Metagenomic Insights into the Phylogenetic and Metabolic Diversity of the Prokaryotic Community Dwelling in Hypersaline Soils from the Odiel Saltmarshes (SW Spain)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling Site and Sample Collection
2.2. Physico-Chemical Characterization of Soils
2.3. DNA Extraction and Sequencing
2.4. Bioinformatics Analysis of the Databases
2.5. Data Availability
3. Results and Discussion
3.1. Environmental Data
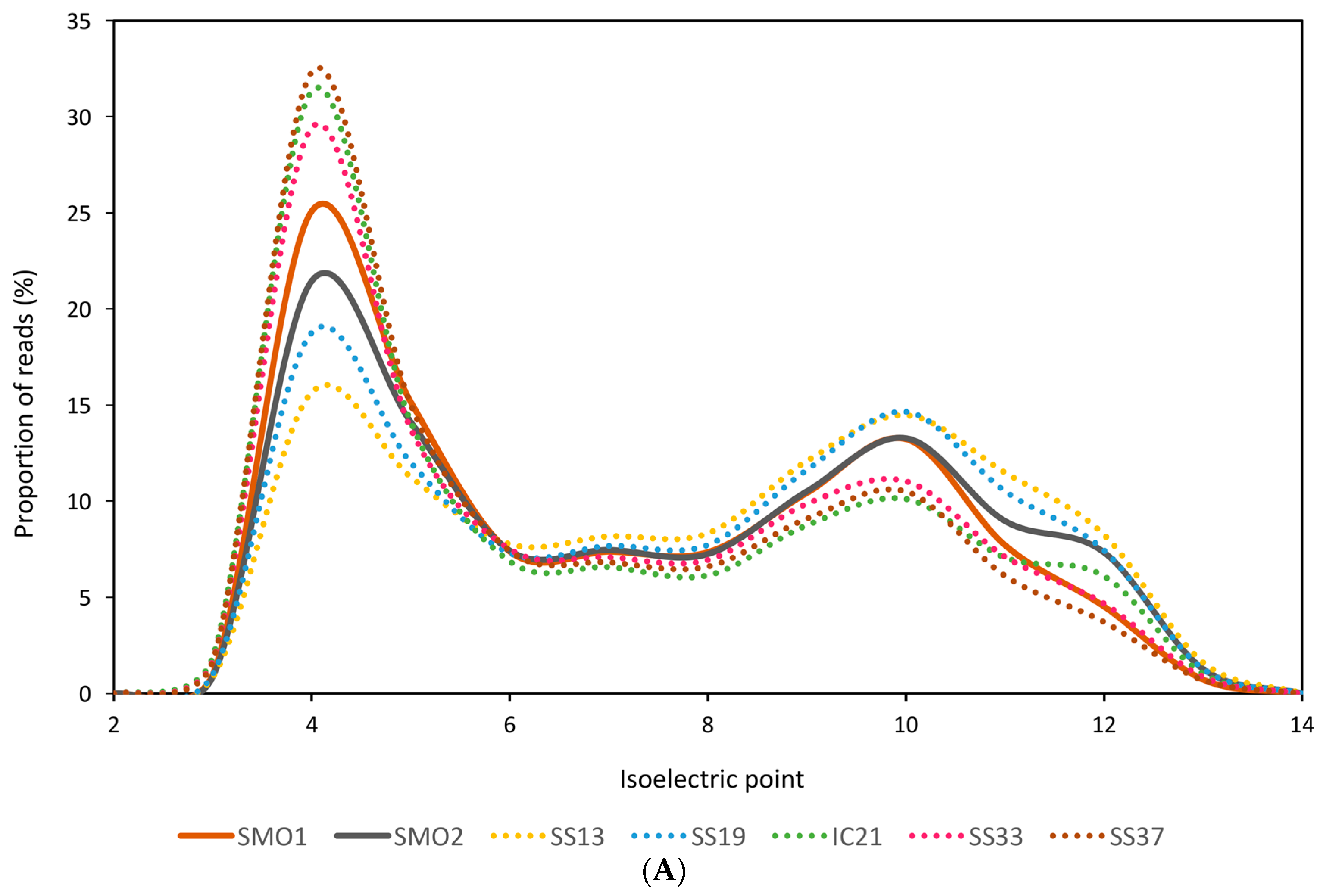
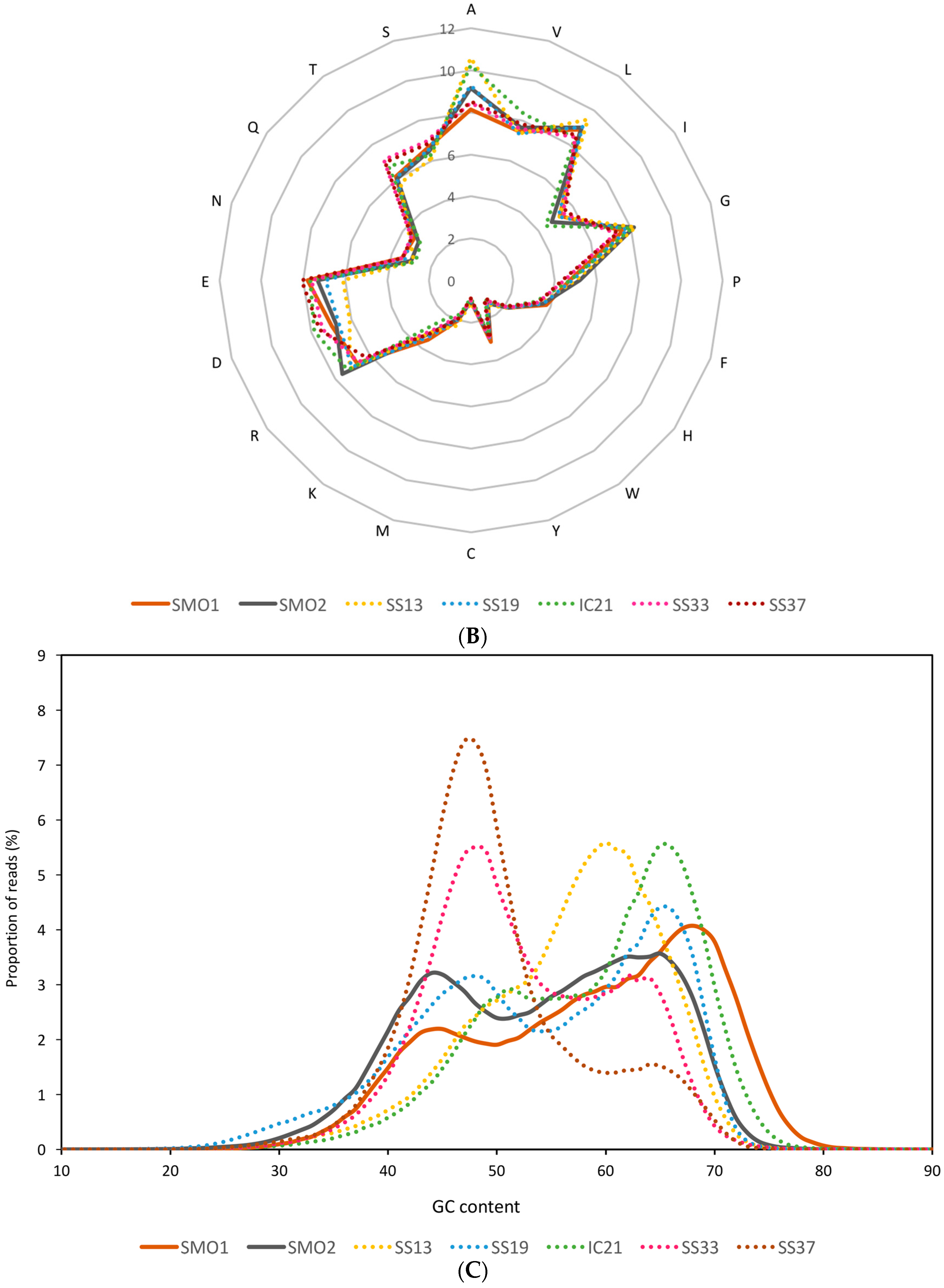
3.2. General Characteristics and Halophilic Traits of the Databases
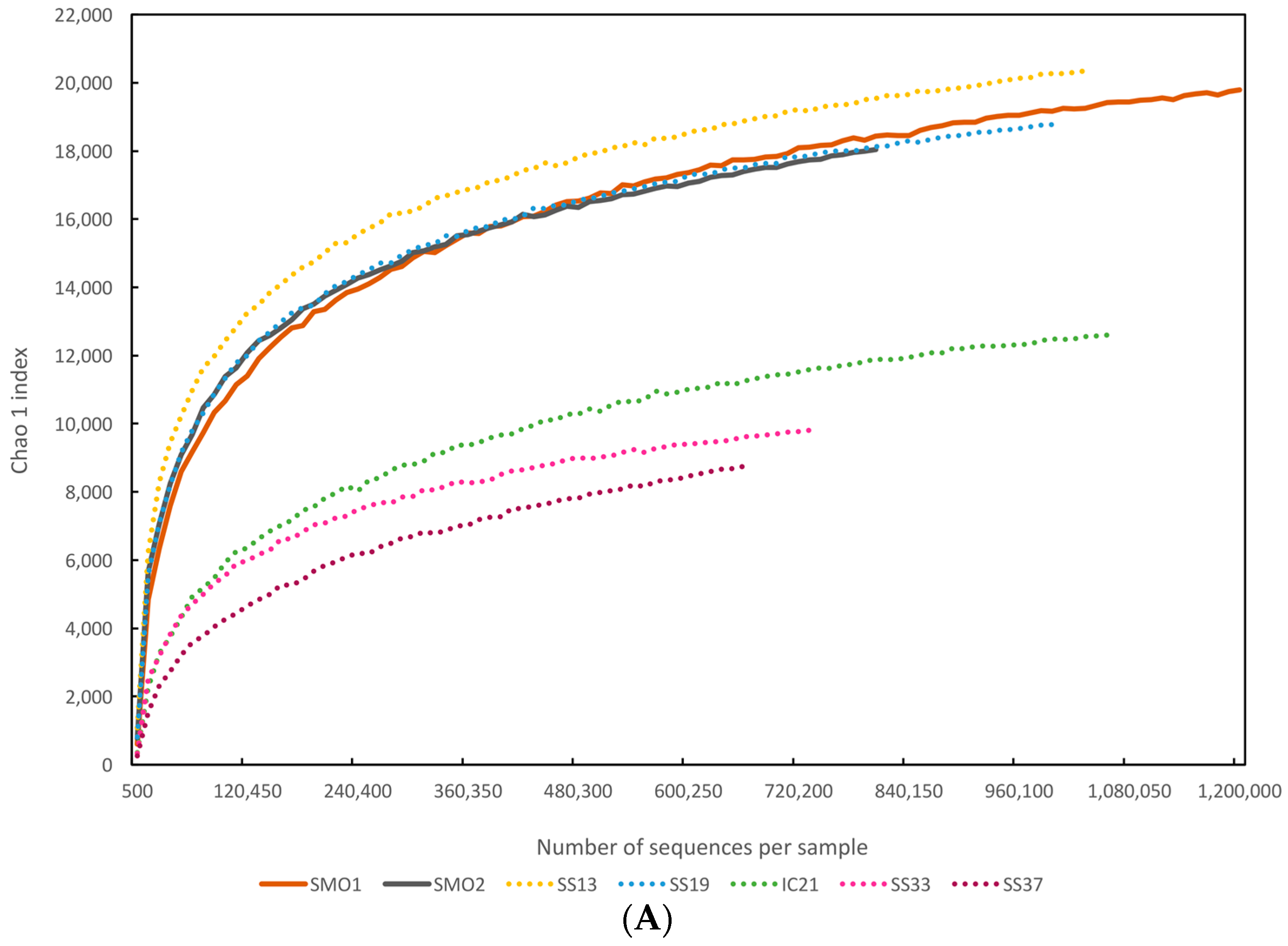
3.3. Diversity Estimates
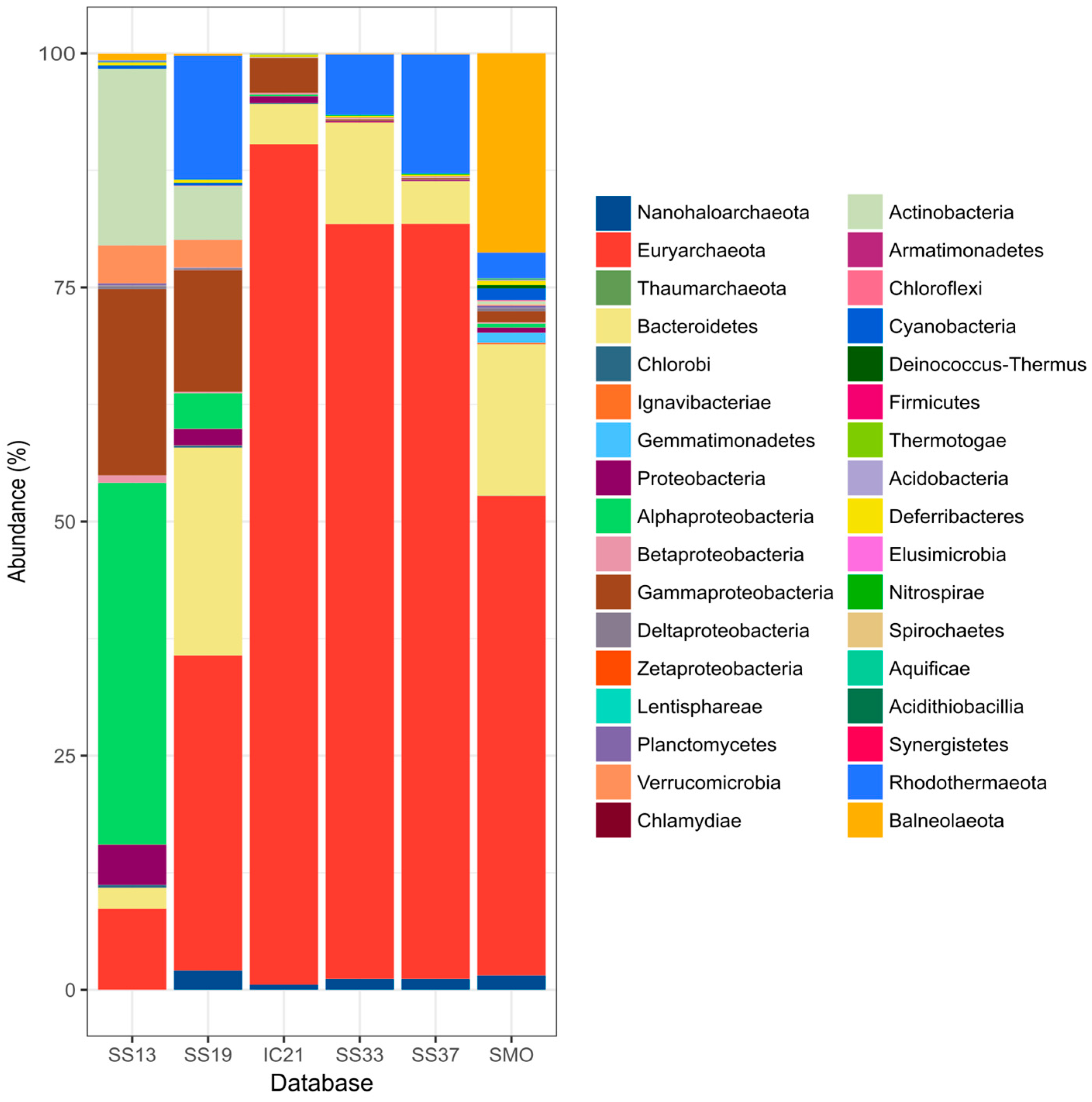
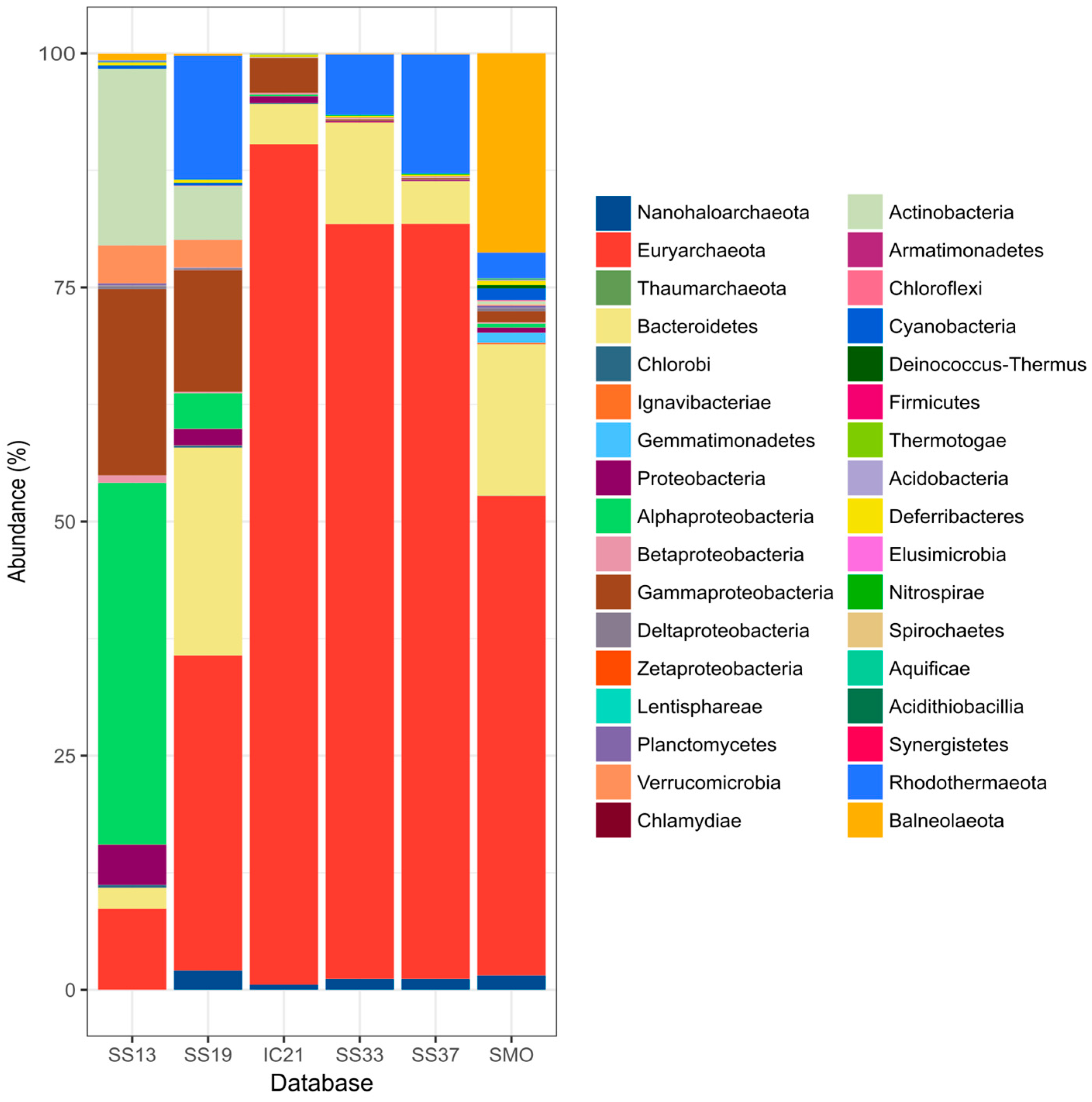
3.4. Microbial Community Composition
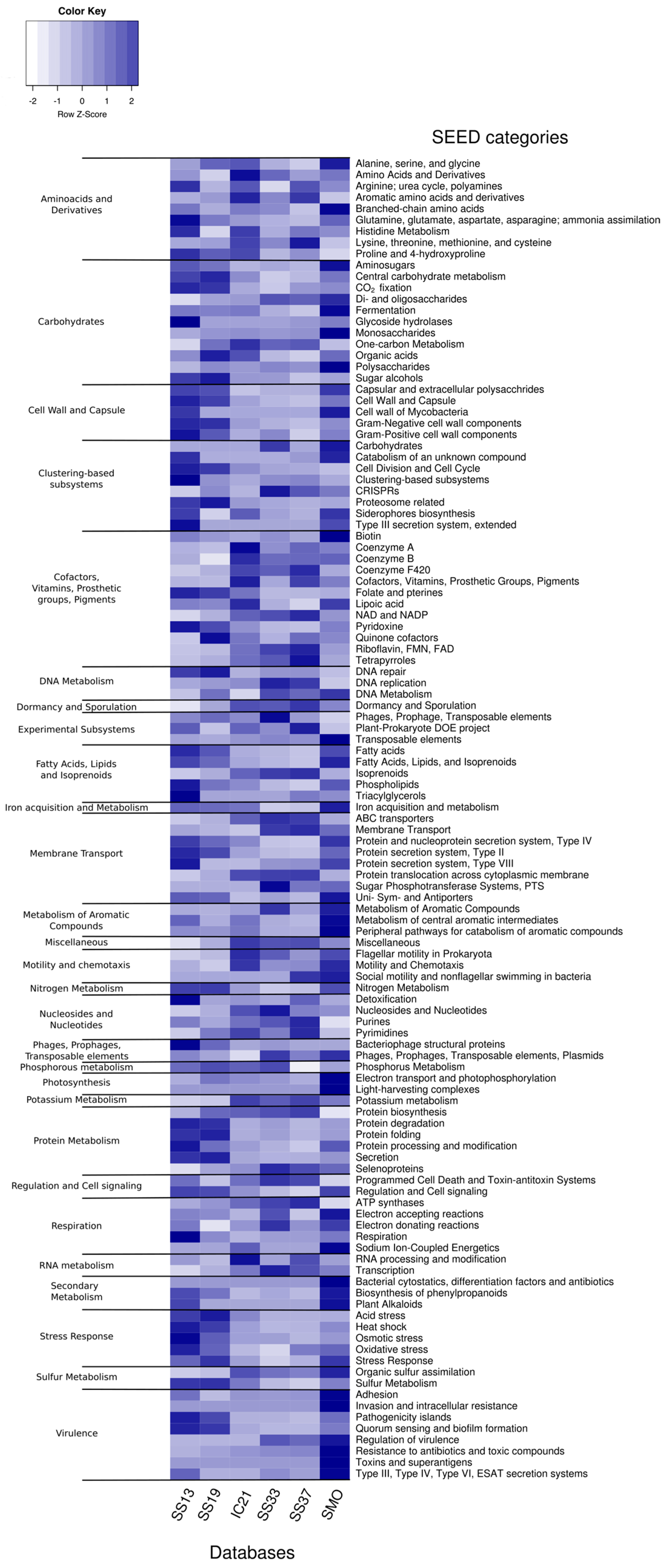
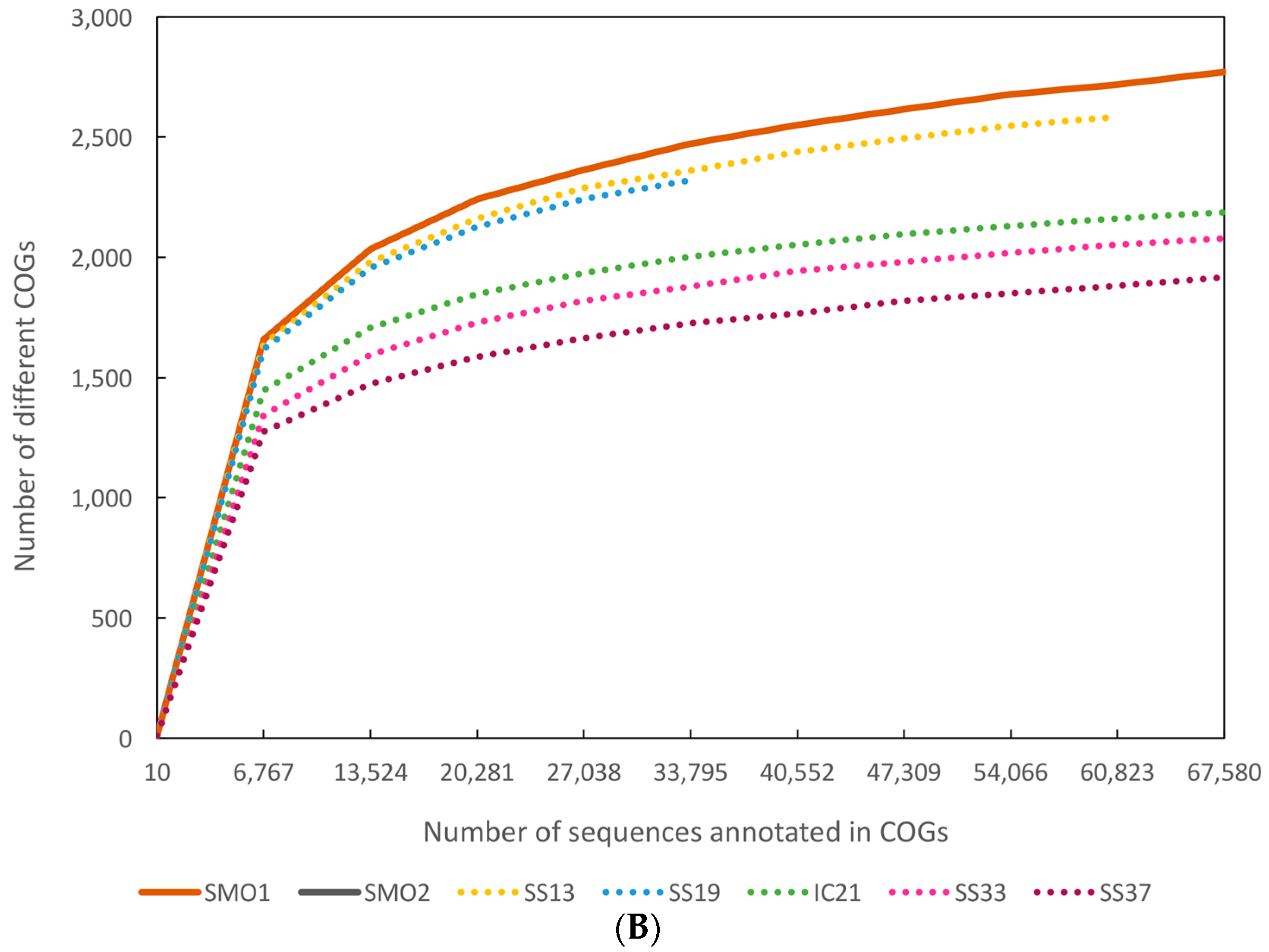
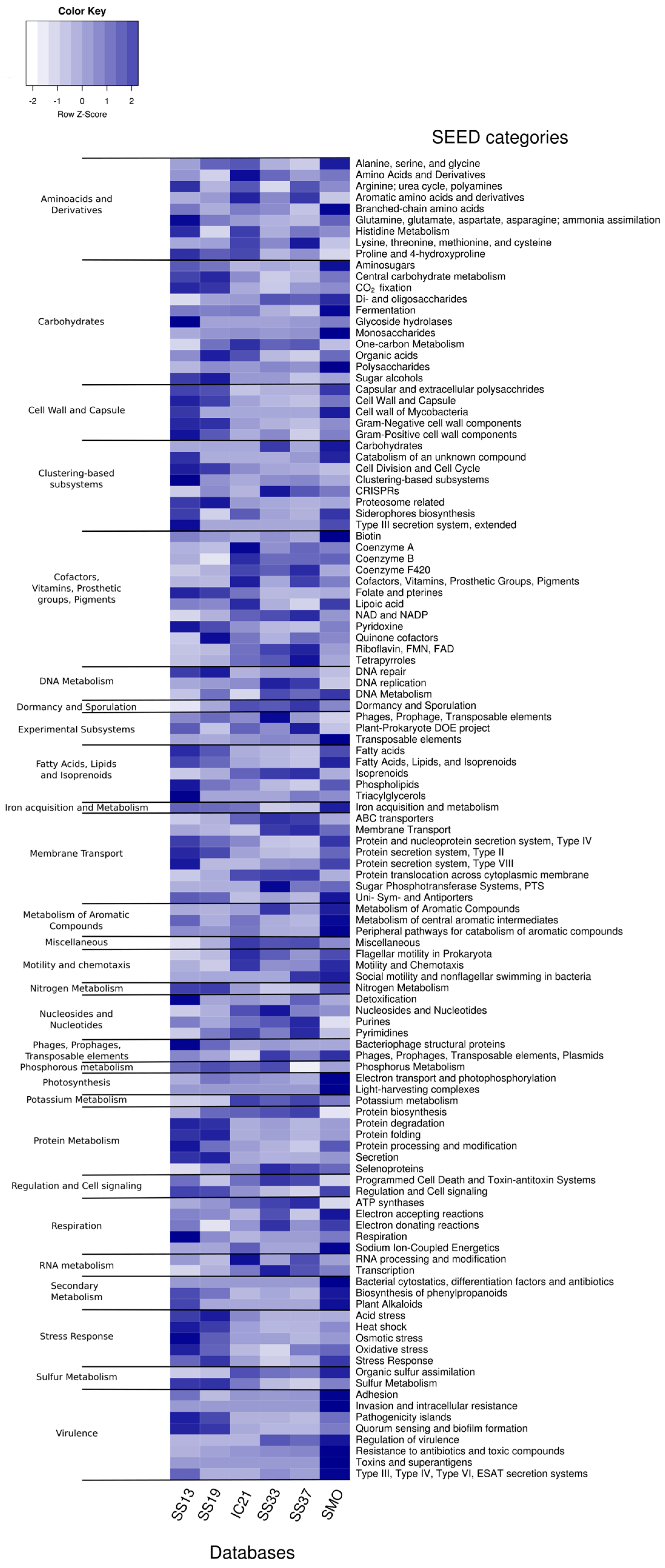
3.5. Functional Diversity
3.6. Insights into Untapped Genomic Diversity through Metagenomic Binning
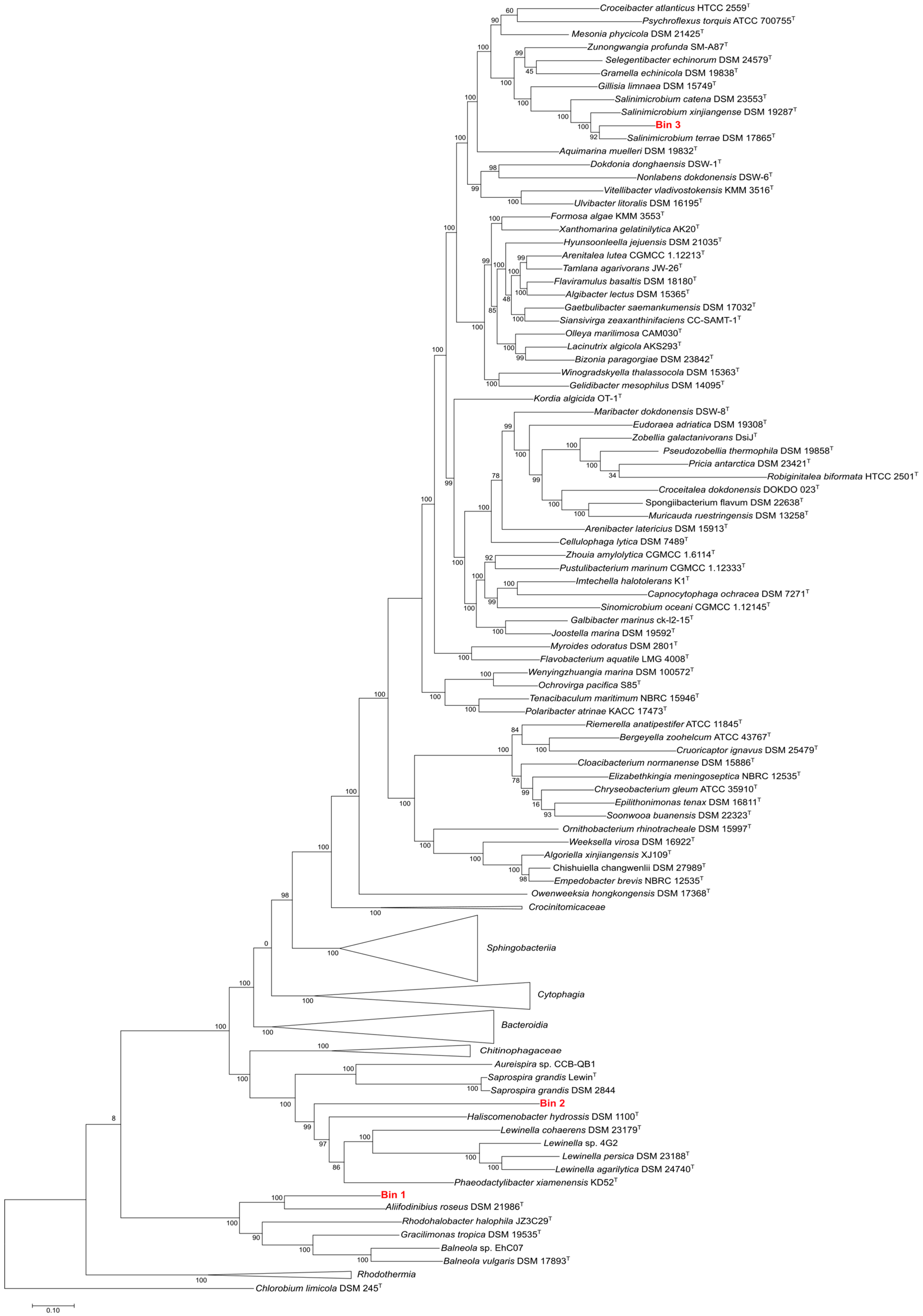
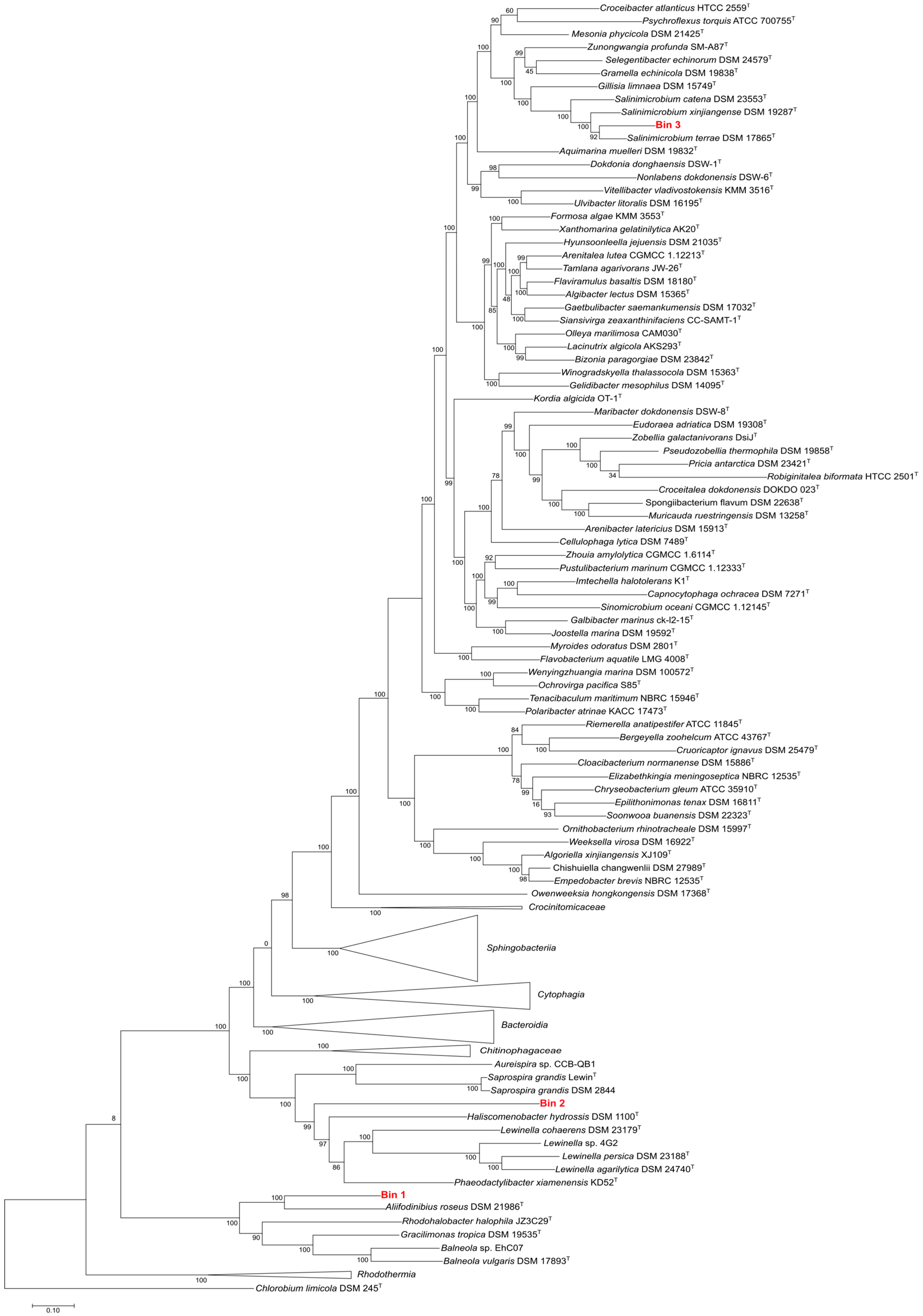
3.6.1. Estimation of the Phylogenetic Affiliation of Assembled MAGs
3.6.2. Genomic Analysis of Assembled MAGs
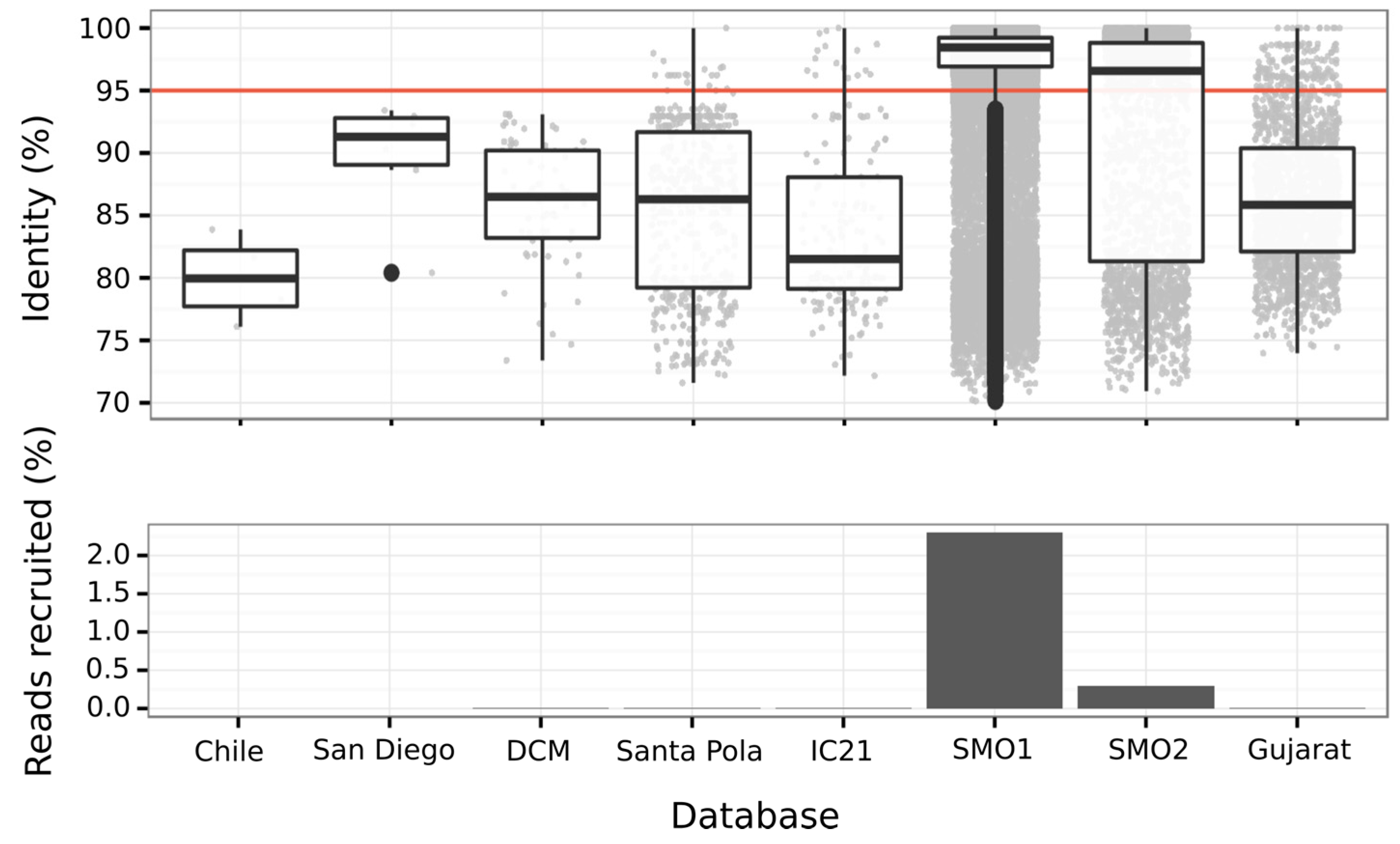
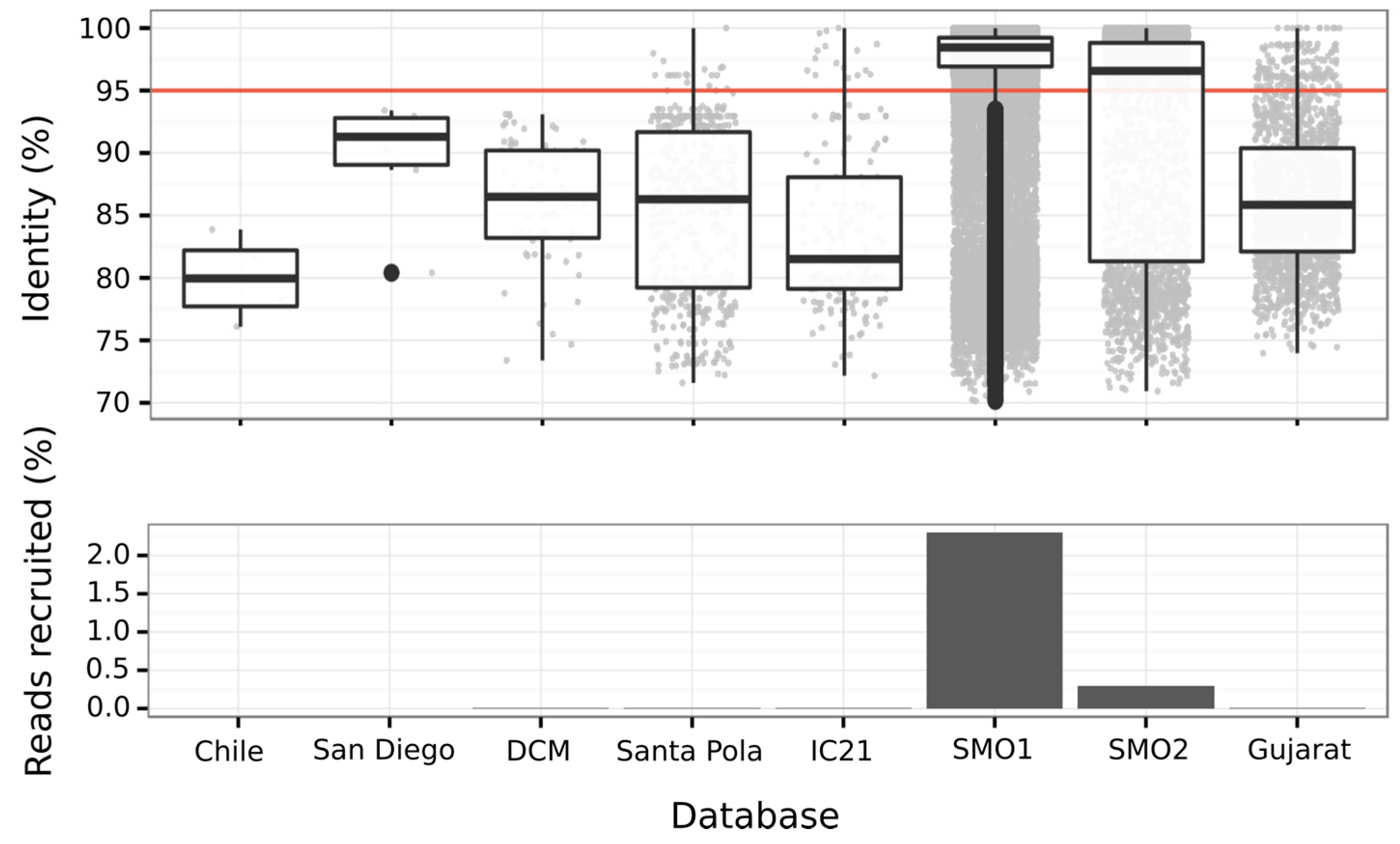
3.6.3. Abundance of Balneolaeota-Related Bin in Hypersaline Environments
