2.1. Seasonal and Diurnal Variations
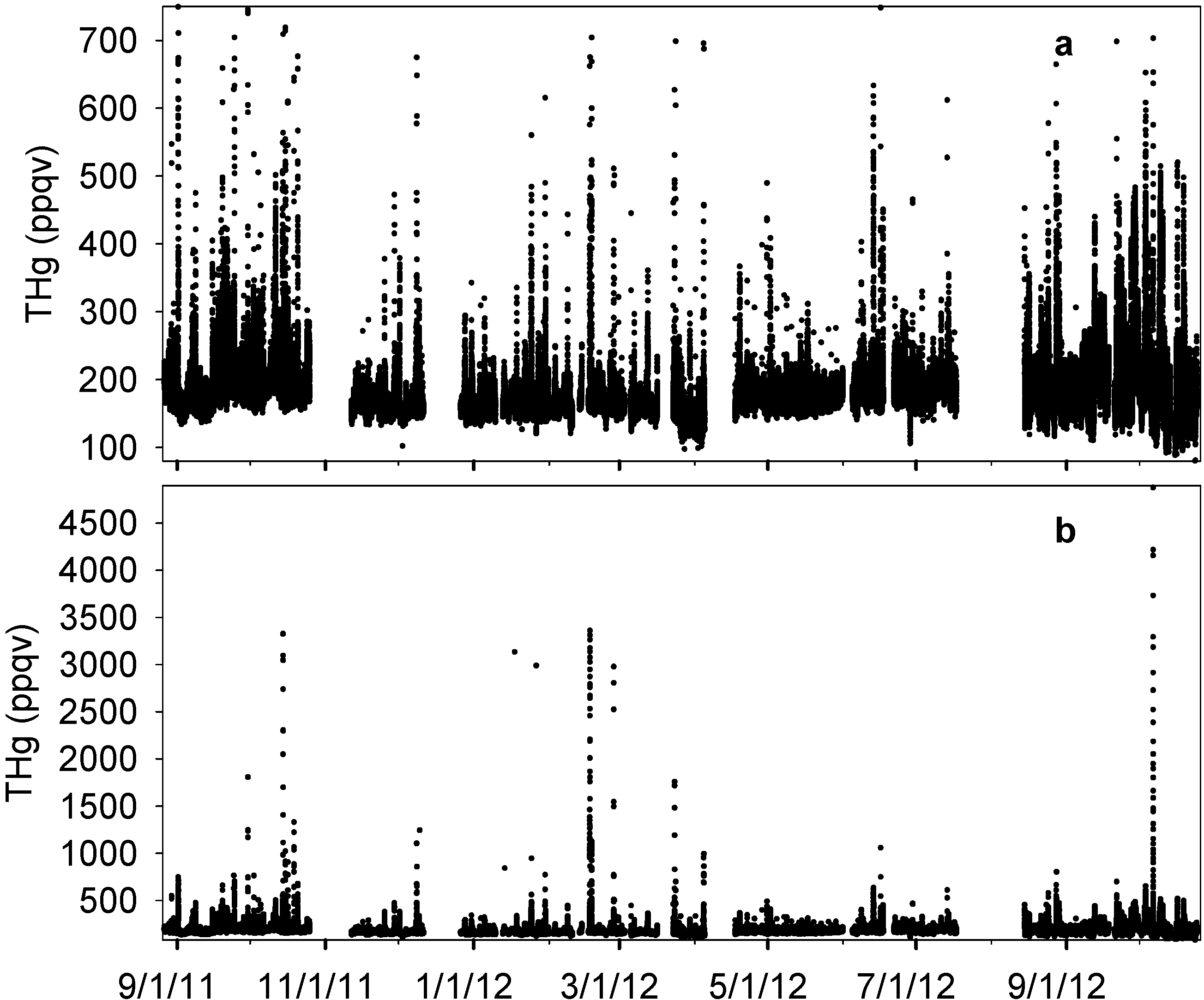
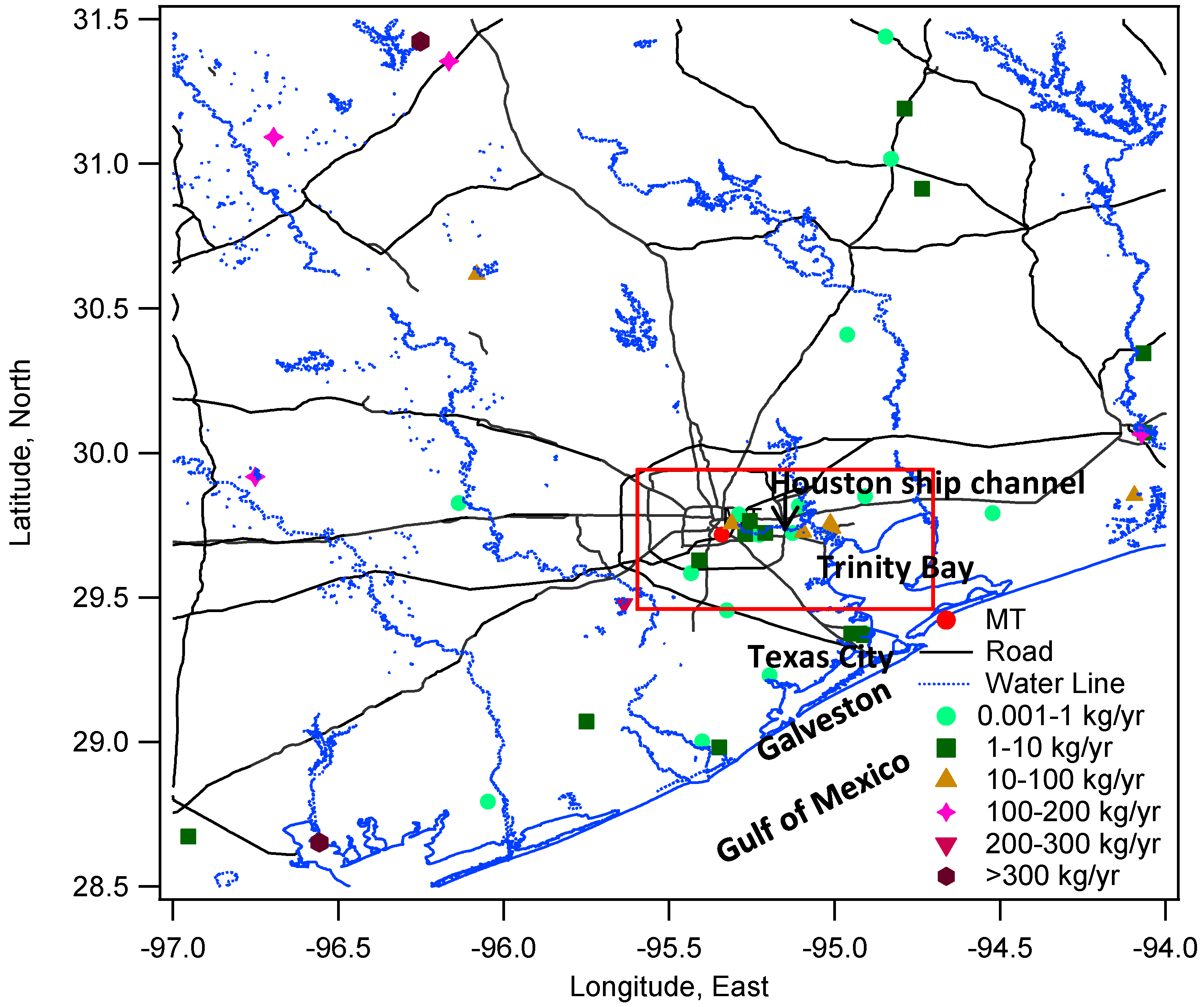
The complete time series of THg is presented in
Figure 2. The median level of THg in Houston was 172 ppqv (181 ± 63 ppqv for mean ± S.D.) during our observation period (see Figure S2 and Table S1 for more statistical details), which was in agreement with the current background level in the Northern Hemisphere [
20], but slightly lower than other urban sites with mercury levels higher than 2 ng∙m
−3 (224 ppqv) [
22,
25,
28,
39,
40]. The majority of THg observations fell within the range of 148 ppqv (10th percentile) to 215 ppqv (90th percentile). The maximum THg mixing ratio, however, was as high as 4876 ppqv, exceeding 25 times the current global background level. The minimum level was 80 ppqv, which occurred with southerly wind that brought cleaner air from the Gulf of Mexico into urban Houston. A prominent feature of THg in the Houston area was the frequent occurrence of large THg spikes (
Figure 2b). From 14-month measurements, we documented 81 well-developed spikes with THg levels higher than 300 ppqv, 34 of which were higher than 500 ppqv. Extremely large peaks with THg levels higher than 1000 ppqv were observed 12 times, and six of them were higher than 3000 ppqv. The time scale of elevated mercury in these spikes ranged from 30 minutes to a few hours. As a consequence of the THg spikes, the standard deviations of our data reached 63 ppqv, which was comparable with other urban sites measurements, such as Salt Lake City (95 ppqv [
22]) and Reno (45–90 ppqv [
39]).
Figure 2.
Complete time series of THg from MT measurements. (a) and (b) show the same data with different ranges in y axis.
Figure 2.
Complete time series of THg from MT measurements. (a) and (b) show the same data with different ranges in y axis.
