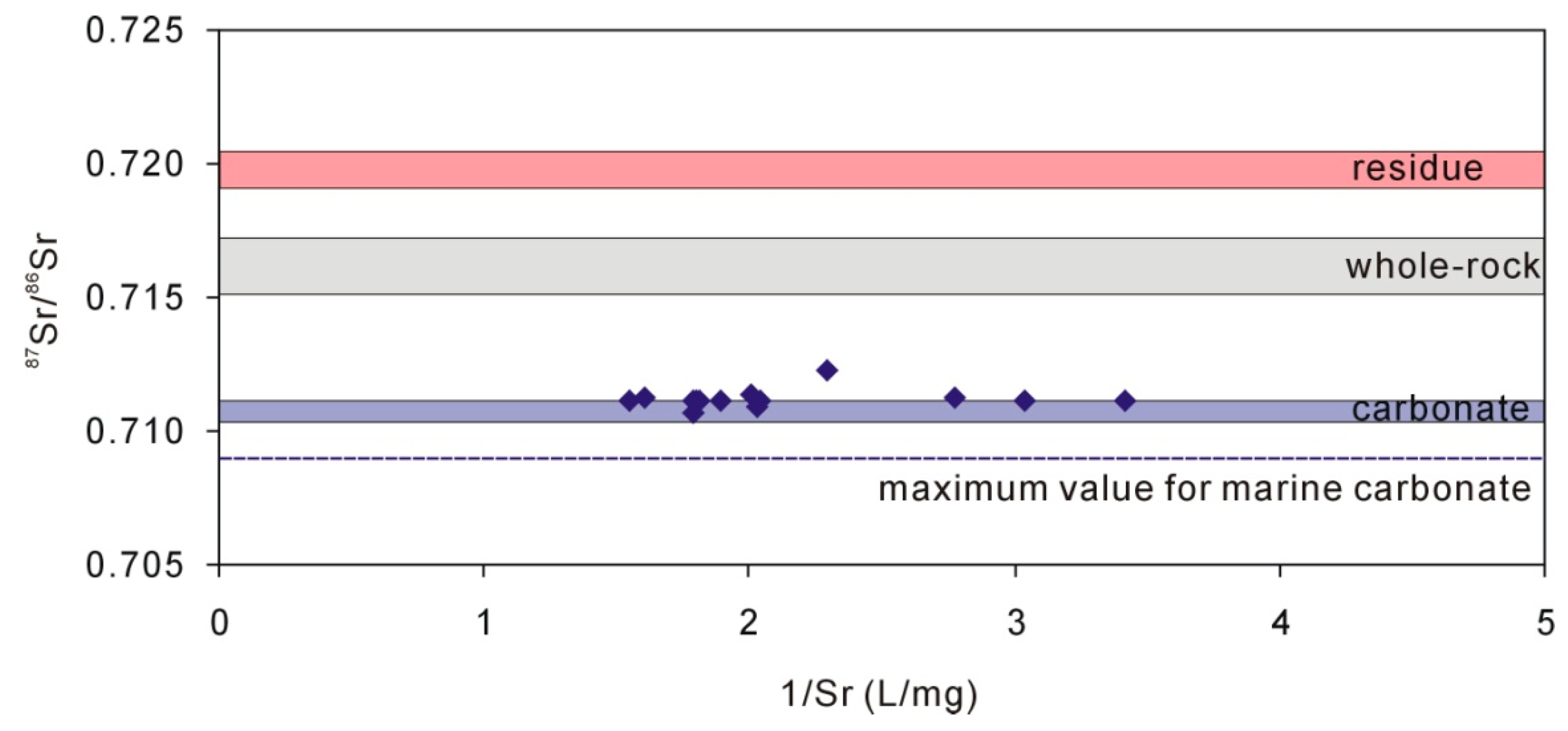
Figure 6.
Plot of 87Sr/86Sr and δ13C for multiple endmembers in the loess aquifer. The blue diamonds present groundwater DIC (mainly as HCO3). Geochemical processes, as indicated (1), (2), and (3) are discussed in the text.
Figure 6.
Plot of 87Sr/86Sr and δ13C for multiple endmembers in the loess aquifer. The blue diamonds present groundwater DIC (mainly as HCO3). Geochemical processes, as indicated (1), (2), and (3) are discussed in the text.