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ventosa, A. Unusual micro-organisms from unusual habitats: Hypersaline environments. In Prokaryotic Diversity: Mechanism and Significance; Logan, N., Lappin-Scott, H., Oyston, P., Eds.; Cambridge University Press: Cambridge, UK, 2006; pp. 223–253. ISBN 9780511754913. [Google Scholar]
- Ventosa, A.; de la Haba, R.R.; Sánchez-Porro, C.; Papke, R.T. Microbial diversity of hypersaline environments: A metagenomic approach. Curr. Opin. Microbiol. 2015, 25, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. Halophilic microbial communities and their environments. Curr. Opin. Biotechnol. 2015, 33, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Ventosa, A.; Fernández, A.B.; León, M.J.; Sánchez-Porro, C.; Rodriguez-Valera, F. The Santa Pola saltern as a model for studying the microbiota of hypersaline environments. Extremophiles 2014, 18, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Deng, Y.; Zhang, S.; Zhang, W.; Liu, J.; Xie, Y.; Zhang, X.; Huang, H. Prokaryotic community distribution along an ecological gradient of salinity in surface and subsurface saline soils. Sci. Rep. 2017, 7, 13332. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; House, J.I.; Bustamante, M.; Sobocká, J.; Harper, R.; Pan, G.; West, P.C.; Clark, J.M.; Adhya, T.; Rumpel, C.; et al. Global change pressures on soils from land use and management. Glob. Chang. Biol. 2016, 22, 1008–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccá, M.L.; Barra Caracciolo, A.; Di Lenola, M.; Grenni, P. Ecosystem services provided by soil microorganisms. In Soil Biological Communities and Ecosystem Resilience; Lukac, M., Grenni, P., Gamboni, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 9–24. ISBN 978-3-319-63336-7. [Google Scholar]