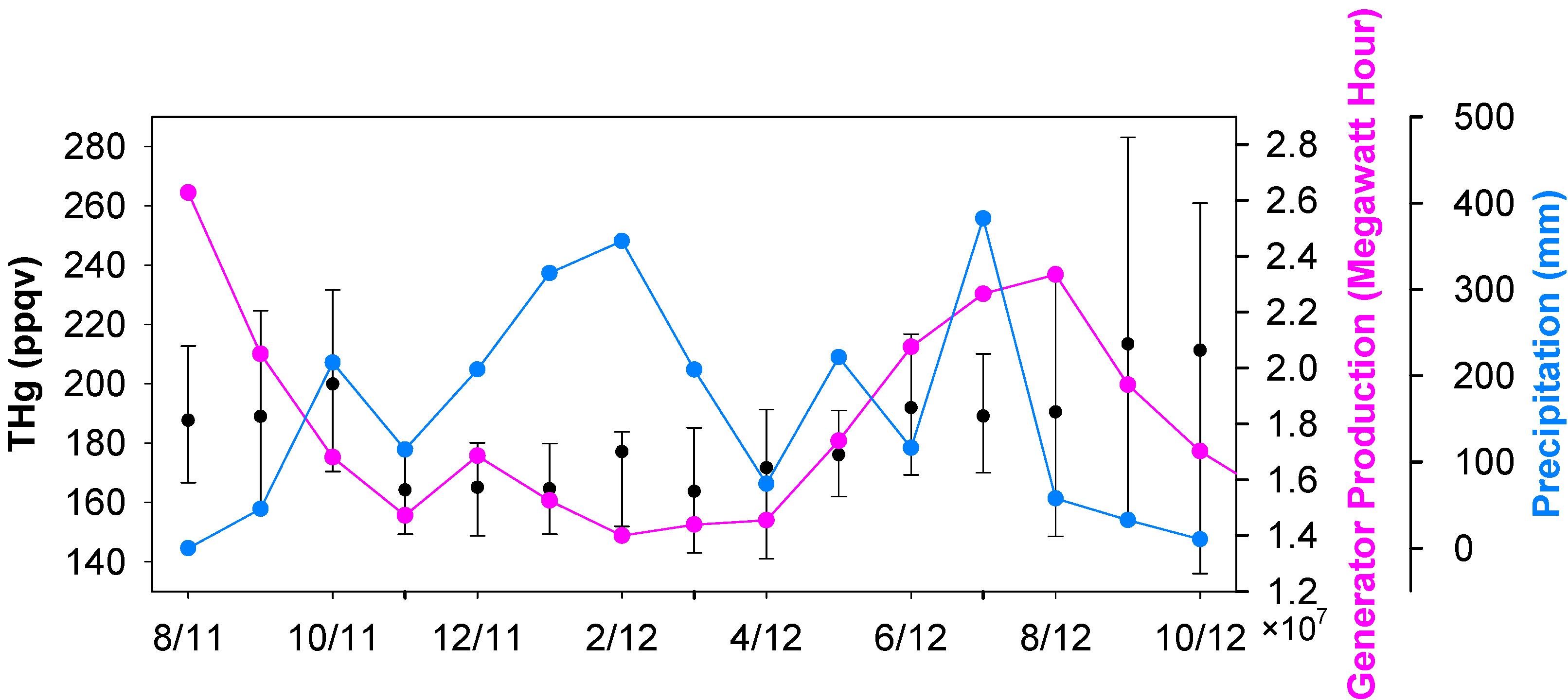
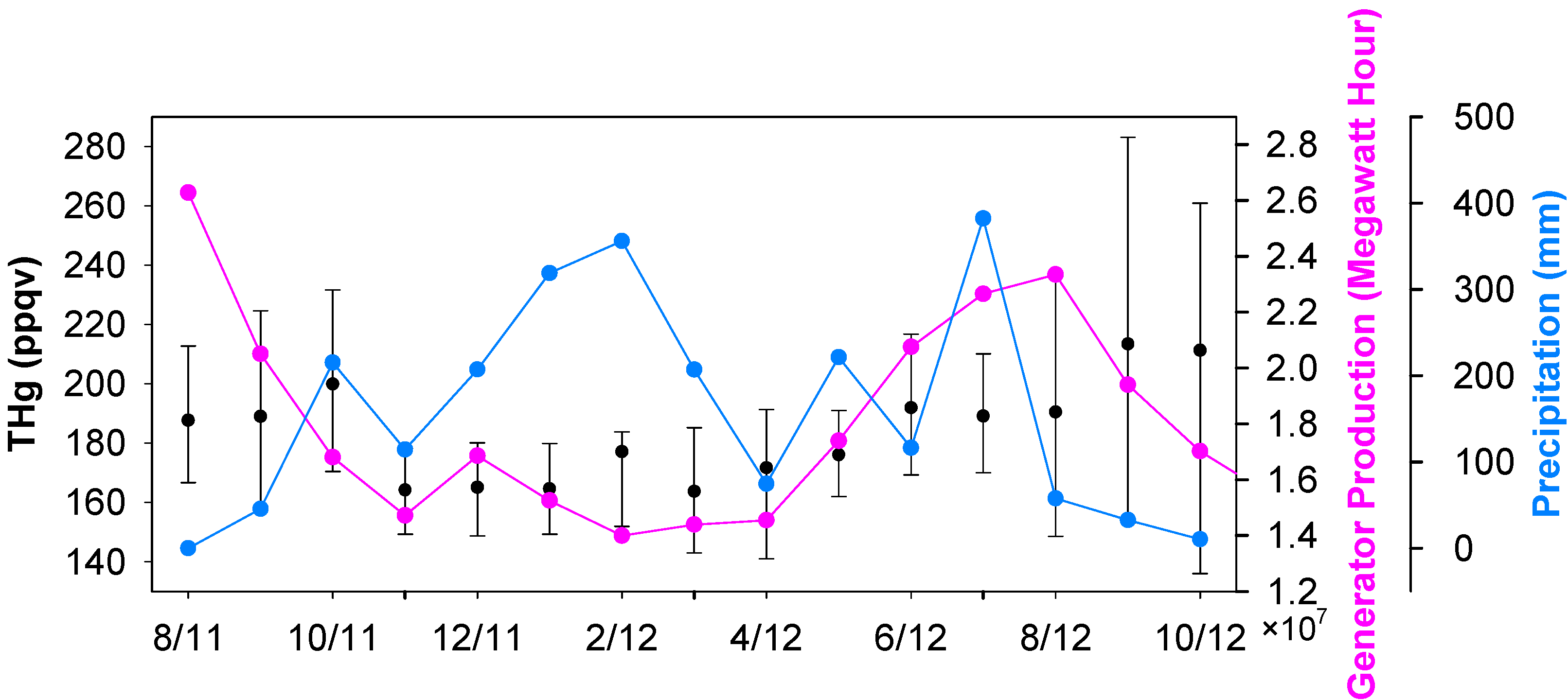
Seasonal median levels of THg were 178 ppqv (179 ± 54 ppqv for mean ± S.D.), 161 ppqv (161 ± 80 ppqv), 172 ppqv (172 ± 26 ppqv), and 185 ppqv (186 ± 32 ppqv) for fall 2011, winter 2011, spring 2012 and summer 2012, respectively. The monthly median THg values are displayed in
Figure 3. Unlike many other ambient mercury measurements, which show higher mercury in winter time [
21,
22], high THg in Houston area occurred in the warm seasons (June to October). It is obvious that the frequent occurrences of large THg spikes during the warm seasons, especially in August, September and October, contributed to the elevated THg levels. The great enrichments in episodic THg spikes suggested that the large pollutant plumes originated from the nearby industrial/urban emission sources. Mercury emissions from anthropogenic sources are closely linked to energy production, especially from coal-fired power plants, the largest anthropogenic mercury source in U.S. [
13]. Enhanced energy production is expected in the Houston area in summer and fall. The high ambient air temperature requires energy to operate air-conditioning units. As a consequence, the ambient THg levels may increase. To illustrate this point, the state-level energy data achieved from Energy Information Administration (EIA) is presented in
Figure 3. The monthly total energy produced in warm months was 50%–100% higher than in the cold months (The correlation coefficient between monthly median THg and monthly median energy production is 0.735, which is statistically significant (p ≤ 0.005)). Besides the fluctuations in mercury sources, we noticed some changes in potential sinks. Mercury deposition in rain water has been commonly reported [
41,
42,
43], indicating rainfall as an important removal mechanism for atmospheric mercury. The precipitation in Houston was observed to be consistently higher in the period from December 2011 to March 2012 (
Figure 3), which coincided with low wintertime THg.
Figure 3.
Monthly medians of THg, energy production and precipitation.
Figure 3.
Monthly medians of THg, energy production and precipitation.
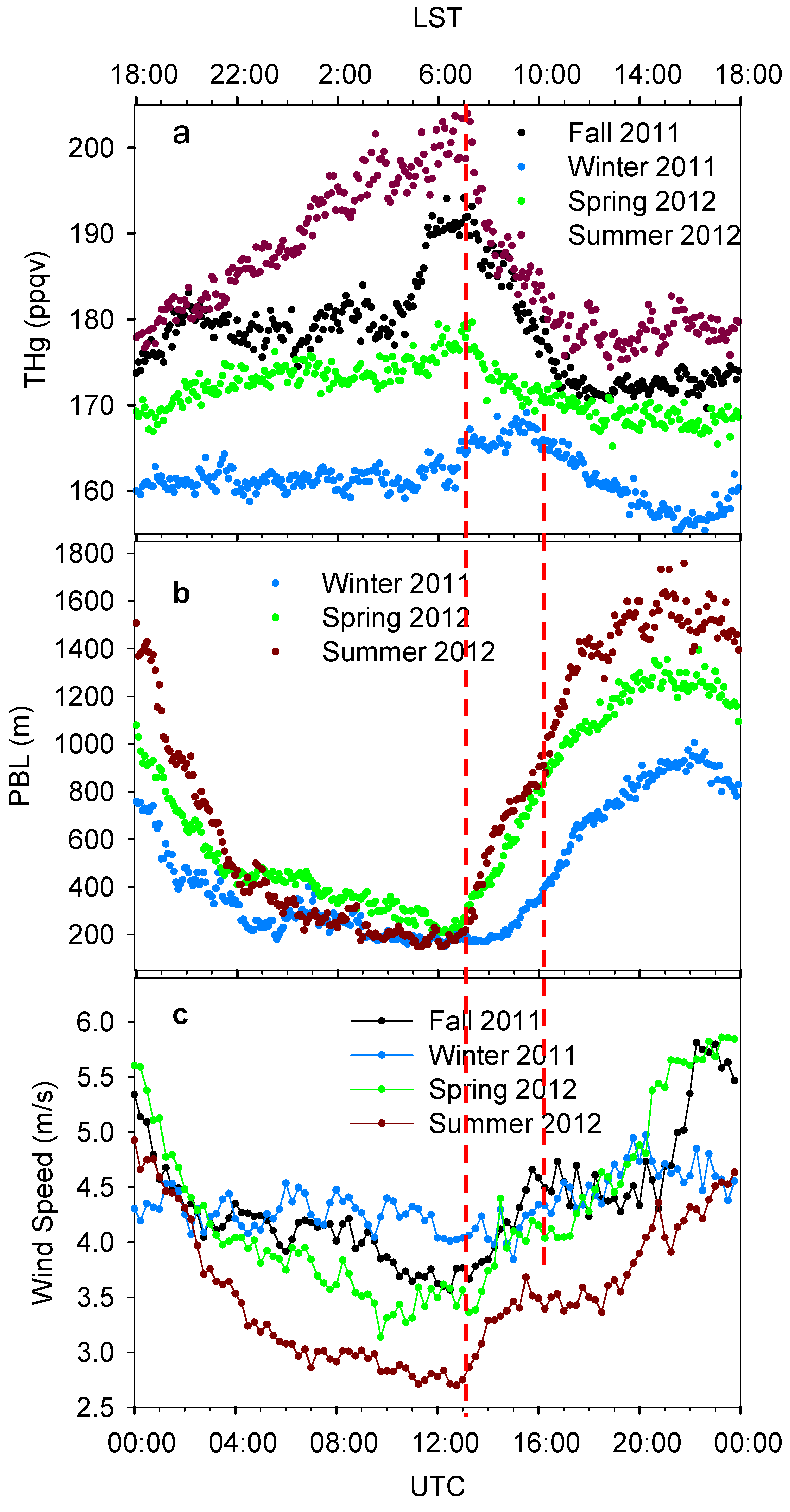
Precipitous day-to-day peaks and valleys variations were also observed as another outstanding feature of THg signal. Hourly median THg mixing ratios are displayed in
Figure 4a. Despite the differences in variation amplitudes, diurnal variation patterns in fall, winter and spring were generally similar. Their diurnal patterns show a period with increasing THg levels starting around 16:00–18:00 local standard time (LST) and lasting for 2–4 hours, followed by a period with relatively constant THg levels. A sharp enhancement of THg levels started at around 04:00 LST, and then the THg reached a daily maximum at 07:00–09:00 LST. The summer diurnal pattern, however, appeared to be very different from other seasons. The THg level in summer gradually increased throughout the whole night and reached its maximum at about 07:00 LST, indicating an accumulation process of THg. Summertime had the highest THg mixing ratios during a day, and the largest diurnal variation amplitude (about 30 ppqv) among the four seasons. To conclude, the THg levels were higher at night than during the daytime. The maximum levels appeared right before sunrise, followed by rapid decreases after sunrise, and daily minimum shortly after noon. Similar diurnal patterns were observed in other urban cities, such as Guiyang, China [
27], Detroit, U.S. [
28], and Toronto, Canada [
29].
