Recent Selection on a Class I ADH Locus Distinguishes Southwest Asian Populations Including Ashkenazi Jews

Abstract
:1. Introduction
2. Materials and Methods
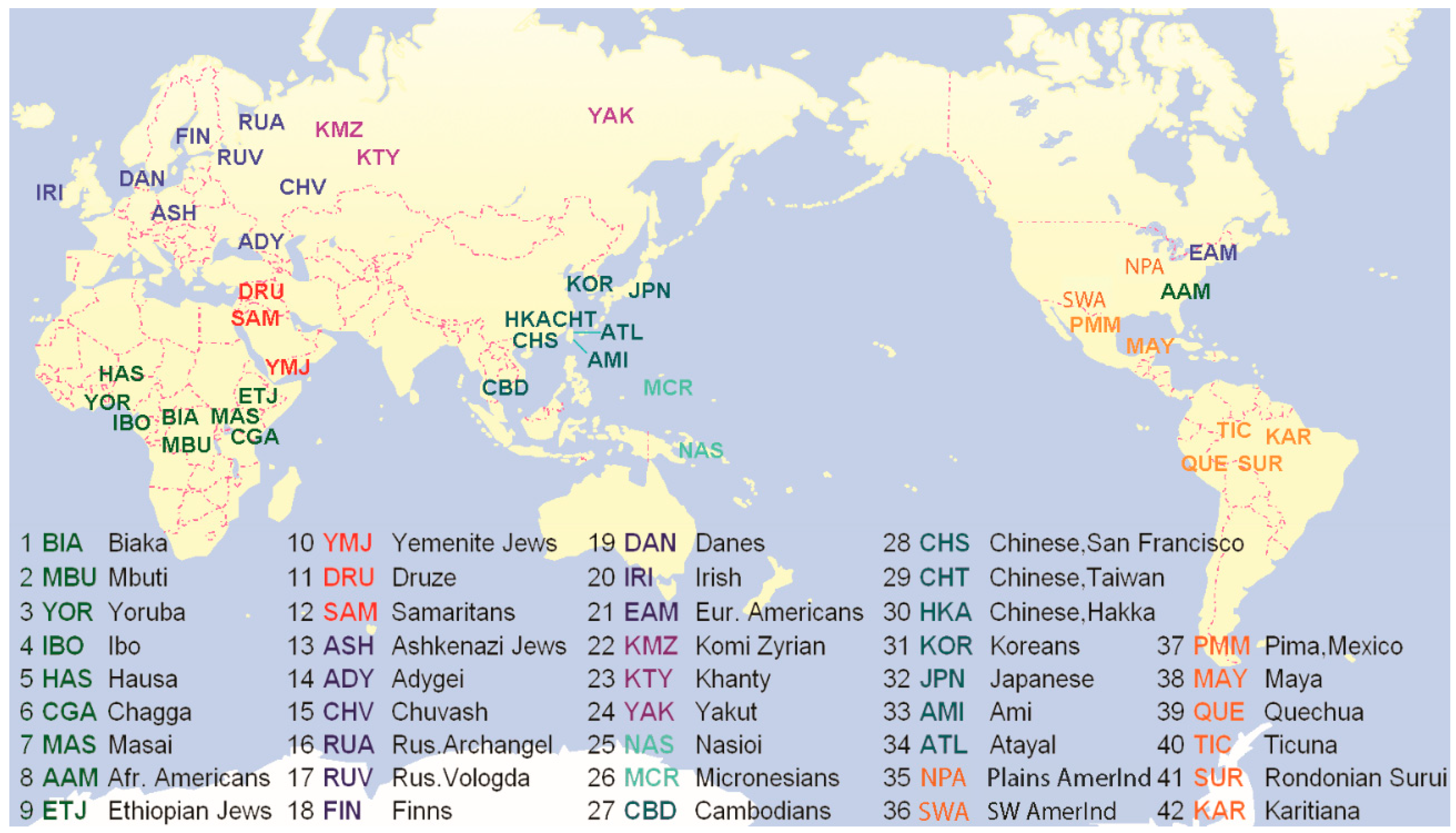
2.1. Subjects
2.2. Polymorphic Sites around ADH Clusters
2.3. Linkage Disequilibrium Pattern
2.4. Haplotype Inference
2.5. Test for Recent Positive Selection
2.6. Inference on Human Evolution
2.7. Simulations
3. Results
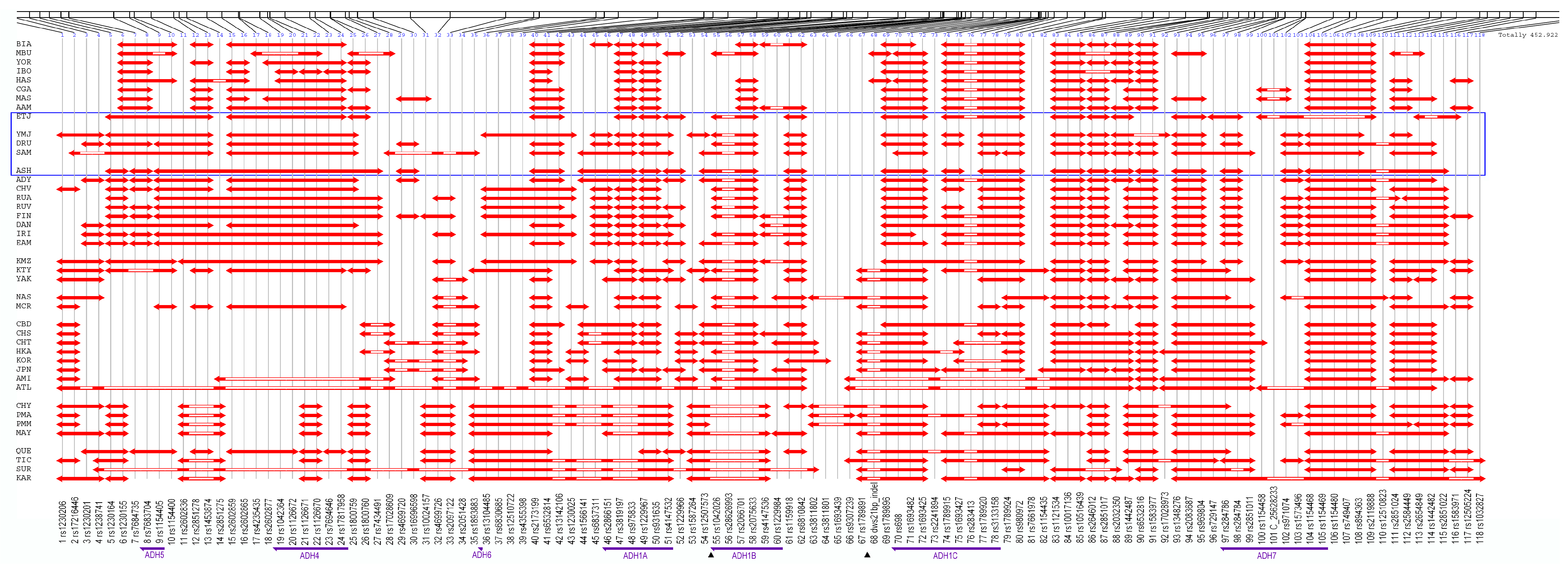
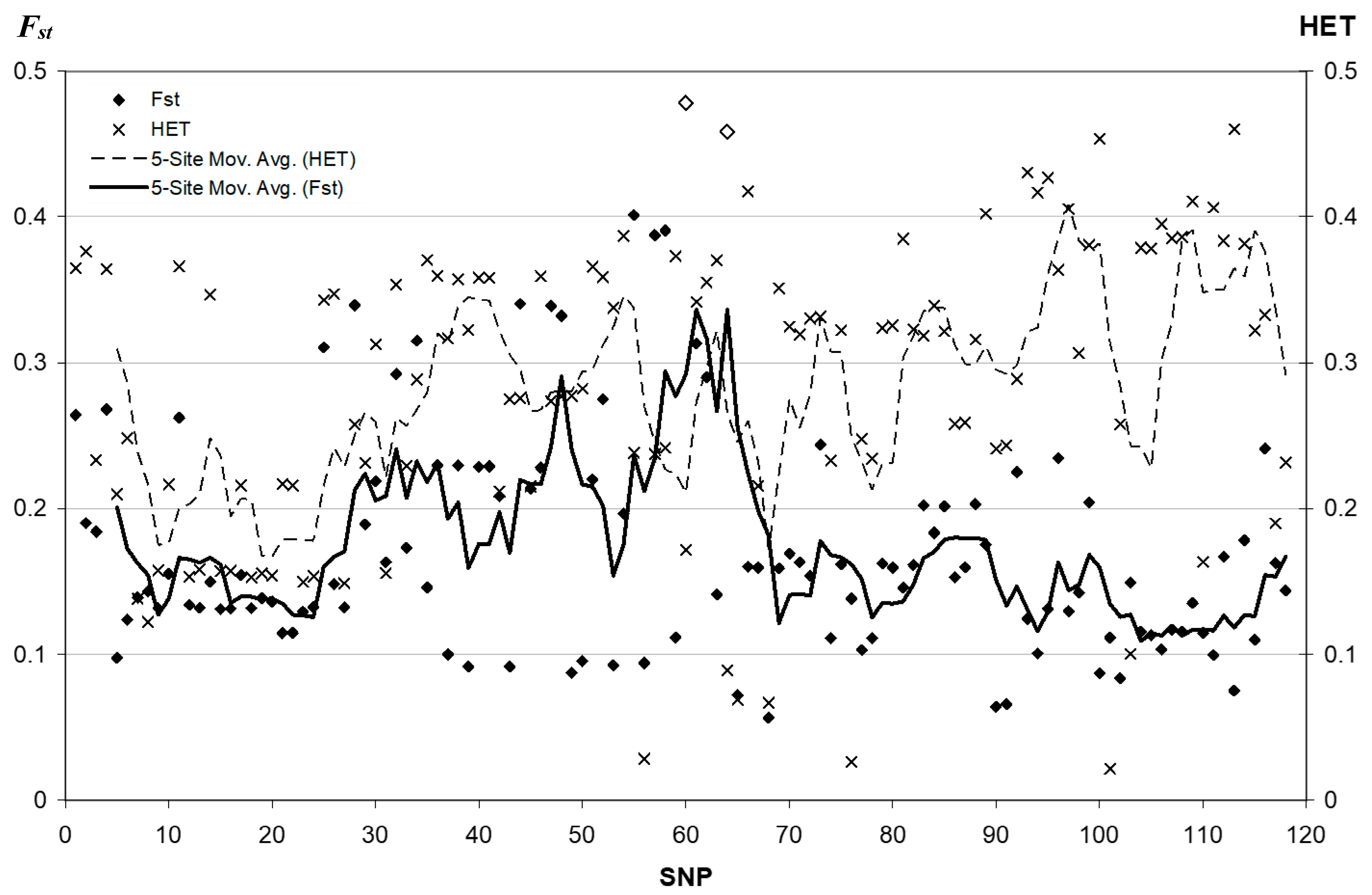
3.1. Linkage Disequilibrium Pattern
3.2. Fixation Index Distribution
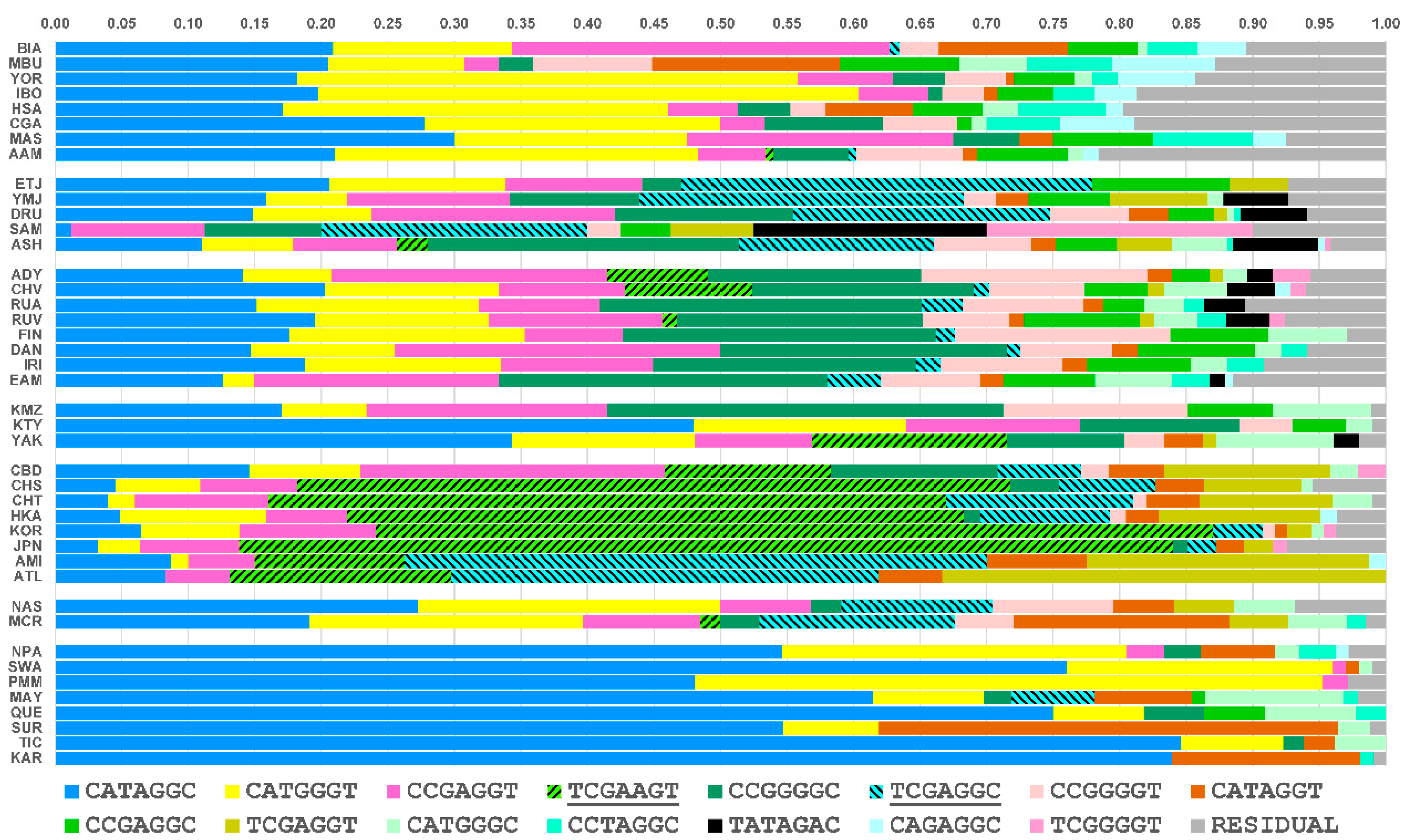
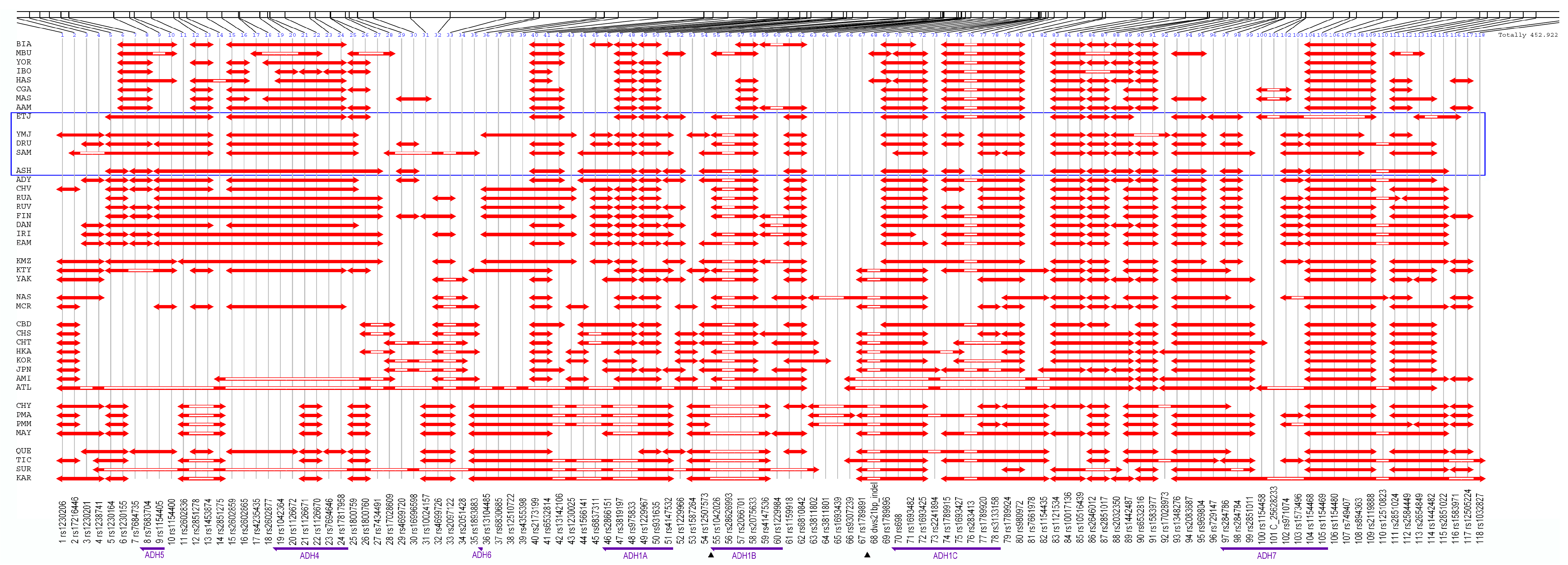
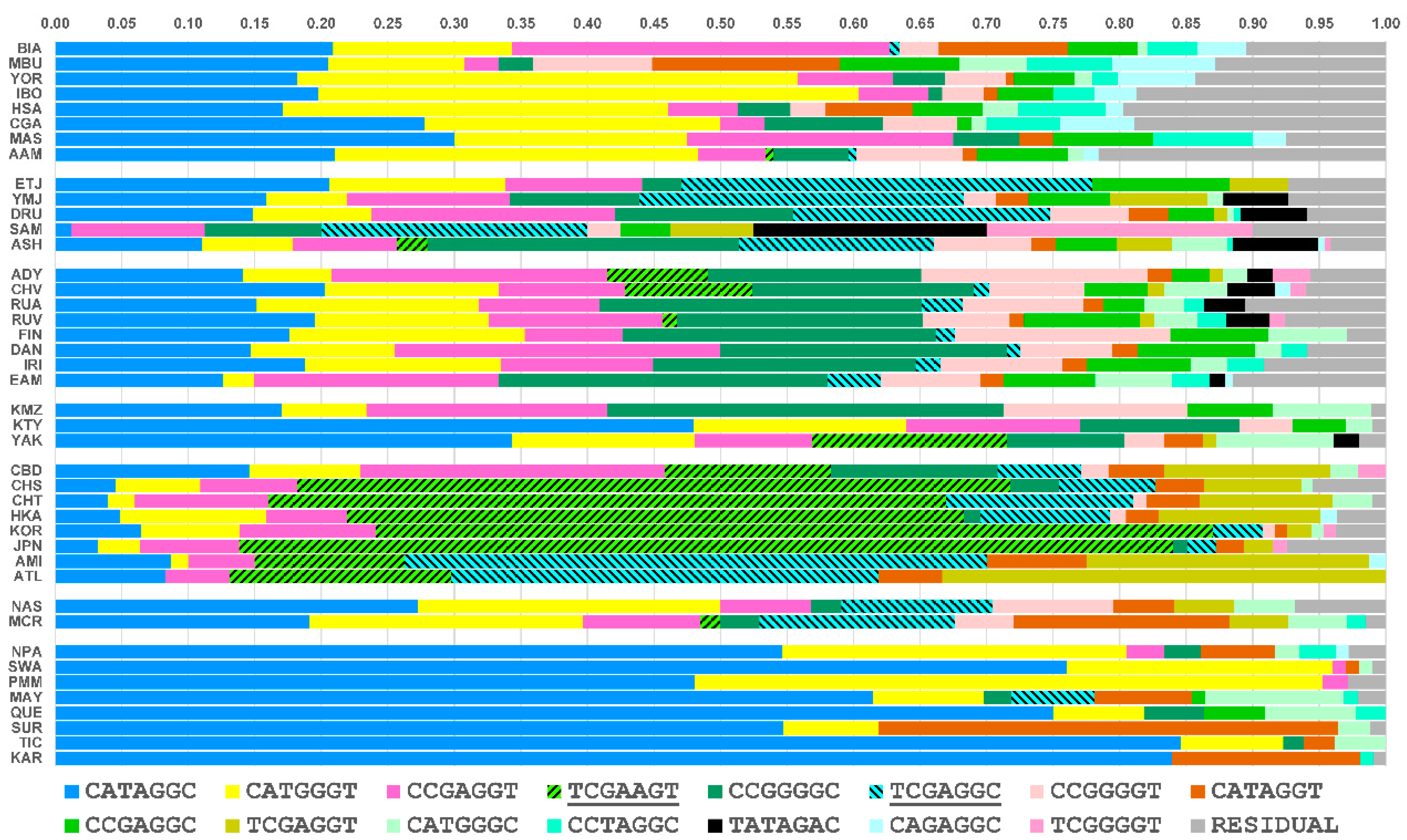
3.3. Core Haplotype Pattern
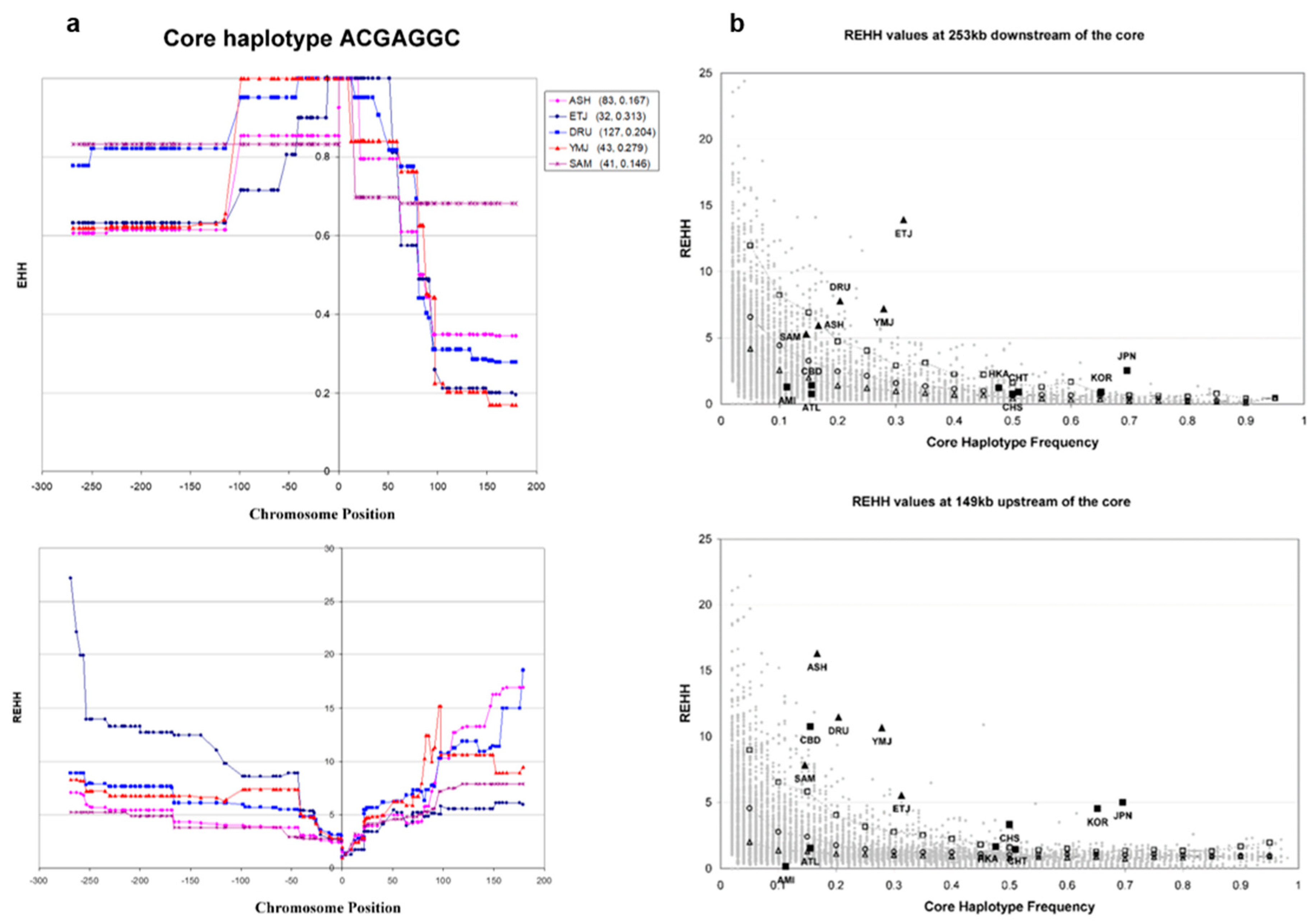
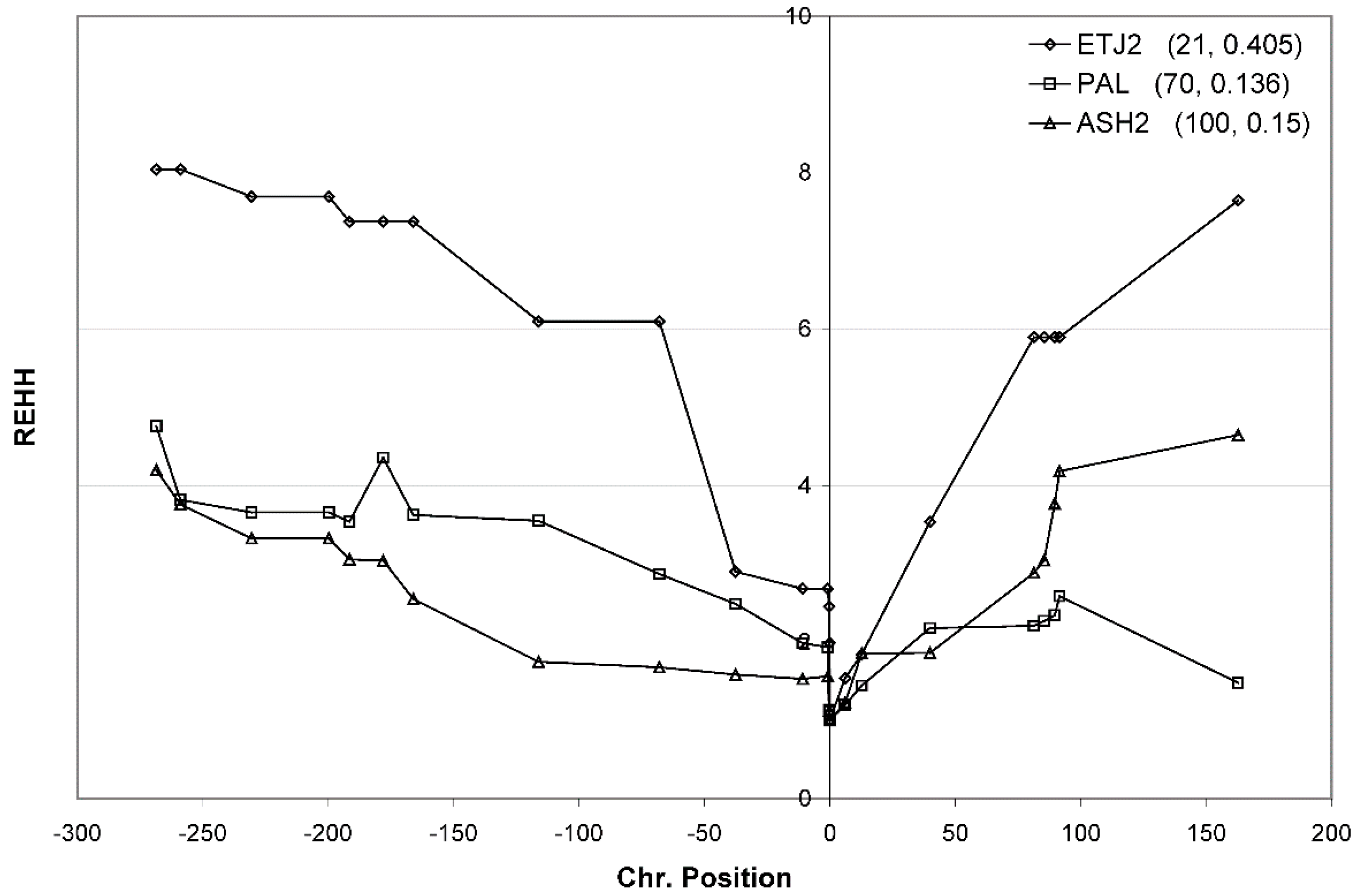
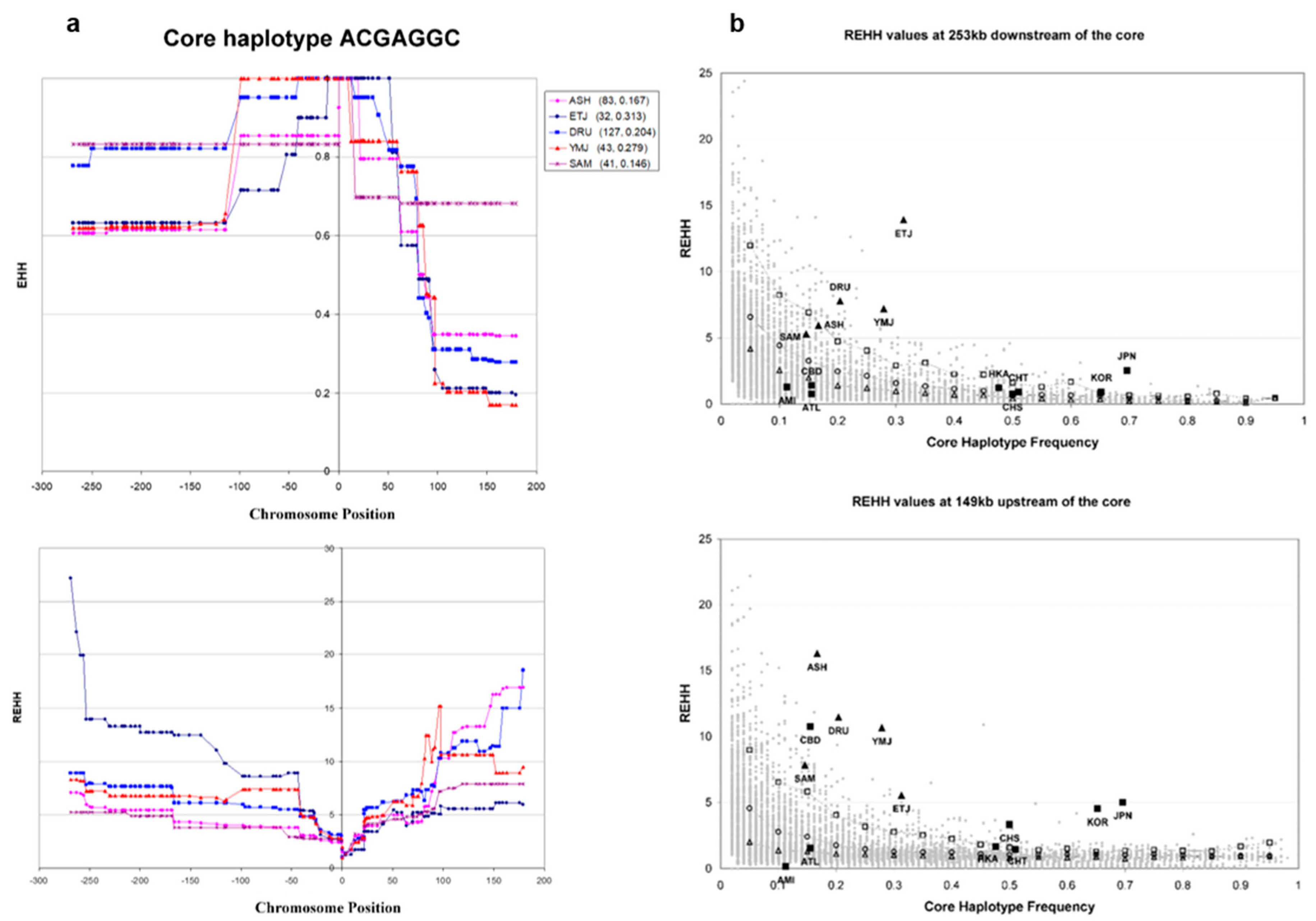
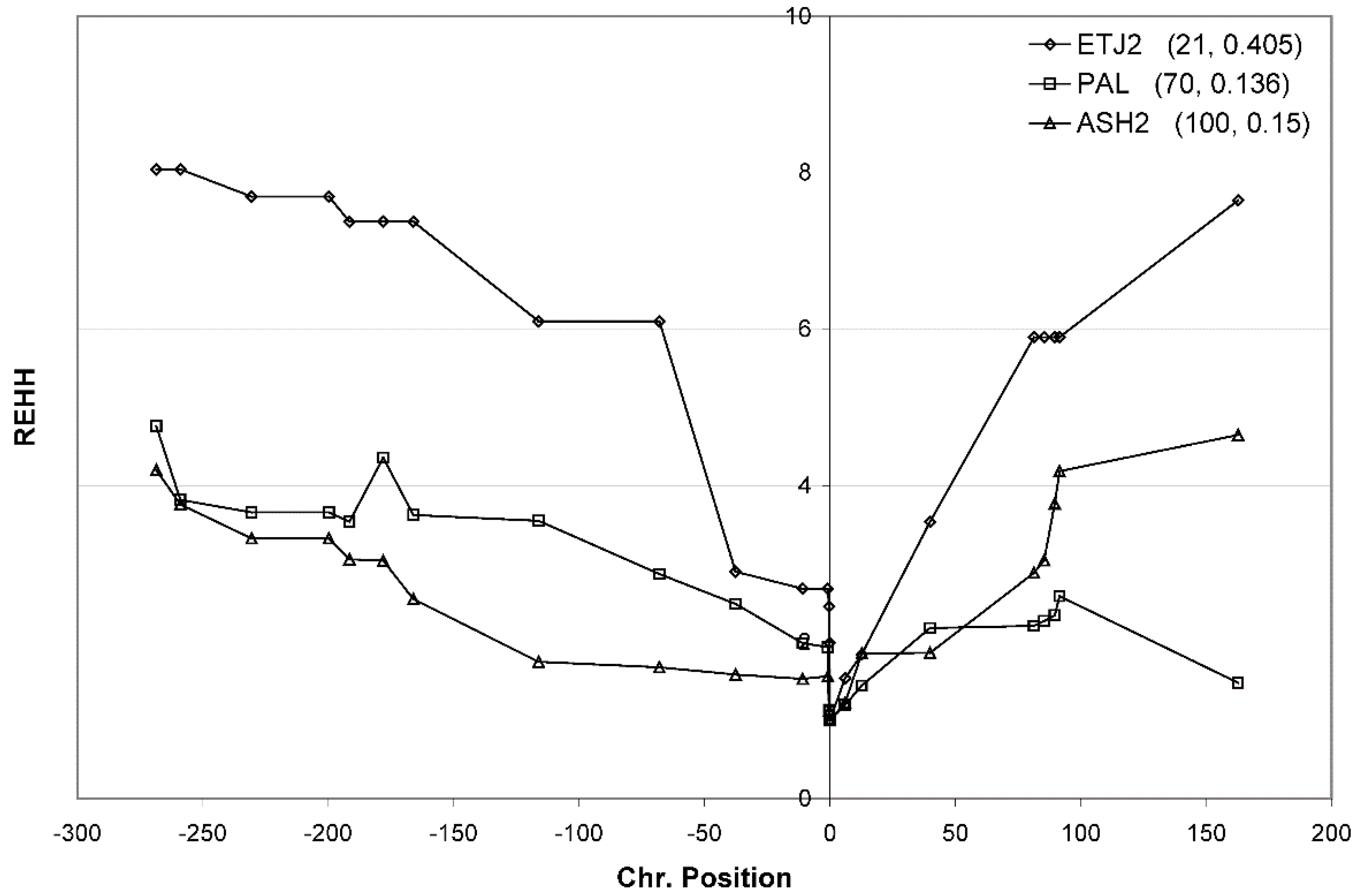
3.4. Haplotype Homozygosity and Relative Haplotype Homozygosity
3.5. Independent Selection
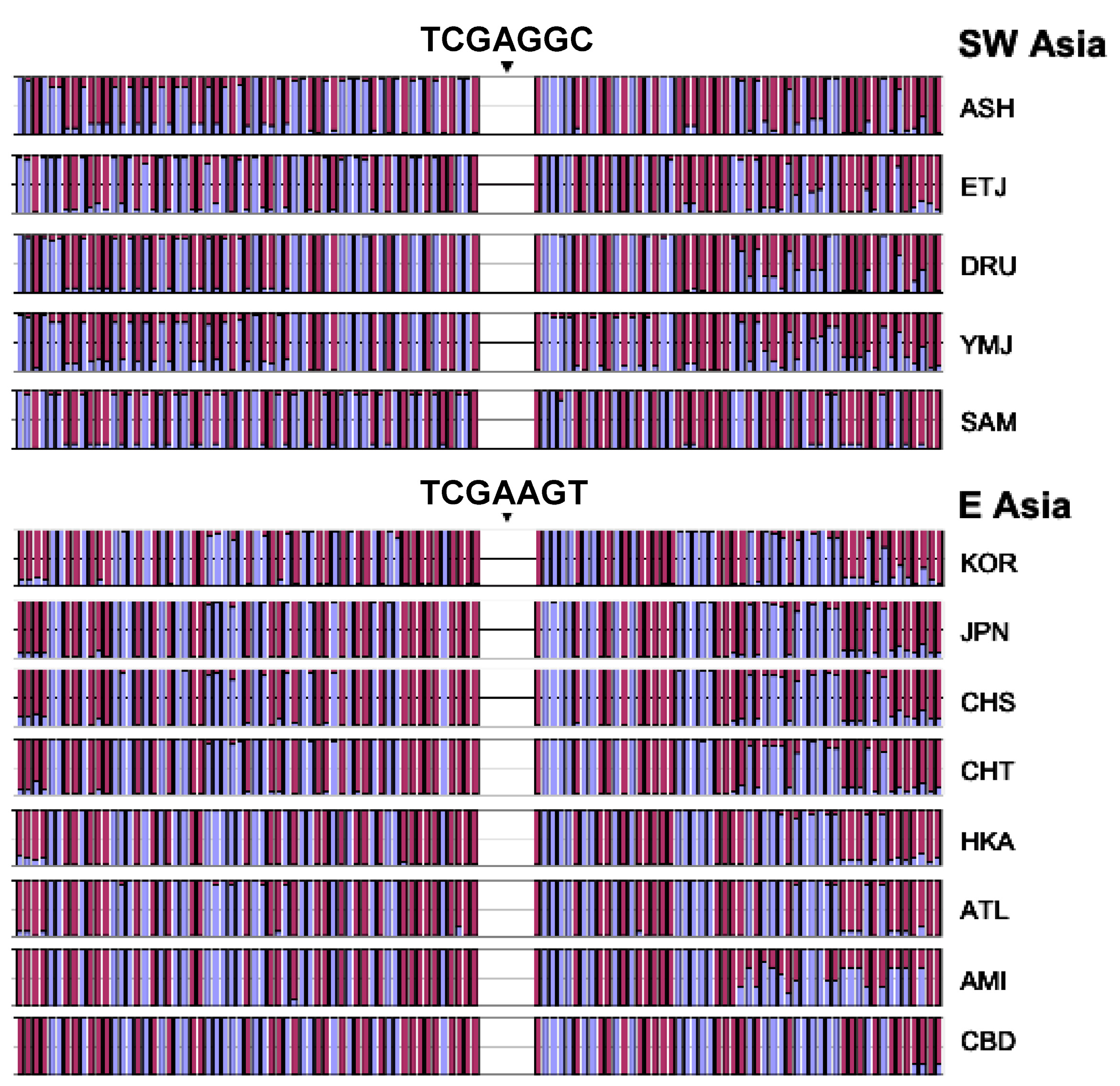
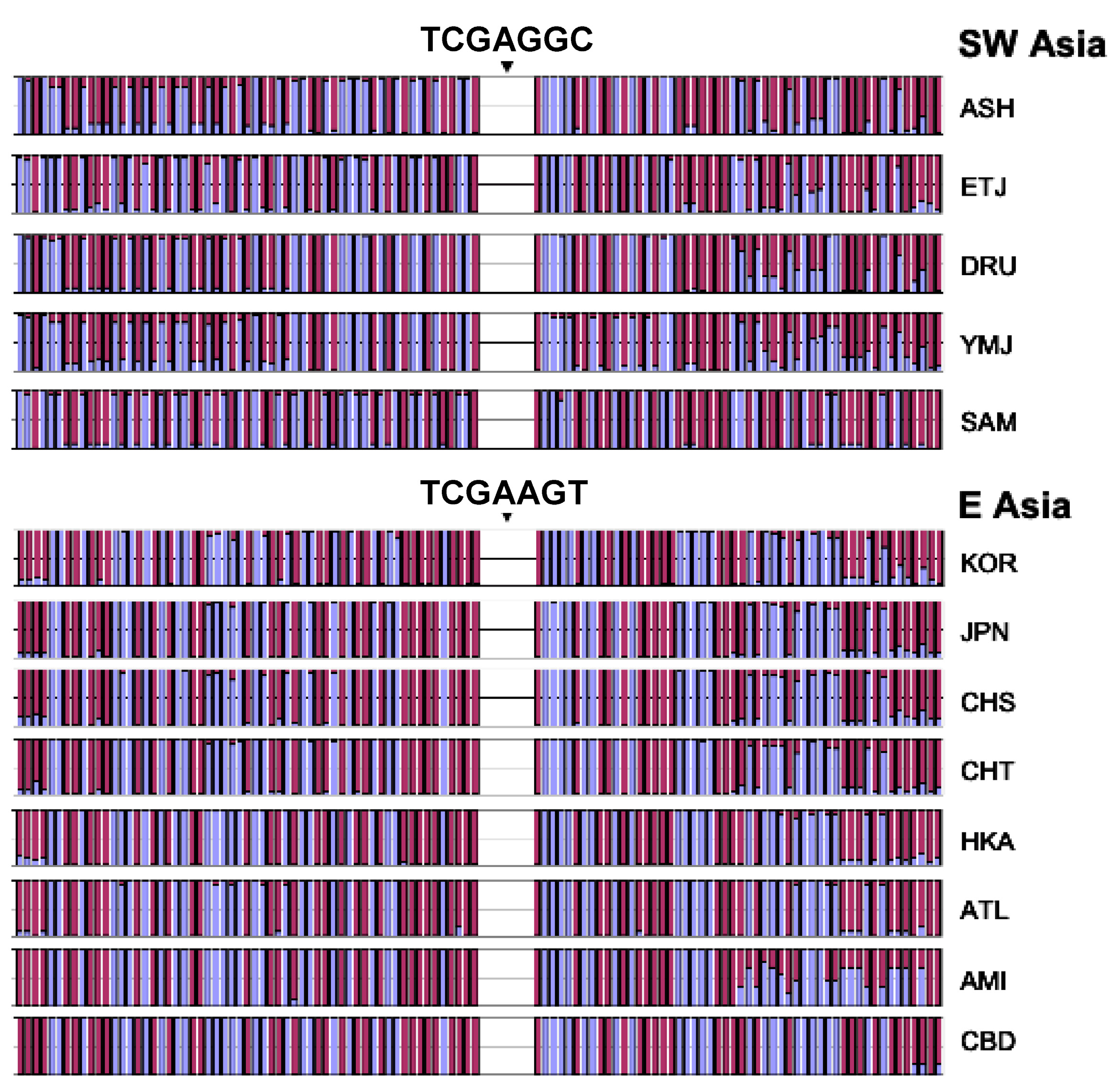
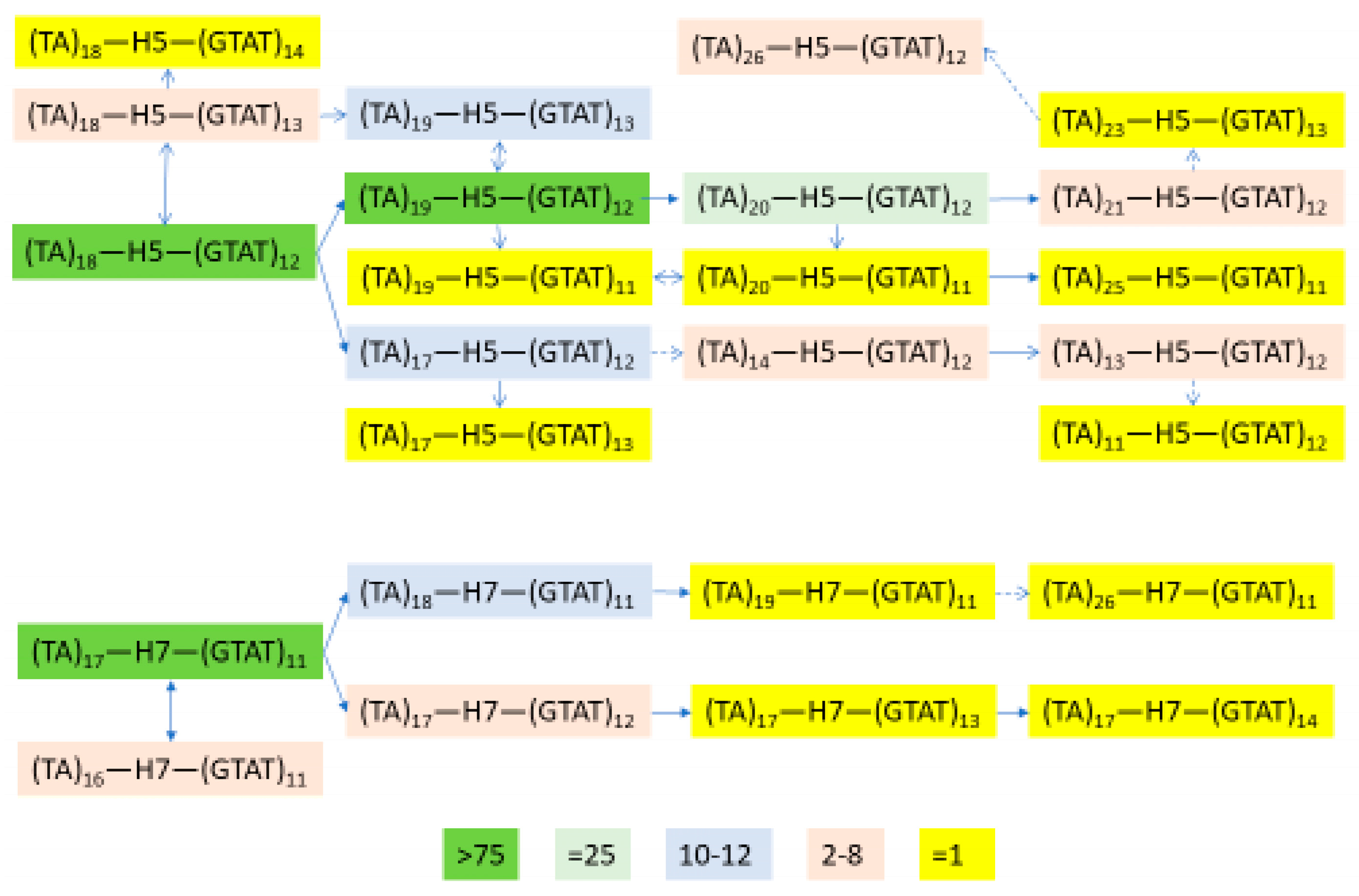
3.6. Independent Mutation