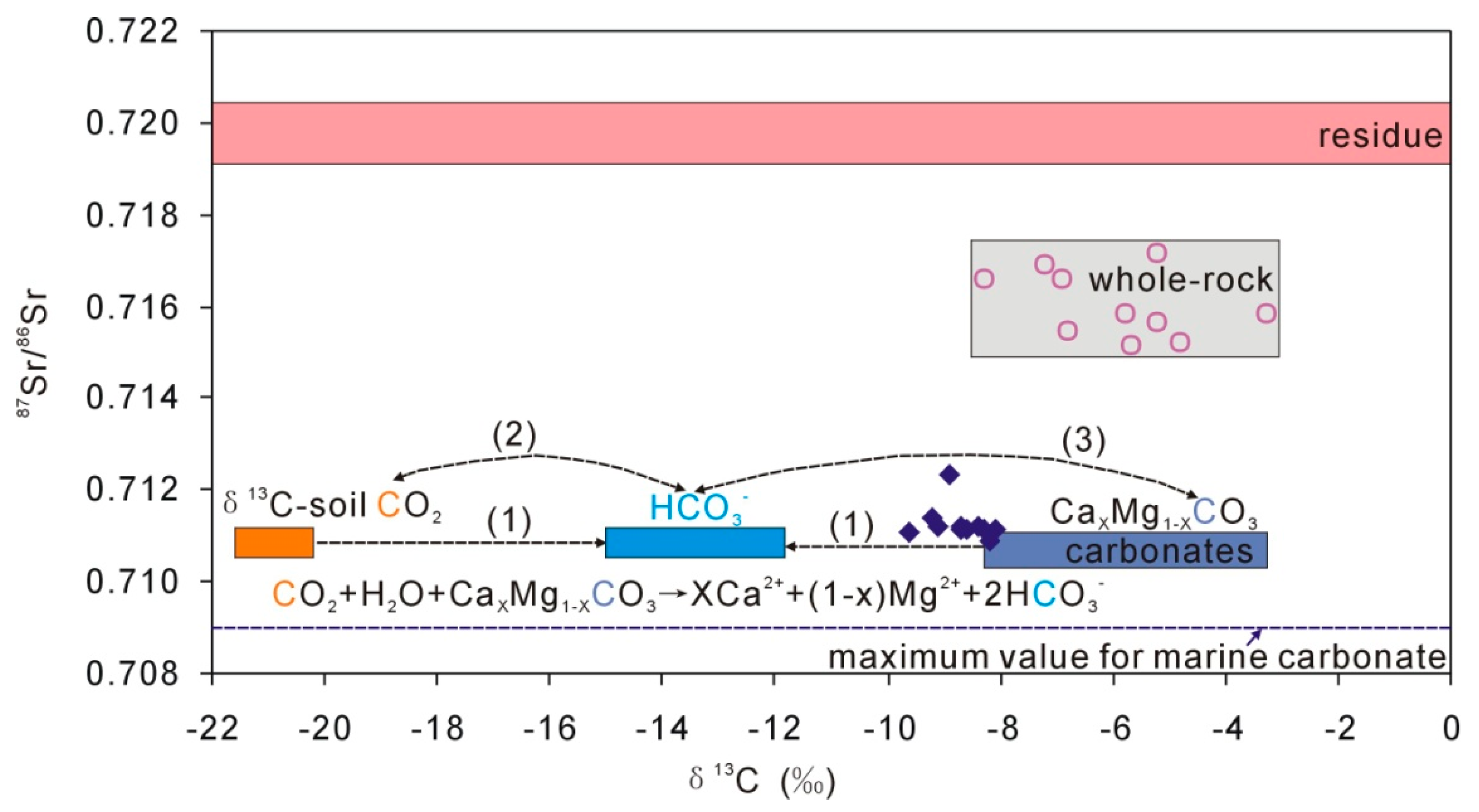
Figure 7.
Plot of groundwater Na against Ca + Mg and Ca + Mg + Na (a) and plot of corrected groundwater 14C age against Na, Ca + Mg, and Ca + Mg + Na (b).
Figure 7.