It is interesting to note that the THg mixing ratio reached its diurnal maximum at about 07:00 LST in fall, spring and summer, while it was two hours later in winter. This phenomenon was related to the diurnal development of the PBL (
Figure 4b). The PBL heights started increasing at about 06:00–07:00 LST for spring and summer and two hours later for winter. The rapid enhancements of PBL height in early morning can facilitate air mixing in three-dimensions and dilute the ambient THg within the boundary layer, and thus caused striking decreases of THg at the same time. The rate of decrease in summer was the largest (7.0 ppqv∙h
−1), compared to those in winter (1.9 ppqv∙h
−1), spring (2.6 ppqv∙h
−1), and fall (4.6 ppqv∙h
−1). The high PBL height in daytime then contributed to the low THg mixing ratios, especially in the afternoon. This phenomenon suggested that the residual layer was not a significant source for THg in Houston; instead, the industrial/urban emissions from the surface were more prominent.
Figure 4.
Seasonally diurnal variations of THg (a), PBL height (b) and horizontal wind speed (c). Data shows the median values with 5 min. time interval. The top x axis shows time in LST and the bottom x axis shows time in UTC. LST = UTC − 6:00.
Figure 4.
Seasonally diurnal variations of THg (a), PBL height (b) and horizontal wind speed (c). Data shows the median values with 5 min. time interval. The top x axis shows time in LST and the bottom x axis shows time in UTC. LST = UTC − 6:00.
However, some features of the diurnal THg variations cannot only be explained by the PBL height propagations, for example, the summertime THg mixing ratios were the highest during all times of the day even though the daytime PBL heights were the highest. It was observed that the diurnal variations of horizontal wind speeds were anti-correlated with THg levels most of the time (
Figure 4c, median wind speeds were calculated using scalar values). High horizontal wind speeds can enhance horizontal mixing of polluted air with cleaner ambient air, and effectively advect the pollutants away from the urban area. The summertime wind speed was the lowest of all seasons, and the especially low wind speeds at night supported the accumulation of THg and caused high summertime THg mixing ratios. In spring, low wind speeds were observed at 04:00–07:00 LST, which probably contributed to the high THg mixing ratios in this period.
The energy production/consumption, precipitation, PBL height, and horizontal wind speed played important roles in the seasonal and diurnal variations of THg; however, we cannot quantify the contribution of each factor from our observations. Regional modeling with a good emission inventory and dynamic processing is necessary for further investigation.
It is unclear whether photochemical reactions play an important role in determining THg mixing ratios in the Houston area. Previous research suggests that the main sink of GEM in the atmosphere is oxidation to GOM [
44]. The GOM then can attach to particles and be transformed to PBM. Both GOM and PBM are easily removed from the air via wet and dry deposition [
21,
45], which will eventually reduce the mixing ratio of THg. From our observations, the diurnal THg mixing ratios remained constant from 12:00 LST to 16:00 LST in fall, spring and summer. During these times, photochemical reactions should be actively changing due to the fluctuations of solar radiation and variations in the abundance of oxidants. The effect of photochemical reactions may be obscured due to complex local emissions and meteorological conditions.
Natural sources can also influence the seasonal and diurnal variations of ambient THg levels; however, we are unable to quantify the contributions of natural emissions in metropolitan Houston due to a lack of direct measurements. However, a large portion of vegetation in the Houston area is evergreen; with no snow in winter, we expect small contributions from natural emissions to the THg seasonal variations.
2.2. Wind Induced Influences