3.7. Confirmation of Haplotype Homozygosity and Relative Haplotype Homozygosity Patterns in Independent Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thomasson, H.R.; Edenberg, H.J.; Crabb, D.W.; Mai, X.L.; Jerome, R.E.; Li, T.K.; Wang, S.P.; Lin, Y.T.; Lu, R.B.; Yin, S.J. Alcohol and aldehyde dehydrogenase genotypes and alcoholism in Chinese men. Am. J. Hum. Genet. 1991, 48, 677–681. [Google Scholar] [PubMed]
- Mizuno, Y.; Harada, E.; Morita, S.; Kinoshita, K.; Hayashida, M.; Shono, M.; Morikawa, Y.; Murohara, T.; Nakayama, M.; Yoshimura, M.; et al. East Asian variant of aldehyde dehydrogenase 2 is associated with coronary spastic angina: Possible roles of reactive aldehydes and implications of alcohol flushing syndrome. Circulation 2015, 131, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Polimanti, R.; Gelernter, J. ADH1B: From alcoholism, natural selection, and cancer to the human phenome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Osier, M.V.; Pakstis, A.J.; Soodyall, H.; Comas, D.; Goldman, D.; Odunsi, A.; Okonofua, F.; Parnas, J.; Schulz, L.O.; Bertranpetit, J.; et al. A global perspective on genetic variation at the ADH genes reveals unusual patterns of linkage disequilibrium and diversity. Am. J. Hum. Genet. 