- Wicke, B.; Smeets, E.; Dornburg, V.; Vashev, B.; Gaiser, T.; Turkenburg, W.; Faaij, A. The global technical and economic potential of bioenergy from salt-affected soils. Energy Environ. Sci. 2011, 4, 2669. [Google Scholar] [CrossRef]
- Quesada, E.; Ventosa, A.; Rodriguez-Valera, F.; Ramos-Cormenzana, A. Types and properties of some bacteria isolated from hypersaline soils. J. Appl. Bacteriol. 1982, 53, 155–161. [Google Scholar] [CrossRef]
- Canfora, L.; Bacci, G.; Pinzari, F.; Lo Papa, G.; Dazzi, C.; Benedetti, A. Salinity and bacterial diversity: To what extent does the concentration of salt affect the bacterial community in a saline soil? PLoS ONE 2014, 9, e106662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela-Encinas, C.; Neria-González, I.; Alcántara-Hernández, R.J.; Estrada-Alvarado, I.; Zavala-Díaz de la Serna, F.J.; Dendooven, L.; Marsch, R. Changes in the bacterial populations of the highly alkaline saline soil of the former lake Texcoco (Mexico) following flooding. Extremophiles 2009, 13, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Oueriaghli, N.; González-Domenech, C.M.; Martínez-Checa, F.; Muyzer, G.; Ventosa, A.; Quesada, E.; Béjar, V. Diversity and distribution of Halomonas in Rambla Salada, a hypersaline environment in the southeast of Spain. FEMS Microbiol. Ecol. 2014, 87, 460–474. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Jia, H.; Wang, Q. The effect of land use on bacterial communities in saline–alkali soil. Curr. Microbiol. 