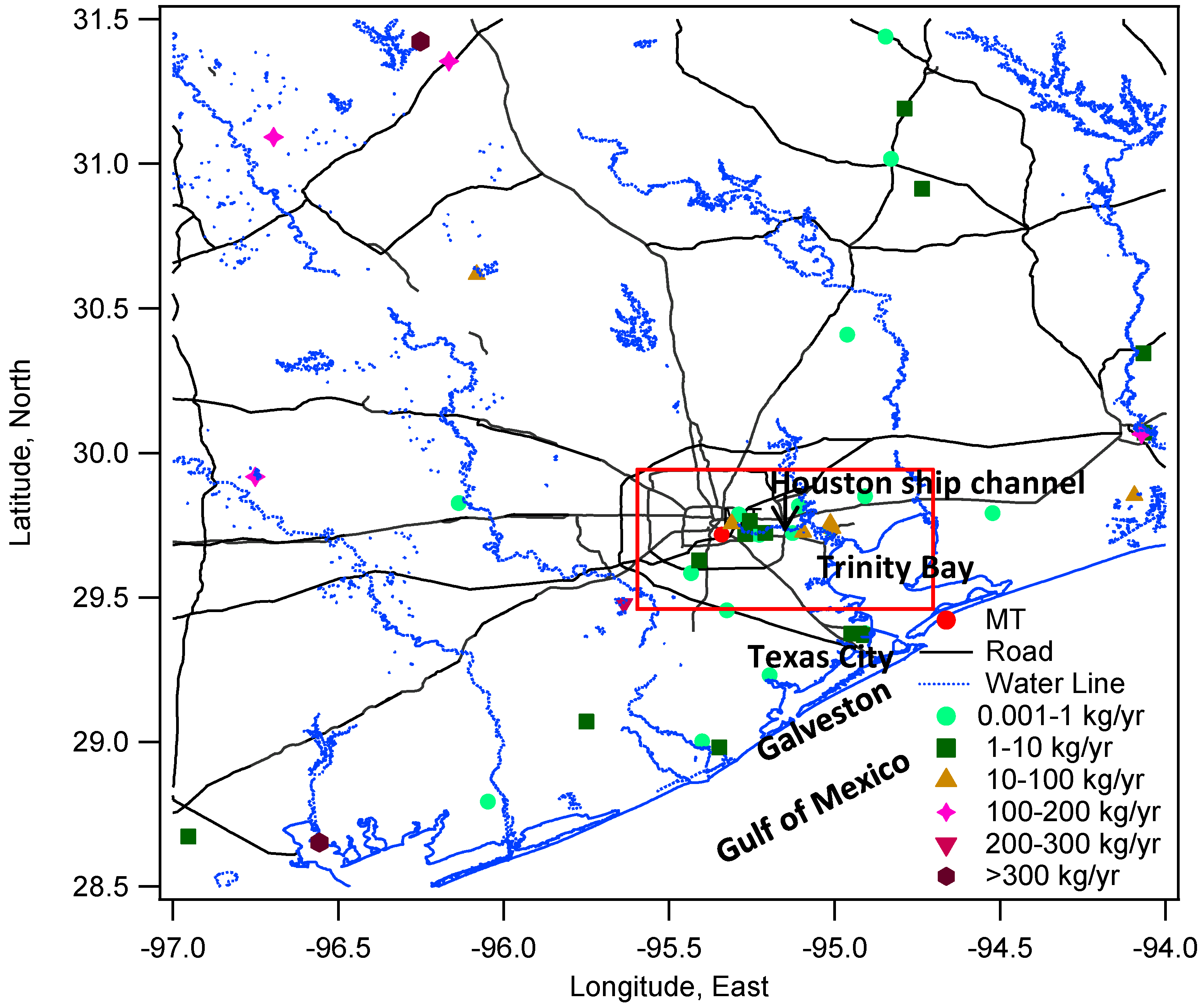
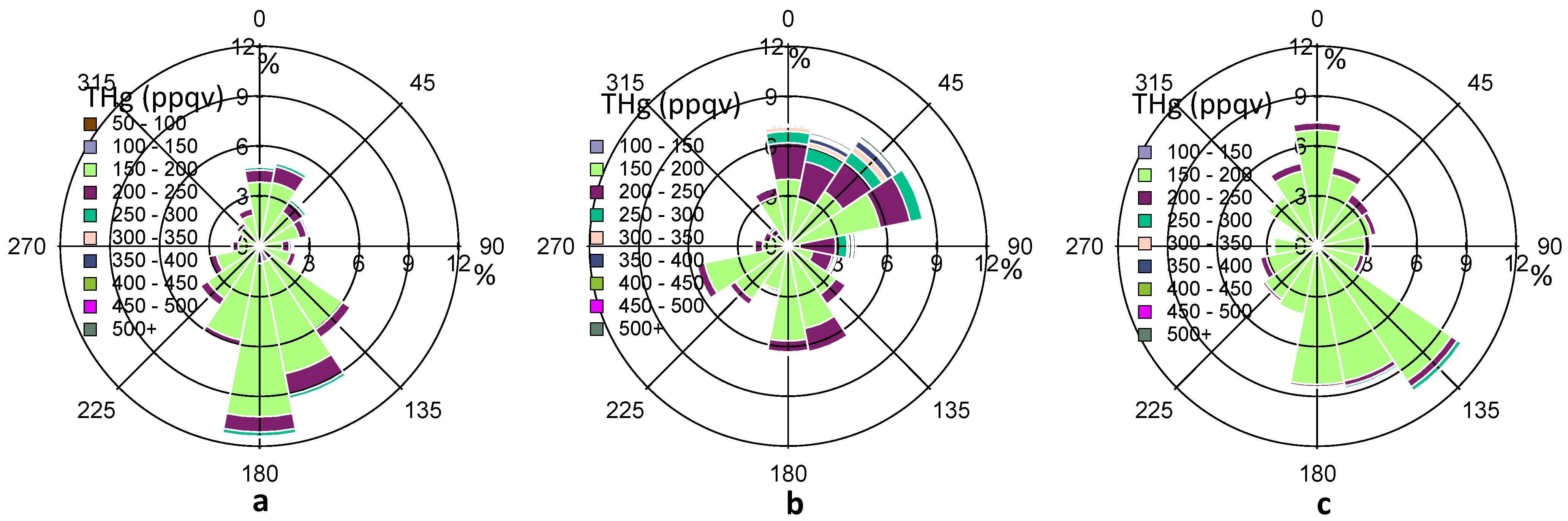
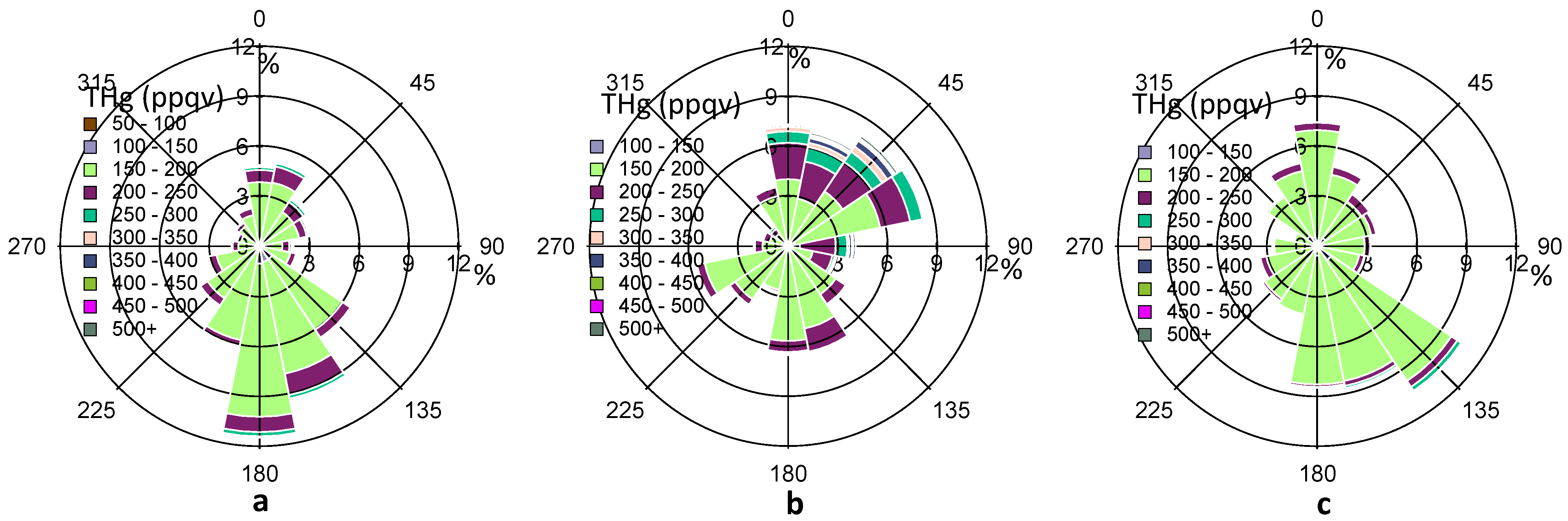
In addition to the wind speed, the wind direction also exerts considerable impacts on THg variations, suggesting the importance of local/regional transport. In
Figure 5, the percentage values on the R axis shows the frequency (%) of THg coming from a certain range (22.5°) of wind directions. The predominant wind directions in our observed period were south and southeast directions (120°–190°) (
Figure 5a), which were from the Gulf of Mexico. The overall frequency from these directions accounts for about 40%, after summing up the percentage values from 120°–190° directions in
Figure 5a. Cleaner air masses with especially low THg levels (<150 ppqv) were observed from these directions. However, the refinery facilities emissions in Texas City may also advected to Houston area as we obtained about 6% of air masses with THg levels higher than 200 ppqv from the same directions.
Figure 5.
Complete THg versus wind direction (a), THg versus wind direction in HMP (b), THg versus wind direction in LMP (c). The color scale shows the ranges of THg mixing ratios. The percentage values on the R axis shows the frequency of THg coming from a certain range (22.5°) of wind directions.
Figure 5.
Complete THg versus wind direction (a), THg versus wind direction in HMP (b), THg versus wind direction in LMP (c). The color scale shows the ranges of THg mixing ratios. The percentage values on the R axis shows the frequency of THg coming from a certain range (22.5°) of wind directions.
Wind direction in Houston varied diurnally. From
Figure 4a, we noticed elevated THg mixing ratios at about 05:00–11:00 LST, and lower mercury mixing ratios at about 12:00–17:00 LST. To evaluate the impacts of shifting wind direction, we defined a daily high mercury period (HMP) of 05:00–11:00 LST and a daily low mercury period (LMP) of 12:00–17:00 LST.
Figure 5b,c depict the wind rose for HMP and LMP in 2011 fall, respectively. In the HMP, air masses with high THg levels (200 ppqv) mainly came from north, northeast, and east sectors, where urban and industrial influences were remarkable, especially in the Houston Ship Channel area. In the LMP, air masses mainly came from the south and southeast sectors, where cleaner marine air seemed to dominate. The median THg level in HMP was statistically higher than that in LMP (14 ppqv higher, p ≤ 0.001). Analysis of other seasons (not shown) also suggested similar influences from shifting source regions. It was also observed that consistent northerly winds with high wind speeds (>4 m/s, occurred in December) yielded THg levels ≥ 150 ppqv, and CO levels ≥ 125 ppbv. In comparison, consistent southerly winds with high wind speeds (occurred in April) produced THg levels as low as 120 ppqv and CO levels as low as 85 ppbv. The large discrepancies between southerly air and northerly air highlight the significance of local urban/industrial influences on elevated THg mixing ratios in the Houston area.
The sea breeze exerted significant and complex impacts on THg levels in the Houston area. The sea breeze is driven by diurnally uneven heating in coastal areas, which produces warmer temperatures over land than over water during the day, and cooler land temperatures at night [
46]. As a result, a sea breeze is produced when the air flows from the sea to the land at low altitude (<500 m). The low level wind vector then rotates through a clockwise (Northern Hemisphere) cycle under the influence of the Coriolis force [
46,
47]. As the sea breeze passes through the shoreline, it can bring cleaner marine air to inland areas that can dilute urban/industrial emissions. The sea breeze can also bring back ashore the aged polluted air that was once transported offshore by other wind systems. A detailed analysis on the wind system in Houston area reported that the afternoon sea breeze was responsible for large O
3 enrichment events, by bringing back aged, polluted air masses to the urban area [
46].