2002, 71, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, C.J.; Robin, R.W.; Osier, M.V.; Sambuughin, N.; Goldfarb, L.G.; Kittles, R.A.; Hesselbrock, D.; Goldman, D.; Long, J.C. Allelic variation at alcohol metabolism genes (ADH1B, ADH1C, ALDH2) and alcohol dependence in an American Indian population. Hum. Genet. 2003, 113, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Voight, B.F. Patterns of shared signatures of recent positive selection across human populations. Nat. Ecol. Evol. 2018, 2, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Jorgenson, E.; Thai, K.K.; Hoffmann, T.J.; Sakoda, L.C.; Kvale, M.N.; Banda, Y.; Schaefer, C.; Risch, N.; Mertens, J.; Weisner, C.; et al. Genetic contributors to variation in alcohol consumption vary by race/ethnicity in a large multi-ethnic genome-wide association study. Mol. Psychiatry 2017, 22, 1359–1367. [Google Scholar] [CrossRef] [Green Version]
- Tsuchihashi-Makaya, M.; Serizawa, M.; Yanai, K.; Katsuya, T.; Takeuchi, F.; Fujioka, A.; Yamori, Y.; Ogihara, T.; Kato, N. Gene-environmental interaction regarding alcohol-metabolizing enzymes in the