2017, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Noya, Y.E.; Valenzuela-Encinas, C.; Sandoval-Yuriar, A.; Jiménez-bueno, N.G.; Marsch, R.; Dendooven, L. Archaeal communities in a heterogeneous hypersaline-alkaline soil. Archaea 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Oueriaghli, N.; Béjar, V.; Quesada, E.; Martínez-Checa, F. Molecular ecology techniques reveal both spatial and temporal variations in the diversity of archaeal communities within the athalassohaline environment of Rambla Salada, Spain. Microb. Ecol. 2013, 66, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A.; Papke, R.T.; Doolittle, W.F. Archaeal diversity along a soil salinity gradient prone to disturbance. Environ. Microbiol. 2005, 7, 1655–1666. [Google Scholar] [CrossRef] [PubMed]
- Caton, T.M.; Caton, I.R.; Witte, L.R.; Schneegurt, M.A. Archaeal diversity at the Great Salt Plains of Oklahoma described by cultivation and molecular analyses. Microb. Ecol. 2009, 58, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Hollister, E.B.; Engledow, A.S.; Hammett, A.J.M.; Provin, T.L.; Wilkinson, H.H.; Gentry, T.J. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J. 2010, 4, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Pandit, A.S.; Joshi, M.N.; Bhargava, P.; Shaikh, I.; Ayachit, G.N.; Raj, S.R.; Saxena, A.K.; Bagatharia, S.B. A snapshot of microbial communities from the Kutch: One of the largest salt deserts in the world. Extremophiles 2015, 19, 973–987. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Mevada, V.; Prajapati, D.; Dudhagara, P.; Koringa, P.; Joshi, C.G. Metagenomic sequence of saline desert microbiota from wild ass sanctuary, Little Rann of Kutch, Gujarat, India. Genom. Data 2015, 3, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R.; Pašić, L.; Fernández, A.B.; Martin-Cuadrado, A.-B.; Mizuno, C.M.; McMahon, K.D.; Papke, R.T.; Stepanauskas, R.; Rodriguez-Brito, B.; Rohwer, F.; et al. New abundant microbial groups in aquatic hypersaline environments. Sci. Rep. 2011, 1, 135. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.B.; Ghai, R.; Martin-Cuadrado, A.B.; Sánchez-Porro, C.; Rodriguez-Valera, F.; Ventosa, A. Prokaryotic taxonomic and metabolic diversity of an intermediate salinity hypersaline habitat assessed by metagenomics. FEMS Microbiol. Ecol. 2014, 88, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Dillon, J.G.; Carlin, M.; Gutierrez, A.; Nguyen, V.; McLain, N. Patterns of microbial diversity along a salinity gradient in the Guerrero Negro solar saltern, Baja CA Sur, Mexico. Front. Microbiol. 2013, 4, 399. [Google Scholar] [CrossRef] [PubMed]
- Vavourakis, C.D.; Ghai, R.; Rodriguez-Valera, F.; Sorokin, D.Y.; Tringe, S.G.; Hugenholtz, P.; Muyzer, G. Metagenomic insights into the uncultured diversity and physiology of microbes in four hypersaline soda lake brines. Front. Microbiol. 2016, 7, 211. [Google Scholar] [CrossRef] [PubMed]