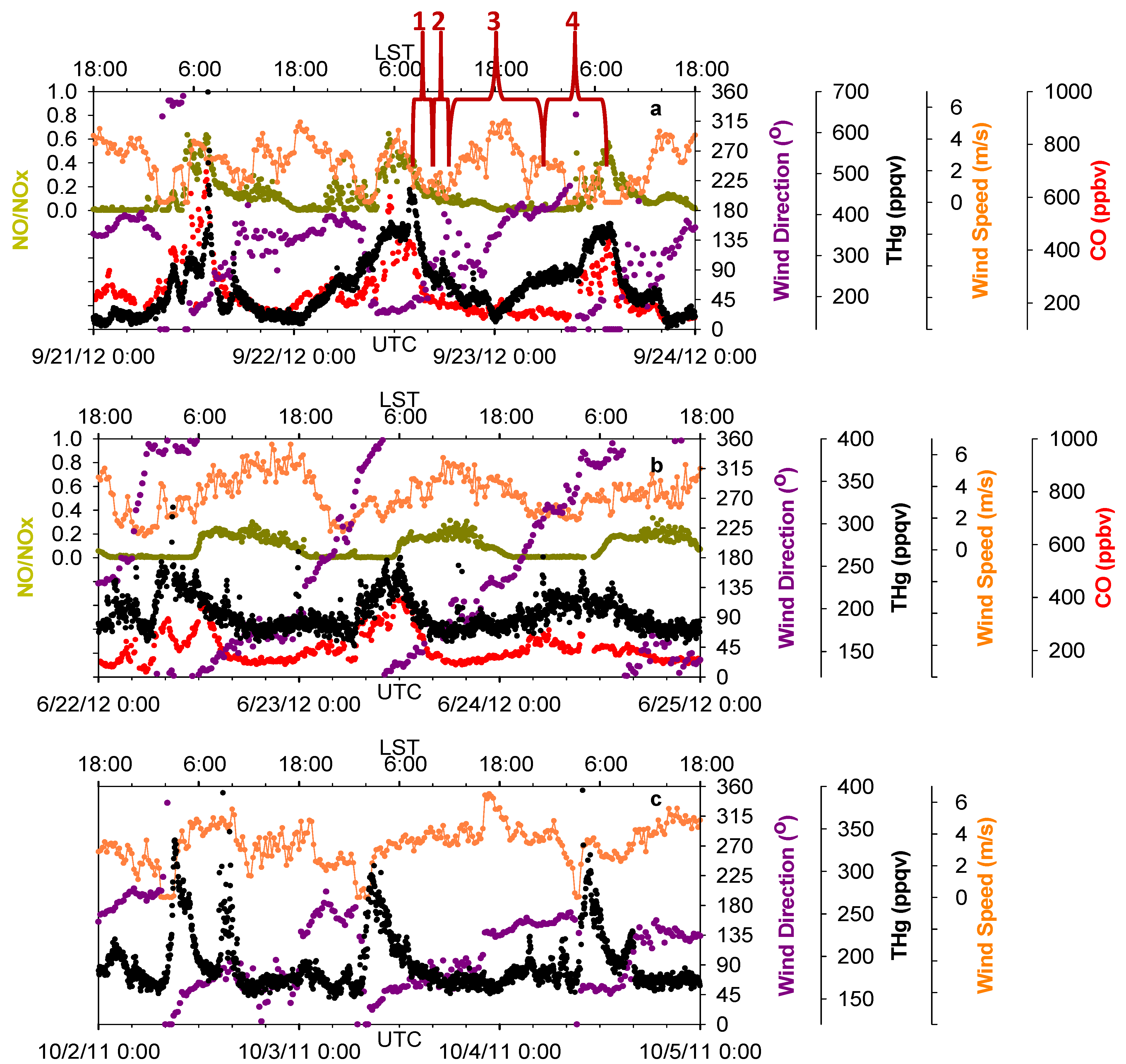
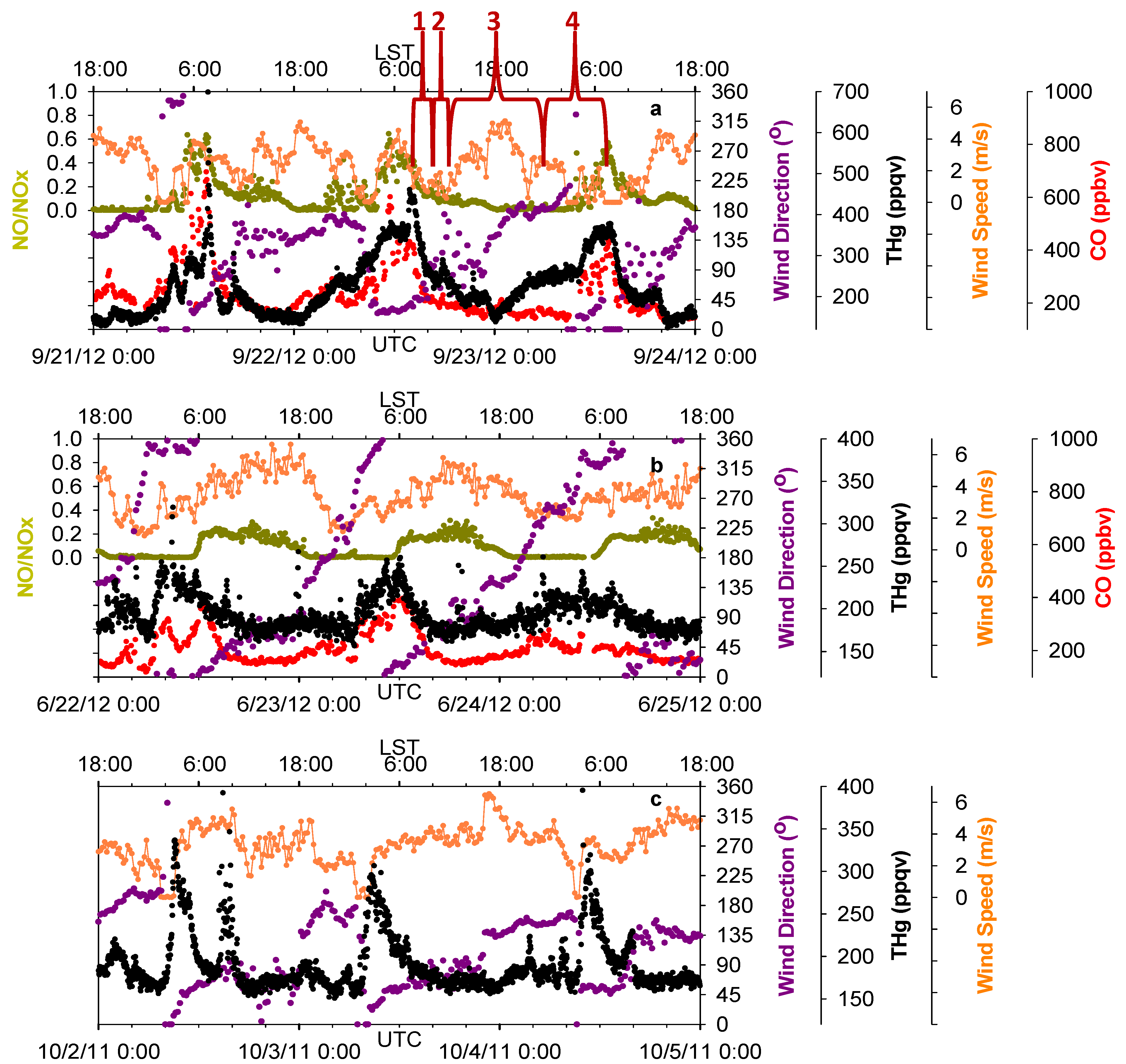
Figure 6.
Time series of THg, CO, NO/NOx, wind speed and wind direction. (a,b,c) show the influences of three common wind patterns on THg levels.
Figure 6.
Time series of THg, CO, NO/NOx, wind speed and wind direction. (a,b,c) show the influences of three common wind patterns on THg levels.
Our observations found similar effects from the bay breezes (from Trinity Bay and Galveston Bay, southeast of MT) and the sea breeze (from the Gulf of Mexico, south of MT).
Figure 6a presents an example of sea breeze impacts on THg diurnal variations. Four stages of THg variations with corresponding wind shifts are marked for illustration. In stage one, THg mixing ratios decreased significantly, partially because of the increase in PBL height, and also the northerly winds that were controlling this area and pushing the urban pollutants offshore. In the second stage, the bay breeze and sea breeze built up and intruded inland as southerly winds. The bay breeze and sea breeze then moved into urban Houston, where they met the northerly flow and formed a sea breeze front. With the influence of a sea breeze front, the horizontal wind speeds decreased to less than 2 m/s, causing a stagnant period, which would not favor vigorous air diffusion or transport. In addition, the polluted flow transported offshore then returned back to Houston with the southerly winds. We observed slightly elevated THg and CO in this period. It is known that NO/NO
x can serve as an indicator for the relative age of air masses, because of the short life time of NO. The NO/NO
x values in this stage were less than 0.3, indicating the influences of aged air masses in which NO was oxidized. In this stage, a convergent zone formed where sea breeze front was concentrated. As a result, the updraft can bring polluted air to higher altitudes and enable long-distance transport of THg. In the third stage, the sea breeze moved farther inland and cleaner marine air behind the sea breeze front controlled air quality in the Houston area. The THg levels were observed to be the lowest of the day. In the final stage, the sea breeze faded out at night and northerly wind again controlled this area. Significant enhancement of THg occurred within the nocturnal boundary layer, along with high CO and NO/NO
x (~0.6), indicating the influences of fresh urban/industrial emissions. These four stages occurred frequently day-to-day during the warm season.
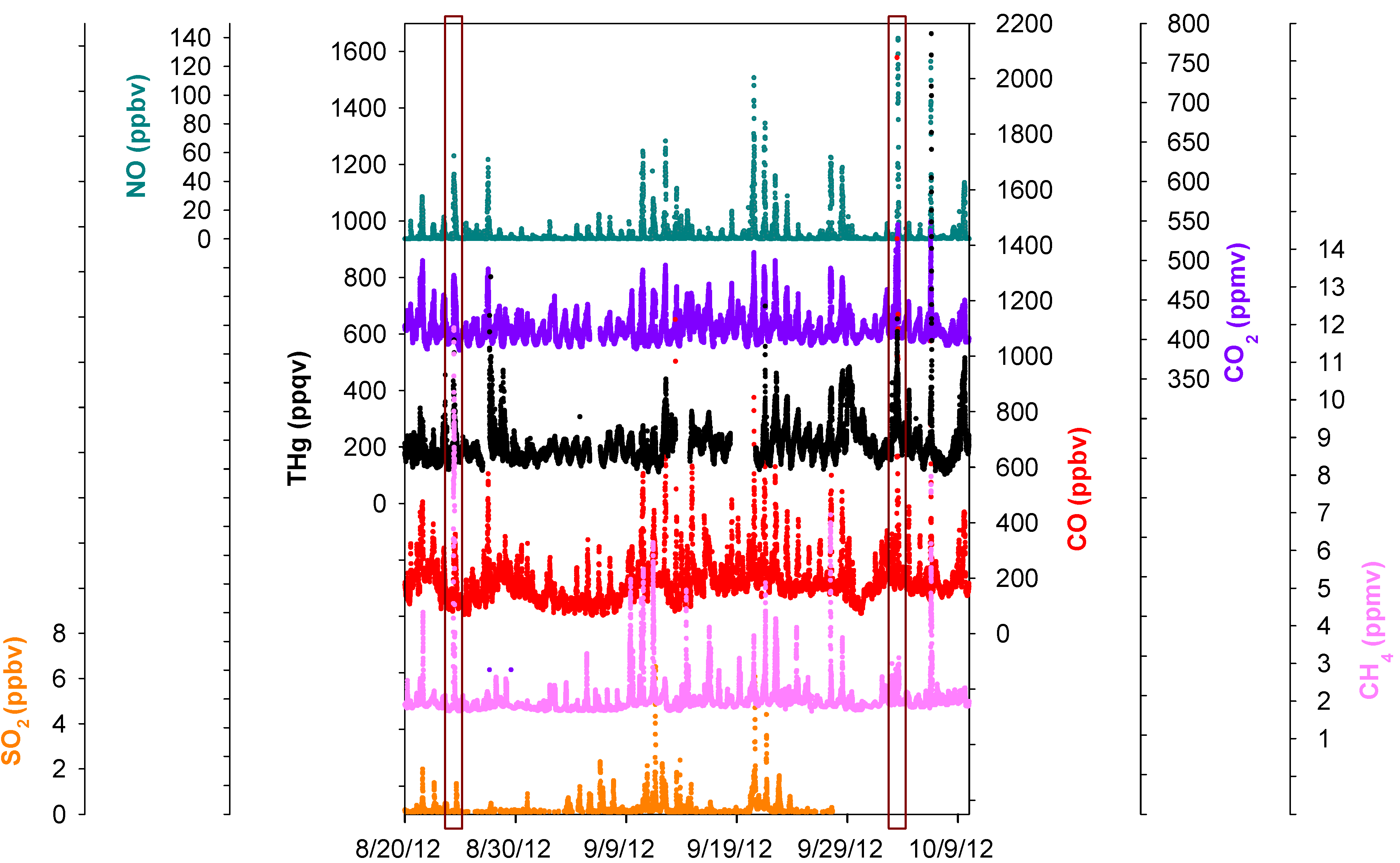
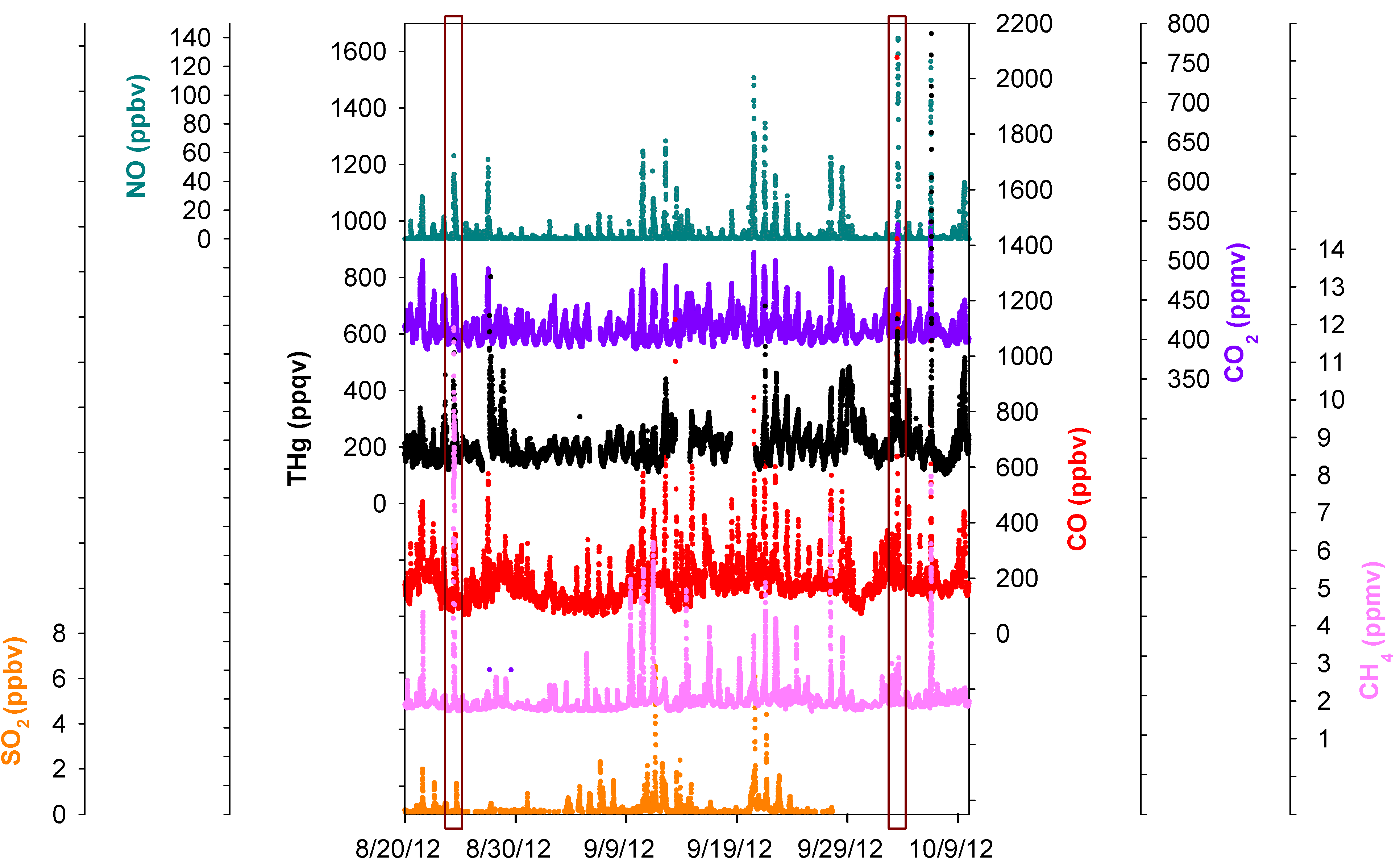
Figure 7.
Time series of THg, CO, CO2, NO, SO2, and CH4. The two boxes are comparisons of two episodes with different CH4 mixing ratios.
Figure 7.
Time series of THg, CO, CO2, NO, SO2, and CH4. The two boxes are comparisons of two episodes with different CH4 mixing ratios.
Another diurnal pattern of THg that also involved the sea breeze is depicted in