Japanese general population. Hypertens. Res. 2009, 32, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Uhl, G.R. Molecular genetics of substance abuse vulnerability: Remarkable recent convergence of genome scan results. Ann. N. Y. Acad. Sci. 2004, 1025, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Thomasson, H.R.; Crabb, D.W.; Edenberg, H.J.; Li, T.K.; Hwu, H.G.; Chen, C.C.; Yeh, E.K.; Yin, S.J. Low frequency of the ADH2*2 allele among atayal natives of Taiwan with alcohol use disorders. Alcohol. Clin. Exp. Res. 1994, 18, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Edenberg, H.J. The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res. Health 2007, 30, 5–13. [Google Scholar] [PubMed]
- Nakamura, K.; Iwahashi, K.; Matsuo, Y.; Miyatake, R.; Ichikawa, Y.; Suwaki, H. Characteristics of Japanese alcoholics with the atypical aldehyde dehydrogenase 2*I. A comparison of the genotypes of ALDH2, ADH2, ADH3, and cytochrome P-4502E1 between alcoholics and nonalcoholics. Alcohol. Clin. Exp. Res. 1996, 20, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Gu, S.; Oota, H.; Osier, M.V.; Pakstis, A.J.; Speed, W.C.; Kidd, J.R.; Kidd, K.K. Evidence of positive selection on a class I ADH locus. Am. J. Hum. Genet. 