- Narasingarao, P.; Podell, S.; Ugalde, J.A.; Brochier-Armanet, C.; Emerson, J.B.; Brocks, J.J.; Heidelberg, K.B.; Banfield, J.F.; Allen, E.E. De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J. 2012, 6, 81–93. [Google Scholar] [CrossRef] [PubMed]
- León, M.J.; Fernández, A.B.; Ghai, R.; Sánchez-Porro, C.; Rodriguez-Valera, F.; Ventosa, A. From metagenomics to pure culture: Isolation and characterization of the moderately halophilic bacterium Spiribacter salinus gen. nov., sp. nov. Appl. Environ. Microbiol. 2014, 80, 3850–3857. [Google Scholar] [CrossRef] [PubMed]
- Curado, G.; Rubio-Casal, A.E.; Figueroa, E.; Castillo, J.M. Potential of Spartina maritima in restored salt marshes for phytoremediation of metals in a highly polluted estuary. Int. J. Phytoremediation 2014, 16, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Casal, A.E.; Castillo, J.M.; Luque, C.J.; Figueroa, M.E. Nucleation and facilitation in salt pans in Mediterranean salt marshes. J. Veg. Sci. 2001, 12, 761–770. [Google Scholar] [CrossRef]
- Castillo, J.M.; Mateos-Naranjo, E.; Nieva, F.J.; Figueroa, E. Plant zonation at salt marshes of the endangered cordgrass Spartina maritima invaded by Spartina densiflora. Hydrobiologia 2008, 614, 363–371. [Google Scholar] [CrossRef]
- UNESCO (United Nations Educational, Scientific and Cultural Organization). Convention on Wetlands of International Importance Especially as Waterfowl Habitat; UN Treaty Series No. 14583; UNESCO: Ramsar, Iran, 2 February 1971. [Google Scholar]