Figure 6b. Clockwise rotation of wind directions diurnally were accompanied with obvious THg diurnal changes in which daily low THg mixing ratios occurred when winds were southeast and southerly flow dominated. The diurnally 360° changes in wind direction occurred in March through October, but not in November through February. This pattern was documented on about 30 days in our 14-month data span. Another interesting feature we observed in THg diurnal patterns was that THg levels tended to peak right after abrupt changes in wind direction (45 degree change, normally from south winds to north winds), with few exceptions. This condition occurred on 90 days over 14 months, while the wind shifts occurred mostly around 02:00–04:00 LST (
Figure 6c). Please note that while sea breeze constantly corresponds with relatively low levels of THg in the afternoon and early evening in
Figure 6b,c, the aged plumes were not always observed as in
Figure 6a, which may due to the differences in the strength (relative to northerly) and the daily propagation of the sea breeze.
2.3. Relationship of THg with Key Trace Gases
An advantage of this study was that multiple trace gas species were sampled at the same location (MT) along with THg with high time resolutions. Investigating the relationships between THg and other key trace gases can provide important information concerning mercury sources.
Figure 7 displaced the time series of SO
2, NO, CO, CO
2, CH
4, and THg. A significant feature we noticed was the common co-occurrence of THg peaks with SO
2, NO, CO, CO
2, CH
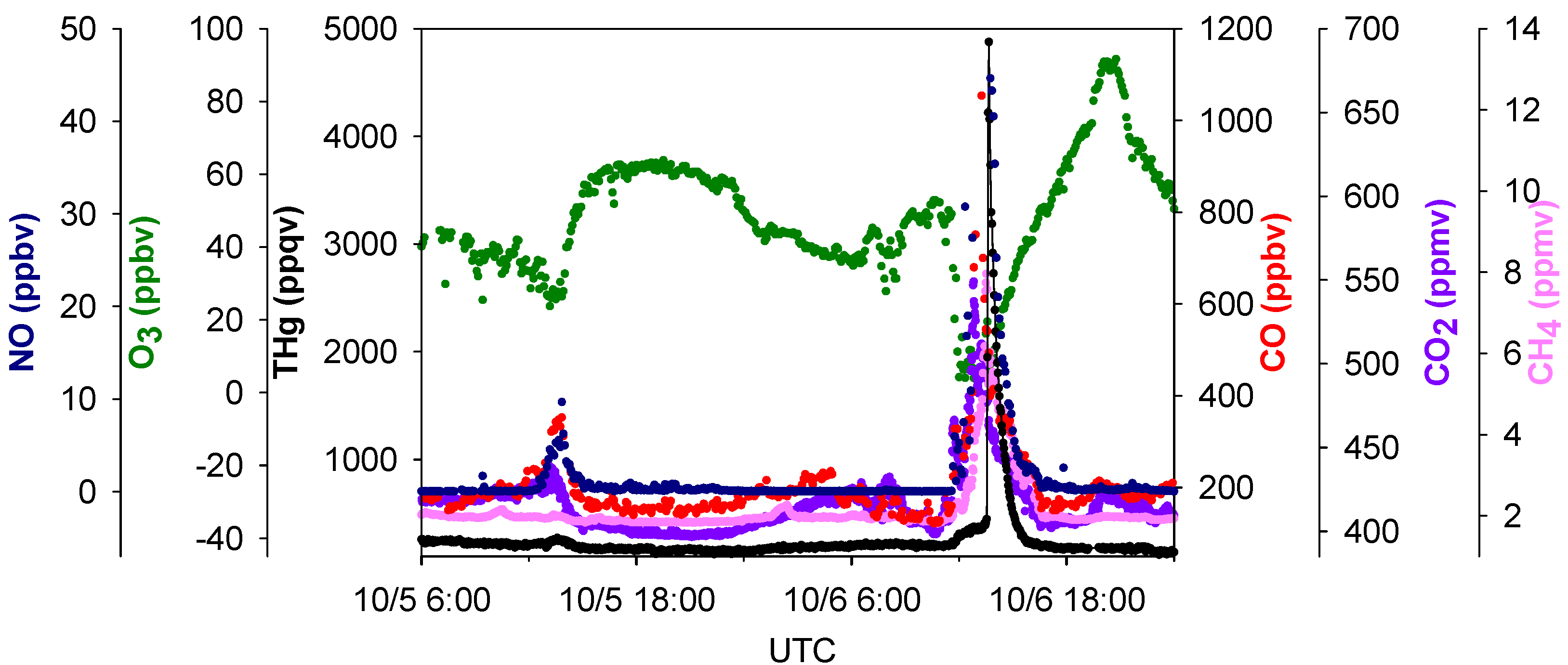
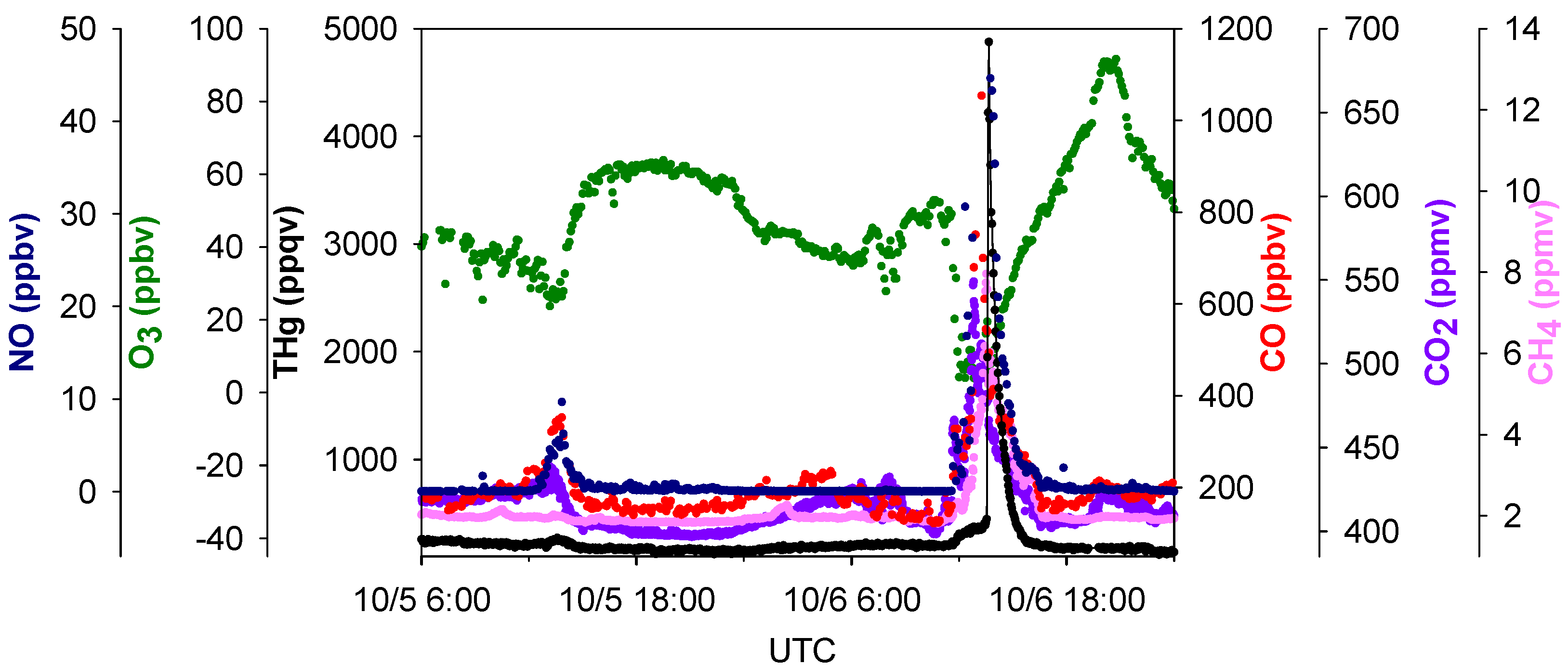
4 peaks in pollution plumes. A zoom-in picture depicting the maximum THg spike observed during this study is presented in
Figure 8. This episode corresponded with air masses coming from the northeast of our sampling site, which is the direction of Houston Ship Channel. This highly concentrated pollution plume with THg mixing ratios as large as 4876 ppqv was related to combustion tracers, such as CO, CO
2, and NO. In addition, this THg peak occurred coincidentally with a peak in CH
4, which is presumably released from oil and natural gas operations, landfills, and waste treatment facilities. In this episode significant enhancements of NO, THg, CO, CO
2, and CH
4 started at 04:30 LST and peaked at 06:45–07:45 LST. The maximum levels of each species in this plume were 4876 ppqv for THg, 1053 ppbv for CO, 45 ppbv for NO, 549 ppmv for CO
2 and 8.0 ppmv for CH
4. The close correspondence between THg and other species in the THg episodes is clearly evident, and thus it is possible that THg came from similar sources as some of the other trace gases. However, the peaks signatures varied greatly in different pollution plumes. Extreme THg peaks sometimes were in conjunction with exceptional large peaks of CH
4 (e.g.,
Figure 7 left box, 11.8 ppmv), but sometimes with slightly elevated CH
4 (e.g.,
Figure 7 right box, 3.1 ppmv). Correlations of THg with other trace gases were diffuse overall; correlation coefficients (R values) between THg and those species were low (R < 0.43, see Table S2) albeit their correlations were statistically significant (95%), suggesting the possible contributions from diverse source types in the Houston area.
Figure 8.
Time series of THg, CO, CO2, NO, O3 and CH4 in the largest THg plume.
Figure 8.
Time series of THg, CO, CO2, NO, O3 and CH4 in the largest THg plume.
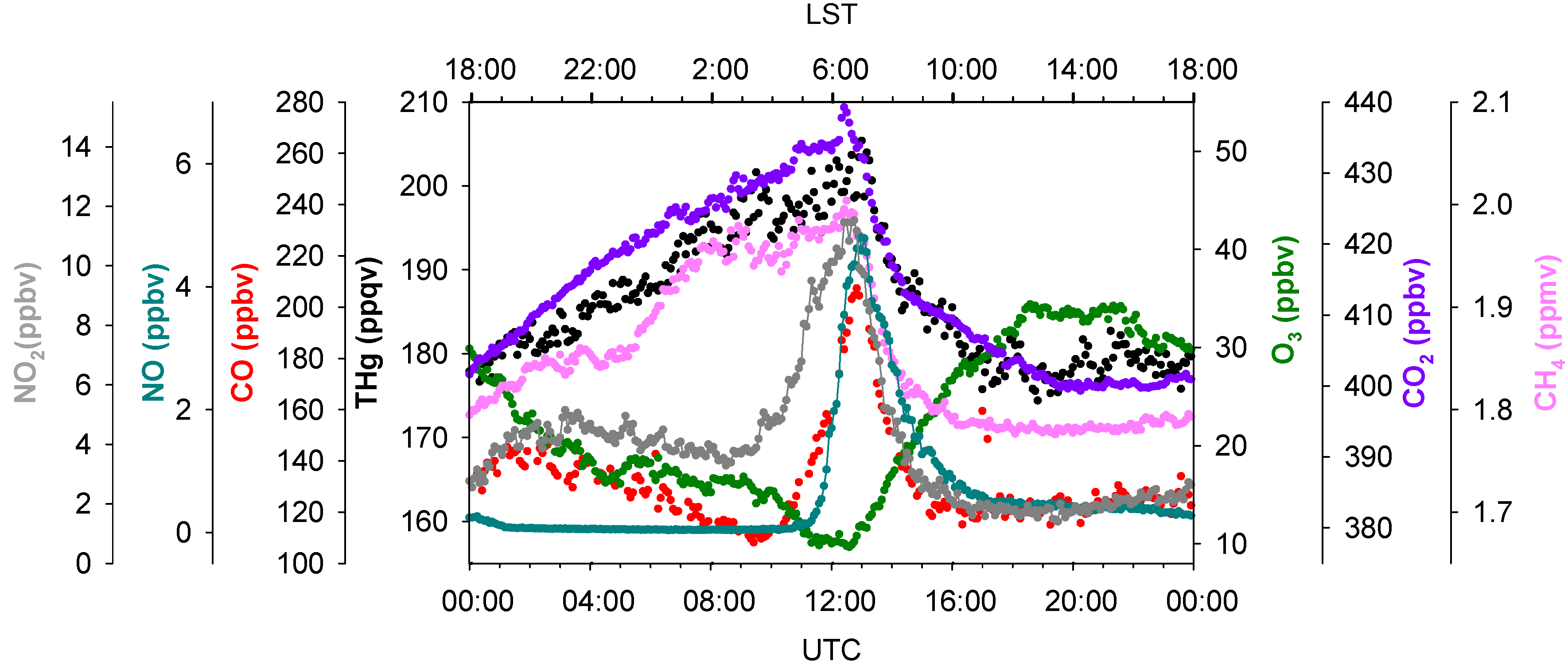
Detailed analyses of diurnal variations of THg and other species can also provide valuable information on mercury sources and sinks.