Plot of groundwater Na against Ca + Mg and Ca + Mg + Na (a) and plot of corrected groundwater 14C age against Na, Ca + Mg, and Ca + Mg + Na (b).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The mineral composition for loess (%).
| Sample | Quartz | Potassium Feldspar | Plagioclase | Calcite | Dolomite | Clay |
|---|---|---|---|---|---|---|
| S1 | 43.0 | 3.4 | 16.5 | 9.1 | 0.0 | 28.0 |
| S2 | 44.7 | 2.9 | 11.8 | 13.6 | 0.0 | 27.0 |
| S3 | 44.5 | 4.3 | 15.3 | 16.4 | 3.4 | 16.1 |
| S4 | 33.1 | 2.8 | 9.8 | 22.1 | 0.0 | 32.2 |
Table 2.
The mineral composition for clay in loess (%).
| Sample | Smectite | Illite-Smectite | Illite | Kaolinite | Chlorite |
|---|---|---|---|---|---|
| S1 | 0 | 32 | 49 | 5 | 14 |
| S2 | 0 | 20 | 60 | 7 | 13 |
| S3 | 0 | 21 | 58 | 9 | 12 |
| S4 | 0 | 13 | 68 | 7 | 12 |
Table 3.
Major element content for loess samples (%). LOI, loss on ignition.
| Sample | SiO2 | Al2O3 | Fe2O3 | MgO | CaO | Na2O | K2O | MnO | TiO2 | P2O5 | LOI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | 59.14 | 12.02 | 4.51 | 2.11 | 7.05 | 1.78 | 2.34 | 0.087 | 0.649 | 0.141 | 9.77 |
| S2 | 58.98 | 12.56 | 4.89 | 2.05 | 6.36 | 1.58 | 2.38 | 0.087 | 0.677 | 0.139 | 9.72 |
| S3 | 59.51 | 11.83 | 4.35 | 2.15 | 7.28 | 1.72 | 2.28 | 0.083 | 0.637 | 0.167 | 9.79 |
| S4 | 54.73 | 11.89 | 4.57 | 1.95 | 9.42 | 1.39 | 2.26 | 0.085 | 0.636 | 0.204 | 12.37 |
Table 4.
Isotopic compositions for loess in Xifeng. The values of δ13C are from [21].
Table 4.
Isotopic compositions for loess in Xifeng. The values of δ13C are from [21].
| Location | Depth Range (m) | δ13C (‰) | 87Sr/86Sr |
|---|---|---|---|
| XZ1 | 9.75–10 (S1) | −6.8 | 0.715481 |
| XZ1 | 19.75–20 | −4.8 | 0.715244 |
| XZ1 | 29.5–30 (S2) | −6.9 | 0.716614 |
| XZ1 | 39.5–40 | −5.2 | 0.715639 |
| XZ1 | 52–52.5 | −5.8 | 0.715851 |
| XZ1 | 54.5–55 (S3) | −3.3 | 0.715873 |
| XZ2 | 9.75–10 | −8.3 | 0.716586 |
| XZ2 | 19.75–20 | −5.7 | 0.715136 |
| XZ2 | 29.5–30 | −5.2 | 0.717155 |
| XZ2 | 44.5–45 (S4) | −7.2 | 0.716909 |
Table 5.
Hydrogeochemical and strontium isotopic composition for groundwater. The values labeled with * are from [21].
Table 5.
Hydrogeochemical and strontium isotopic composition for groundwater. The values labeled with * are from [21].