- Castellanos, E.M.; Figueroa, M.E.; Davy, A.J. Nucleation and facilitation in saltmarsh succession: Interactions between Spartina maritima and Arthrocnemum perenne. J. Ecol. 1994, 82, 239–248. [Google Scholar] [CrossRef]
- Beltrán, R.; de la Rosa, J.D.; Santos, J.C.; Beltrán, M.; Gómez-Ariza, J.L. Heavy metal mobility assessment in sediments from the Odiel River (Iberian Pyritic Belt) using sequential extraction. Environ. Earth Sci. 2010, 61, 1493–1503. [Google Scholar] [CrossRef]
- Hierro, A.; Olías, M.; Ketterer, M.E.; Vaca, F.; Borrego, J.; Cánovas, C.R.; Bolivar, J.P. Geochemical behavior of metals and metalloids in an estuary affected by acid mine drainage (AMD). Environ. Sci. Pollut. Res. Int. 2014, 21, 2611–2627. [Google Scholar] [CrossRef] [PubMed]
- Bouyoucos, G.J. Directions for making mechanical analysis of soils by the hydrometer method. Soil Sci. 1936, 4, 225–228. [Google Scholar] [CrossRef]
- Soil Science Division Staff. Soil Survey Manual; USDA Handbook; Government Printing Office: Washington, DC, USA, 2017.
- Walkley, A.; Black, I.A. An examination of Degtjareff method for determining soil organic matter, and proposed modification of the chromic acid tritation method. Soil Sci. 1934, 37, 29–38. [Google Scholar] [CrossRef]
- Kjeldahl, J. Neue methode zur bestimmung des stickstoffs in organischen körpern. Zeitschrift für Anal. Chemie 1883, 22, 366–383. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Isolation of high-molecular-weight DNA using organic solvents. Cold Spring Harb. Protoc. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 February 2018).