Figure 9 exhibits the summertime diurnal variations of THg, NO, NO
2, CO, O
3, CO
2, and CH
4. Interestingly, THg, CO
2 and CH
4 had similar diurnal patterns; however, it is still difficult to determine whether these patterns were resulting from meteorological forcing, such as PBL height and winds, and/or their similar emission sources. The diurnal patterns of NO, NO
2, and CO, were ubiquitously distinguished from those of THg, CO
2, and CH
4. Vehicle emissions are believed to be the dominant sources of NO, NO
2, and CO in urban areas [
48,
49]. We noticed that the mixing ratios of CO and NO
2 started increasing simultaneously at 03:30 LST (04:30 Local Daylight Saving Time) and reached their maximums at 06:00–07:00 LST. The mixing ratios of NO started increasing after sunrise, about two hours later compared to NO
2 and CO. Ozone titration was also observed at the same time when elevated CO and NO
2 occurred (note that O
3 photochemistry in this site is limited by VOCs, instead of NO
x [
50]). We believe these were traffic signals; NO was emitted by vehicles subsequently titrating O
3 and being converted to NO
2, but without sunlight NO
2 builds up. The diurnal pattern of THg tracked those of CO
2 and CH
4, instead of CO and NO
2, suggested that vehicle emissions may not exert outstanding impacts on THg levels in summer. The importance of vehicle emissions as a THg source is yet to be further investigated.
Figure 9.
Diurnal variations of THg, CO, CO2, NO, NO2, O3 and CH4 in summer 2012. Data shows the median values within a 5 min. time interval.
Figure 9.
Diurnal variations of THg, CO, CO2, NO, NO2, O3 and CH4 in summer 2012. Data shows the median values within a 5 min. time interval.
2.4. Mercury Episodes
As we reported previously, 81 spikes were documented with THg mixing ratios greater than 300 ppqv. We calculated the enhancement ratios (ER) in these plumes to retrieve information about point sources. Enhancement ratios are obtained by dividing the excess species (THg in this study) concentrations measured in a pollution plume by the excess concentration of a reference gas, for example, CO
2, CO and CH
4 in this study. Enhancement ratios are commonly expressed in molar ratios, and the ambient background levels of each gas must be subtracted to get the “excess” values. For example, the ER for THg relative to CO is:
Enhancement ratio is a different parameter compared to emission ratio because the measurement is conducted from a downwind location instead of at the source. Since THg, CO, CO
2 and CH
4 are not highly reactive species, it is possible that ERs remain close to emission ratios of a specific source in short-distant transport. Further we may assume that pollution plumes coming from the same direction were from the same emission source if they have similar ERs. For ER calculation, we characterized the air masses with the lower 25th percentiles of CO mixing ratios as background air [
23], and the corresponding medians values for this part of data were used as the background levels so as to minimize the possible influence of local sinks. Those values were 172 ppqv for THg, 112.6 ppbv for CO, 1.79 ppmv for CH
4, and 403.3 ppmv for CO
2.