| Item | Unit | Min. | Max. | Average | Median |
|---|---|---|---|---|---|
| TDS * | mg/L | 225 | 316 | 288 | 289 |
| pH * | - | 7.6 | 8.2 | 7.9 | 7.8 |
| Cl * | mg/L | 4.0 | 10.9 | 5.5 | 5.3 |
| SO4 * | mg/L | 3.2 | 13.1 | 6.8 | 6.9 |
| NO3 * | mg/L | 6.5 | 19.2 | 12.4 | 11.9 |
| HCO3 * | mg/L | 246 | 357 | 322 | 324 |
| F * | mg/L | 0.3 | 0.6 | 0.4 | 0.4 |
| Br * | mg/L | 0.019 | 0.029 | 0.025 | 0.027 |
| Na * | mg/L | 18 | 84 | 35 | 28 |
| K * | mg/L | 0.5 | 1.8 | 1.0 | 1.0 |
| Mg * | mg/L | 11 | 28 | 21 | 23 |
| Ca * | mg/L | 22 | 56 | 45 | 49 |
| δ13C-DIC * | ‰ | −9.6 | −8.1 | −8.7 | −8.7 |
| 3H * | TU | <0.4 | <0.4 | <0.4 | <0.4 |
| 14C age * | years | 220 | 19220 | 9990 | 12424 |
| saturation indices (SI) | calcite | 0.20 | 0.83 | 0.44 | 0.43 |
| dolomite | 0.36 | 1.67 | 0.83 | 0.78 | |
| fluorite | −2.14 | −1.65 | −1.90 | −1.91 | |
| gypsum | −3.47 | −2.59 | −2.97 | −2.90 | |
| magnesite | −0.41 | 0.26 | −0.17 | −0.21 | |
| strontianite | −1.44 | −0.62 | −1.04 | −1.03 | |
| Sr | mg/L | 0.278 | 0.646 | 0.489 | 0.516 |
| 87Sr/86Sr | - | 0.710677 | 0.712319 | 0.711207 | 0.711157 |
Table 6.
The results for loess dissolution using HAc (−1:40 g soil and 400 mL 0.5 M of HAc; −2:15 g residual soil and 150 mL 1 M of HAc).
Table 6.
The results for loess dissolution using HAc (−1:40 g soil and 400 mL 0.5 M of HAc; −2:15 g residual soil and 150 mL 1 M of HAc).
| Sample | Ca mg/L | Mg mg/L | Na mg/L | K mg/L | Sr μg/L | 87Sr/86Sr | CaCO3 (%) | MgCO3 (%) | CaMg(CO3)2 (%) |
|---|---|---|---|---|---|---|---|---|---|
| S1-1 | 5870 | 161 | 11.7 | 3.7 | 8304 | 0.710750 | 14.68 | 0.56 | 0.76 |
| S1-2 | 485 | 117 | 8.7 | 4.4 | 785 | 0.710818 | |||
| S2-1 | 5260 | 147 | 12.4 | 4.0 | 6394 | 0.711038 | 13.15 | 0.51 | 0.48 |
| S2-2 | 406 | 72 | 8.2 | 4.2 | 683 | 0.710969 | |||
| S3-1 | 5770 | 127 | 11.9 | 5.1 | 8081 | 0.710455 | 14.43 | 0.44 | 1.43 |
| S3-2 | 691 | 219 | 8.2 | 4.3 | 896 | 0.710329 | |||
| S4-1 | 7445 | 128 | 12.7 | 5.5 | 5111 | 0.711085 | 18.61 | 0.45 | 0.47 |
| S4-2 | 1034 | 76 | 8.4 | 6.0 | 996 | 0.710958 |
Table 7.
The mineral composition and 87Sr/86Sr ratios for residual loess after 1 M of HAc dissolution.
Table 7.
The mineral composition and 87Sr/86Sr ratios for residual loess after 1 M of HAc dissolution.
| Sample | Quartz | Potassium Feldspar | Plagioclase | Calcite | Dolomite | Clay | 87Sr/86Sr |
|---|---|---|---|---|---|---|---|
| S1-res | 51.2 | 4.0 | 19.2 | 0 | 0 | 25.6 | 0.719091 |
| S2-res | 54.9 | 2.6 | 13.4 | 0 | 0 | 29.1 | 0.720415 |
| S3-res | 53.5 | 1.9 | 16.2 | 0 | 0 | 28.4 | 0.719876 |
| S4-res | 54.0 | 2.2 | 11.8 | 0 | 0 | 32.0 | 0.720438 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huang, T.; Ma, B. The Origin of Major Ions of Groundwater in a Loess Aquifer. Water 2019, 11, 2464. https://doi.org/10.3390/w11122464
AMA Style
Huang T, Ma B. The Origin of Major Ions of Groundwater in a Loess Aquifer. Water. 2019; 11(12):2464. https://doi.org/10.3390/w11122464
Chicago/Turabian StyleHuang, Tianming, and Baoqiang Ma. 2019. "The Origin of Major Ions of Groundwater in a Loess Aquifer" Water 11, no. 12: 2464. https://doi.org/10.3390/w11122464
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.