2007, 80, 441–456. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gu, S.; Cai, X.; Speed, W.C.; Pakstis, A.J.; Golub, E.I.; Kidd, J.R.; Kidd, K.K. Ethnic related selection for an ADH class I variant within East Asia. PLoS ONE 2008, 3, e1881. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Shi, H.; Qi, X.B.; Xiao, C.J.; Zhong, H.; Ma, R.L.; Su, B. The ADH1B ARG47HIS polymorphism in East Asian populations and expansion of rice domestication in history. BMC Evol. Biol. 2010, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Peter, B.M.; Huerta-Sanchez, E.; Nielsen, R. Distinguishing between selective sweeps from standing variation and from a de novo mutation. PLoS Genet. 2012, 8, e1003011. [Google Scholar] [CrossRef] [PubMed]
- Evsyukov, A.; Ivanov, D. Selection variability for ARG48HIS in alcohol dehydrogenase ADH1B among Asian populations. Hum. Biol. 2013, 85, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Celorrio, D.; Bujanda, L.; Chbel, F.; Sanchez, D.; Martinez-Jarreta, B.; de Pancorbo, M.M. Alcohol-metabolizing enzyme gene polymorphisms in the Basque country, Morocco, and Ecuador. Alcohol. Clin. Exp. Res. 2011, 35, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, J.M.; Geske, J.R.; Schneekloth, T.D.; Frye, M.A.; Cunningham, J.M.; Choi, D.S.; Tapp, C.L.; Lewis, B.R.; Drews, M.S.L.; Pietrzak, T.; et al. Replication of genome wide association studies of alcohol dependence: Support for association with variation in ADH1C. PLoS ONE 2013, 8, e58798. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mukherjee, N.; Soundararajan, U.; Tarnok, Z.; Barta, C.; Khaliq, S.; Mohyuddin, A.; Kajuna, S.L.; Mehdi, S.Q.; Kidd, J.R.; et al. Geographically separate increases in the frequency of the derived ADH1B*47HIS allele in Eastern and Western Asia. Am. J. Hum. Genet. 2007, 81, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Borinskaya, S.; Kalina, N.; Marusin, A.; Faskhutdinova, G.; Morozova, I.; Kutuev, I.; Koshechkin, V.; Khusnutdinova, E.; Stepanov, V.; Puzyrev, V.; et al. Distribution of the alcohol dehydrogenase ADH1B*47HIS allele in Eurasia. Am. J. Hum. Genet. 2009, 84, 89–92, author reply 92-84. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Reich, D.E.; Higgins, J.M.; Levine, H.Z.; Richter, D.J.; Schaffner, S.F.; Gabriel, S.B.; Platko, J.V.; Patterson, N.J.; McDonald, G.J.; et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 2002, 419, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Hammer, M.F.; Redd, A.J.; Wood, E.T.; Bonner, M.R.; Jarjanazi, H.; Karafet, T.; Santachiara-Benerecetti, S.; Oppenheim, A.; Jobling, M.A.; Jenkins, T.; et al. Jewish and middle eastern non-Jewish populations share a common pool of Y-chromosome biallelic haplotypes. Proc. Natl. Acad. Sci. USA 2000, 97, 6769–6774. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.K.; Kidd, J.R. Human genetic variation of medical significance. In Evolution in Health and Disease, 2nd ed.; Oxford University Press: New York, NY, USA, 2008; pp. xxi, 374. [Google Scholar]
- Simon, R.S.; Laskier, M.M.; Reguer, S. The Jews of The Middle East and North Africa in Modern Times; Columbia University Press: New York, NY, USA, 2003. [Google Scholar]
- Kleiman, R.Y. DNA Evidence for Common Jewish Origin and Maintenance of the Ancestral Genetic Profile. Available online: http://www.cohen-levi.org/jewish_genes_and_genealogy/jewish_genes_-_dna_evidence.htm (accessed on 8 August 2018).