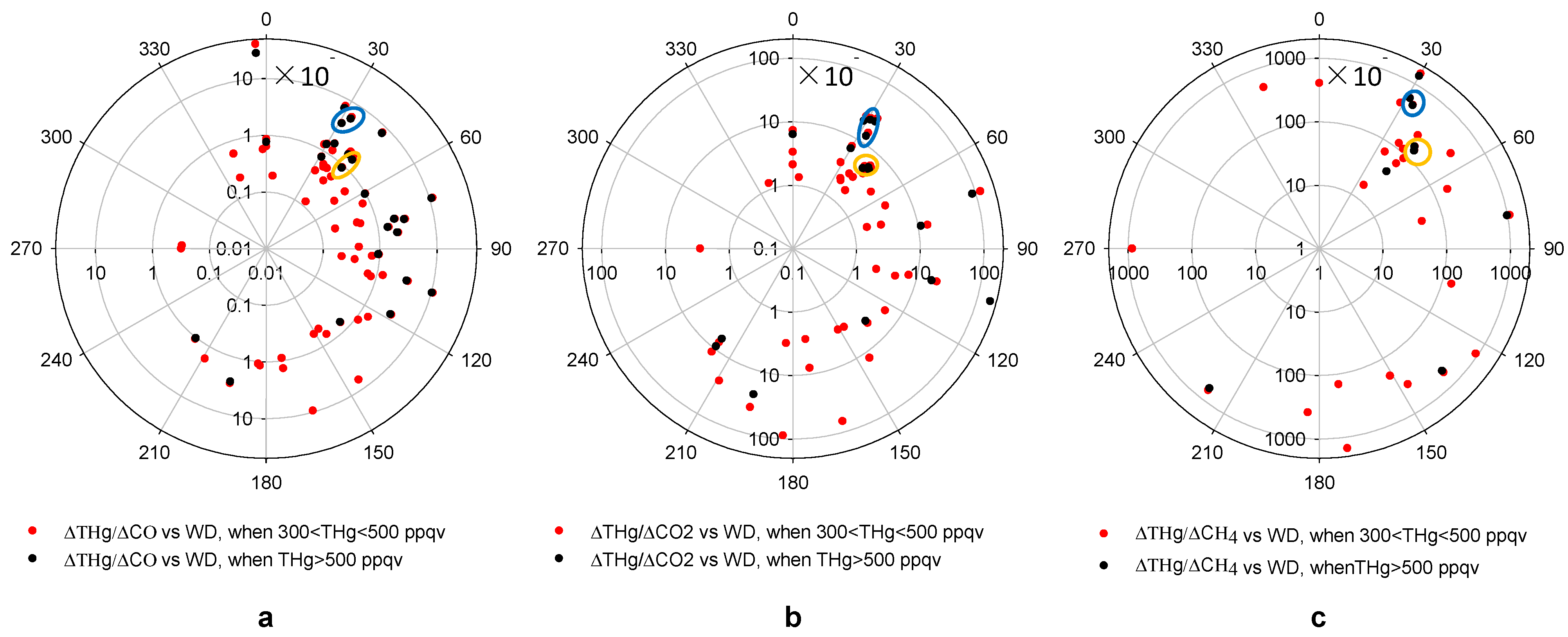
Figure 10 presents the ER values for ∆THg/∆CO, ∆THg/∆CO
2 and ∆THg/∆CH
4,
versus wind directions. The CO
2 measurements are only available from March 2012 and CH
4 measurements from June 2012. Thus, fewer episodes were available for ∆THg/∆CO
2 and ∆THg/∆CH
4 compared to ∆THg/∆CO.
The patterns of ∆THg/∆CO and ∆THg/∆CO
2 versus wind direction shared some similarities (
Figure 10). Pollution plumes with THg mixing ratios higher than 500 ppqv tended to produce higher ERs, compared to those with THg mixing ratios between 300 ppqv and 500 ppqv. A large percentage of THg episodes with high CO or CO
2 mixing ratios originated from the 30°–120° direction, the Houston Ship Channel area. Note that, from
Figure 5a, air masses from this direction only accounted for about 17% of the total air mass measured at MT. This means that most THg episodes originated from directions that were the least favored wind directions.
Figure 10.
Enhancement ratios versus wind direction in high THg episodes.(a,b,c) show ∆THg/∆CO, ∆THg/∆CO2 and ∆THg/∆CH4 versus wind direction, respectively. The yellow circles point out two ERs potentially from the same source at ~40° direction and the blue circles represent another two episodes from a source at ~30° direction.
Figure 10.
Enhancement ratios versus wind direction in high THg episodes.(a,b,c) show ∆THg/∆CO, ∆THg/∆CO2 and ∆THg/∆CH4 versus wind direction, respectively. The yellow circles point out two ERs potentially from the same source at ~40° direction and the blue circles represent another two episodes from a source at ~30° direction.
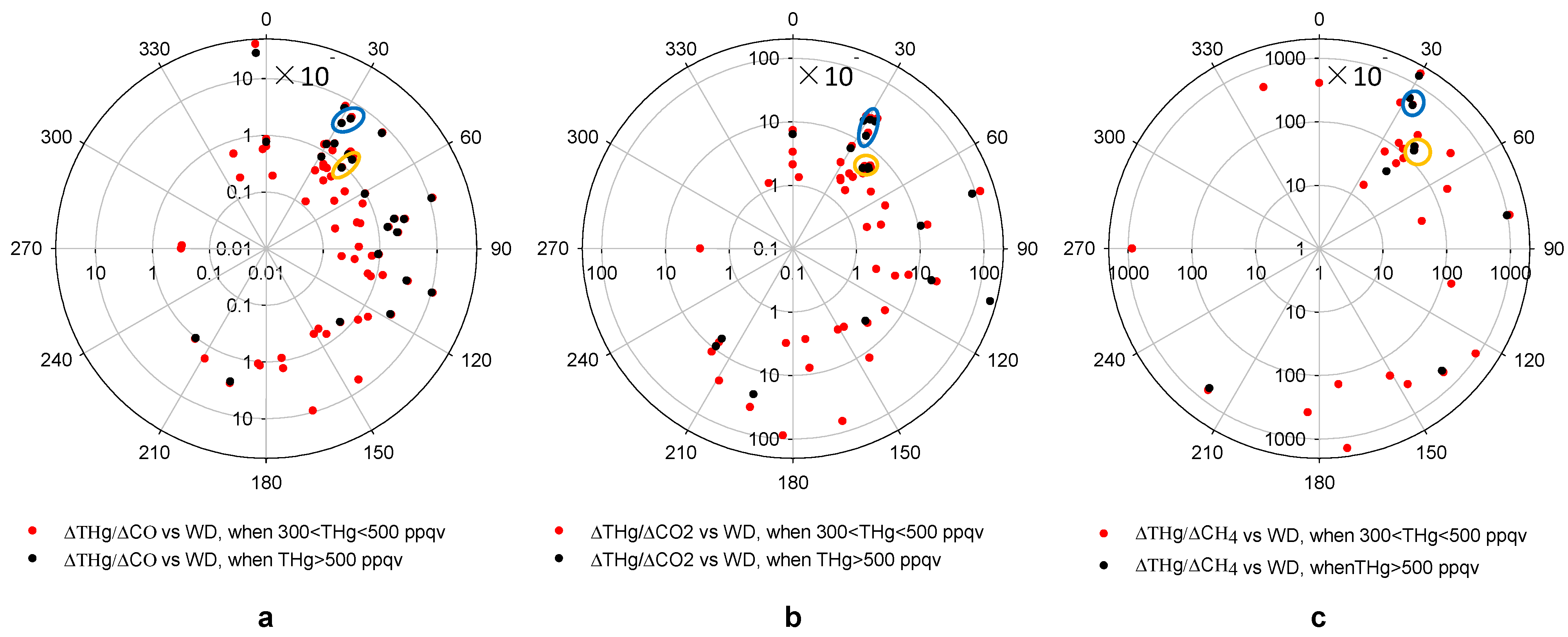
For now, we are unable to correlate these pollution plumes with specific sources due to the absence of detailed and accurate source signatures. The EPA NEI [
13] provides annual total emissions for THg and CO, and the Greenhouse Gases (GHGs) Emissions Data [
51] also provides annual total emissions of CO
2 and CH
4 for significant emission sources. We attempted to compare our ER values with emission ratios calculated from the EPA inventory. In the northeast direction from MT, the direction with a large number of THg episodes, we found only 2 point sources within a short distance (<80 km) from MT that were documented in the 2008 EPA NEI as mercury sources. From the ER
versus wind direction plot, we noticed 2 episodes with similar ∆THg/∆CO, ∆THg/∆CO
2 and ∆THg/∆CH
4 values from the 40° direction (
Figure 10, yellow circles). Considering the large elevations of THg, CO, CO
2 and CH
4 in these episodes, we suspect that they could be attributed to local sources. A waste treatment facility located about 5 km away from MT in that direction was reported by the 2008 NEI and 2010 GHGs Emissions Data as a source for THg, CO, CO
2 and CH
4. The other point source that was likely associated with another 2 episodes with similar ∆THg/∆CO, ∆THg/∆CO
2 and ∆THg/∆CH
4 values were from the 30° direction (
Figure 10, blue circles). We found an iron and steel casting facility from EPA NEI 9 km away from MT.
Table 1 shows the comparisons between ER values calculated from observations and emission ratios calculated from the EPA inventory (please note that this iron and steel casting facility is not listed in the 2010 GHGs Emissions Data, which may be due to the fact that the 2010 GHGs Emissions Data does not include sources that have annual emissions of less than 25,000 metric tons of CO
2e. For the emission ratios calculations, we assume the CO
2 and CH
4 emissions from this facility were 25,000 metric tons of CO
2e). The differences between these two ratios were significant, up to a few orders of magnitude. It is highly possible that the EPA emission inventory may have inaccurate estimations of the total emissions. It is also possible that some emission sources may not be documented in the EPA inventory.
Table 1.
Comparisons between ER values derived from MT observations and emission factors calculated from EPA Inventory.
Table 1.
Comparisons between ER values derived from MT observations and emission factors calculated from EPA Inventory.
| Facility | ∆THg/∆CO | ∆THg/∆CO2 | ∆THg/∆CH4 |
|---|
| EPA | Observation | EPA | Observation | EPA | Observation |
|---|
30°, Iron and steel
casting (blue circles) | 1.77 × 10−5 | 8.9 × 10−7~1.49 × 10−6 | 5.04 × 10−7 | 5.3 × 10−9 | 9.78 × 10−3 | 1.40 × 10−7 |
| 40°, Waste treatment (yellow circles) | 5.70 × 10−8 | 3.7 × 10−6 ~5.4 × 10−6 | 1.77 × 10−10 | 1.3 × 10−8~2.3 × 10−8 | 1.35 × 10−9 | 5.0 × 10−7~5.8 × 10−7 |



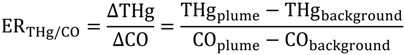

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}