- Wright, S. Evolution in mendelian populations. Genetics 1931, 16, 97–159. [Google Scholar] [PubMed]
- Beaumont, M.A.; Balding, D.J. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 2004, 13, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M.; Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 1974, 23, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.; Williamson, S.; Casals, F.; Bosch, E.; Navarro, A.; Calafell, F.; Bertranpetit, J.; Comas, D. Extreme individual marker Fst values do not imply population-specific selection in humans: The NRG1 example. Hum. Genet. 2007, 121, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Osier, M.V.; Cheung, K.H.; Kidd, J.R.; Pakstis, A.J.; Miller, P.L.; Kidd, K.K. Alfred: An allele frequency database for diverse populations and DNA polymorphisms—An update. Nucleic Acids Res. 2001, 29, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Osier, M.V.; Cheung, K.H.; Kidd, J.R.; Pakstis, A.J.; Miller, P.L.; Kidd, K.K. Alfred: An allele frequency database for anthropology. Am. J. Phys. Anthropol. 2002, 119, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Pakstis, A.J.; Kidd, K.K. Haplot: A graphical comparison of haplotype blocks, tagSNP sets and SNP variation for multiple populations. Bioinformatics 2005, 21, 3938–3939. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Pakstis, A.J.; Li, H.; Speed, W.C.; Kidd, J.R.; Kidd, K.K. Significant variation in haplotype block structure but conservation in tagSNP patterns among global populations. Eur. J. Hum. Genet. 2007, 15, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheet, P.; Stephens, M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef] [PubMed]
- Hawley, M.E.; Kidd, K.K. Haplo: A program using the EM algorithm to estimate the frequencies of multi-site haplotypes. J. Hered. 1995, 86, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.R. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 2002, 18, 337–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidd, K.K.; Pakstis, A.J.; Speed, W.C.; Kidd, J.R. Understanding human DNA sequence variation. J. Hered. 2004, 95, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Mukherjee, N.; Pakstis, A.J.; Feuk, L.; Kidd, J.R.; Brookes, A.J.; Kidd, K.K. Linkage disequilibrium patterns vary substantially among populations. Eur. J. Hum. Genet. 2005, 13, 677–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Gu, S.; Han, Y.; Xu, Z.; Pakstis, A.J.; Jin, L.; Kidd, J.R.; Kidd, K.K. Diversification of the ADH1B gene during expansion of modern humans. Ann. Hum. Genet. 2011, 75, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Kobayashi, S.; Shibata, H.; Furuumi, H.; Endo, T.; Fucharoen, S.; Hamano, S.; Acharya, G.P.; Kawasaki, T.; Fukumaki, Y. Molecular analysis of α-thalassemia in Nepal: Correlation with malaria endemicity. J. Hum. Genet. 2000, 45, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Norman, P.J.; Cook, M.A.; Carey, B.S.; Carrington, C.V.; Verity, D.H.; Hameed, K.; Ramdath, D.D.; Chandanayingyong, D.; Leppert, M.; Stephens, H.A.; et al. SNP haplotypes and allele frequencies show evidence for disruptive and balancing selection in the human leukocyte receptor complex. Immunogenetics 2004, 56, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Oota, H.; Pakstis, A.J.; Bonne-Tamir, B.; Goldman, D.; Grigorenko, E.; Kajuna, S.L.; Karoma, N.J.; Kungulilo, S.; Lu, R.B.; Odunsi, K.; et al. The evolution and population genetics of the ALDH2 locus: Random genetic drift, selection, and low levels of recombination. Ann. Hum. Genet. 2004, 68, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.C.; Sabeti, P.; Hutcheson, H.B.; Fry, B.; Schaffner, S.F.; de Bakker, P.I.; Varilly, P.; Palma, A.A.; Roy, J.; Cooper, R.; et al. Searching for signals of evolutionary selection in 168 genes related to immune function. Hum. Genet. 2006, 119, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Lu, R.B.; Chen, Y.C.; Wang, M.F.; Chang, Y.C.; Li, T.K.; Yin, S.J. Interaction between the functional polymorphisms of the alcohol-metabolism genes in protection against alcoholism. Am. J. Hum. Genet. 1999, 65, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Pollinger, J.P.; Bustamante, C.D.; Fledel-Alon, A.; Schmutz, S.; Gray, M.M.; Wayne, R.K. Selective sweep mapping of genes with large phenotypic effects. Genome Res. 2005, 15, 1809–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Oota, H.; Osier, M.V.; Pakstis, A.J.; Speed, W.C.; Odunsi, A.; Okonofua, F.; Kajuna, S.L.; Karoma, N.J.; Kungulilo, S.; et al. Considerable haplotype diversity within the 23kb encompassing the ADH7 gene. Alcohol. Clin. Exp. Res. 2005, 29, 2091–2100. [Google Scholar] [CrossRef] [PubMed]
- Osier, M.; Pakstis, A.J.; Kidd, J.R.; Lee, J.F.; Yin, S.J.; Ko, H.C.; Edenberg, H.J.; Lu, R.B.; Kidd, K.K. Linkage disequilibrium at the ADH2 and ADH3 loci and risk of alcoholism. Am. J. Hum. Genet. 1999, 64, 1147–1157. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position in Figure 2 | Chromosome 4 SNPs, STRPs, Indel near ADH1B | Build 38 Nucleotide Position (Start-End for STRP, Indel) | SNP Alleles: Ancestral, Derived | |
|---|---|---|---|---|
| Start | End | |||
| #54 | rs12507573 | 99305167 | A, C | |
| centromeric (TA)n | 99305562 | 99305609 | ||
| #55 | rs1042026 | 99307309 | T, C | |
| #60 | rs1229984 | 99318162 | C, T | |
| #61 | rs1159918 | 99321852 | A, C | |
| #62 | rs6810842 | 99322288 | G, T | |
| #63 | rs3811802 | 99323064 | A, G | |
| #64 | rs3811801 | 99323162 | G, A | |
| #65 | rs1693439 | 99324332 | G, A | |
| #66 | rs9307239 | 99325780 | C, T | |
| #67 | rs1789891 | 99329262 | C, A | |
| telomeric (GTAT)n | 99331657 | 99331709 | ||
| #68 | rs36207960 dws 21 bp indel | 99332232 | 99332252 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, S.; Li, H.; Pakstis, A.J.; Speed, W.C.; Gurwitz, D.; Kidd, J.R.; Kidd, K.K. Recent Selection on a Class I ADH Locus Distinguishes Southwest Asian Populations Including Ashkenazi Jews. Genes 2018, 9, 452. https://doi.org/10.3390/genes9090452
Gu S, Li H, Pakstis AJ, Speed WC, Gurwitz D, Kidd JR, Kidd KK. Recent Selection on a Class I ADH Locus Distinguishes Southwest Asian Populations Including Ashkenazi Jews. Genes. 2018; 9(9):452. https://doi.org/10.3390/genes9090452
Chicago/Turabian StyleGu, Sheng, Hui Li, Andrew J. Pakstis, William C. Speed, David Gurwitz, Judith R. Kidd, and Kenneth K. Kidd. 2018. "Recent Selection on a Class I ADH Locus Distinguishes Southwest Asian Populations Including Ashkenazi Jews" Genes 9, no. 9: 452. https://doi.org/10.3390/genes9090452
APA StyleGu, S., Li, H., Pakstis, A. J., Speed, W. C., Gurwitz, D., Kidd, J. R., & Kidd, K. K. (2018). Recent Selection on a Class I ADH Locus Distinguishes Southwest Asian Populations Including Ashkenazi Jews. Genes, 9(9), 452. https://doi.org/10.3390/genes9090452