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Aislabie, J.M.; Chhour, K.L.; Saul, D.J.; Miyauchi, S.; Ayton, J.; Paetzold, R.F.; Balks, M.R. Dominant bacteria in soils of Marble Point and Wright Valley, Victoria Land, Antarctica. Soil Biol. Biochem. 2006, 38, 3041–3056. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2006, 441, 120. [Google Scholar] [CrossRef]
- Huson, D.; Auch, A.; Qi, J.; Schuster, S. MEGAN analysis of metagenome data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Bose, T.; Haque, M.M.; Cvsk, R.; Mande, S.S. COGNIZER: A framework for functional annotation of metagenomic datasets. PLoS ONE 2015, 10, e0142102. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Laczny, C.C.; Sternal, T.; Plugaru, V.; Gawron, P.; Atashpendar, A.; Margossian, H.H.; Coronado, S.; van der Maaten, L.; Vlassis, N.; Wilmes, P. VizBin—An application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Jospin, G.; Lowe, E.; Matsen, F.A.; Bik, H.M.; Eisen, J.A. PhyloSift: Phylogenetic analysis of genomes and metagenomes. PeerJ 2014, 2, e243. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek 2017, 110, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.B.; Vera-Gargallo, B.; Sánchez-Porro, C.; Ghai, R.; Papke, R.T.; Rodriguez-Valera, F.; Ventosa, A. Comparison of prokaryotic community structure from Mediterranean and Atlantic saltern concentrator ponds by a metagenomic approach. Front. Microbiol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R.; Martin-Cuadrado, A.-B.; Molto, A.G.; Heredia, I.G.; Cabrera, R.; Martin, J.; Verdú, M.; Deschamps, P.; Moreira, D.; López-García, P.; et al. Metagenome of the Mediterranean deep chlorophyll maximum studied by direct and fosmid library 454 pyrosequencing. ISME J. 2010, 4, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Plominsky, A.M.; Delherbe, N.; Ugalde, J.A.; Allen, E.E.; Blanchet, M.; Ikeda, P.; Santibañez, F.; Hanselmann, K.; Ulloa, O.; De la Iglesia, R.; et al. Metagenome sequencing of the microbial community of a solar saltern crystallizer pond at Cáhuil Lagoon, Chile. Genome Announc. 2014, 2, e01172-14. [Google Scholar] [CrossRef] [PubMed]
- Vera-Gargallo, B.; Navarro-Sampedro, L.; Carballo, M.; Ventosa, A. Metagenome sequencing of prokaryotic microbiota from two hypersaline soils of the Odiel salt marshes in Huelva, southwestern Spain. Genome Announc. 2018, 6, e00140-18. [Google Scholar] [CrossRef] [PubMed]
- IUSS Working Group WRB. World Reference Base for Soil Resources 2006, First Update; IUSS Working Group WRB: Rome, Italy, 2007. [Google Scholar]
- Soil Science Division Staff. Keys to Soil Taxonomy, 11th ed.; USDA-Natural Resources Conservation Service: Washington, DC, USA, 2010.
- Richards, L. Diagnosis and Improvement of Saline and Alkali Soils. US Salinity Lab.; Agriculture Handbook No. 60; US Govt. Printing Office: Washington, DC, USA, 1954.
- Chowdhury, N.; Marschner, P.; Burns, R. Response of microbial activity and community structure to decreasing soil osmotic and matric potential. Plant Soil 2011, 344, 241–254. [Google Scholar] [CrossRef]
- Vos, M.; Wolf, A.B.; Jennings, S.J.; Kowalchuk, G.A. Micro-scale determinants of bacterial diversity in soil. FEMS Microbiol. Rev. 2013, 37, 936–954. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; DeSutter, T.; Hopkins, D.; Jia, X.; Wysocki, D.A. Predicting ECe of the saturated paste extract from value of EC1:5. Can. J. Soil Sci. 2013, 93, 585–594. [Google Scholar] [CrossRef]
- Clooney, A.G.; Fouhy, F.; Sleator, R.D.; O’Driscoll, A.; Stanton, C.; Cotter, P.D.; Claesson, M.J. Comparing apples and oranges?: Next generation sequencing and its impact on microbiome analysis. PLoS ONE 2016, 11, e0148028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Bag, S.K.; Das, S.; Harvill, E.T.; Dutta, C. Molecular signature of hypersaline adaptation: Insights from genome and proteome composition of halophilic prokaryotes. Genome Biol. 2008, 9, R70. [Google Scholar] [CrossRef] [PubMed]
- Barberán, A.; Fernández-Guerra, A.; Bohannan, B.J.M.; Casamayor, E.O. Exploration of community traits as ecological markers in microbial metagenomes. Mol. Ecol. 2012, 21, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Mongodin, E.F.; Nelson, K.E.; Daugherty, S.; DeBoy, R.T.; Wister, J.; Khouri, H.; Weidman, J.; Walsh, D.A.; Papke, R.T.; Sanchez Perez, G.; et al. The genome of Salinibacter ruber: Convergence and gene exchange among hyperhalophilic bacteria and archaea. Proc. Natl. Acad. Sci. USA 2005, 102, 18147–18152. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Baxter, B.K. Bipyrimidine signatures as a photoprotective genome strategy in G + C-rich halophilic archaea. Life 2016, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Bolhuis, H.; Palm, P.; Wende, A.; Falb, M.; Rampp, M.; Rodriguez-Valera, F.; Pfeiffer, F.; Oesterhelt, D. The genome of the square archaeon Haloquadratum walsbyi: Life at the limits of water activity. BMC Genom. 2006, 7, 169. [Google Scholar] [CrossRef] [PubMed]
- Hahnke, R.L.; Meier-Kolthoff, J.P.; García-López, M.; Mukherjee, S.; Huntemann, M.; Ivanova, N.N.; Woyke, T.; Kyrpides, N.C.; Klenk, H.P.; Göker, M. Genome-based taxonomic classification of Bacteroidetes. Front. Microbiol. 2016, 7, 2003. [Google Scholar] [CrossRef] [PubMed]
- Munoz, R.; Rosselló-Móra, R.; Amann, R. Revised phylogeny of Bacteroidetes and proposal of sixteen new taxa and two new combinations including Rhodothermaeota phyl. nov. Syst. Appl. Microbiol. 2017, 40, 190. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of Actinobacteria: Tracing the evolutionary history of an ancient phylum. Microbiol. Mol. Biol. Rev. 2007, 71, 495–548. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, V.; Øvreås, L.; Thingstad, T.F. Prokaryotic diversity-magnitude, dynamics, and controlling factors. Science 2002, 296, 1064–1066. [Google Scholar] [CrossRef] [PubMed]
- Smucker, A.J.M.; Wang, W.; Kravchenko, A.N.; Dick, W.A. Forms and functions of meso and micro-niches of carbon within soil aggregates. J. Nematol. 2010, 42, 84–86. [Google Scholar] [PubMed]
- Carson, J.K.; Gonzalez-Quiñones, V.; Murphy, D.V.; Hinz, C.; Shaw, J.A.; Gleeson, D.B. Low pore connectivity increases bacterial diversity in soil. Appl. Environ. Microbiol. 2010, 76, 3936–3942. [Google Scholar] [CrossRef] [PubMed]
- Imelfort, M.; Parks, D.; Woodcroft, B.J.; Dennis, P.; Hugenholtz, P.; Tyson, G.W. GroopM: An automated tool for the recovery of population genomes from related metagenomes. PeerJ 2014, 2, e603. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, N.; Xia, F.; Gilbert, J.A. Recovering complete and draft population genomes from metagenome datasets. Microbiome 2016, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.; Grant, W.D.; Jones, B.E.; Kato, C.; Li, L. Novel archaeal phylotypes from an East African alkaline saltern. Extremophiles 1999, 3, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Baricz, A.; Coman, C.; Andrei, A.Ş.; Muntean, V.; Keresztes, Z.G.; Păuşan, M.; Alexe, M.; Banciu, H.L.; Pǎuşan, M.; Alexe, M.; et al. Spatial and temporal distribution of archaeal diversity in meromictic, hypersaline Ocnei Lake (Transylvanian Basin, Romania). Extremophiles 2014, 18, 399–413. [Google Scholar] [CrossRef] [PubMed]
- dC Rubin, S.S.; Marín, I.; Gómez, M.J.; Morales, E.A.; Zekker, I.; San Martín-Uriz, P.; Rodríguez, N.; Amils, R. Prokaryotic diversity and community composition in the Salar de Uyuni, a large scale, chaotropic salt flat. Environ. Microbiol. 2017, 19, 3745–3754. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, L.; Santos, F.; Gomariz, M.; Almansa, C.; López, C.; Antón, J.; Nercessian, D. Seasonal dynamics of extremely halophilic microbial communities in three Argentinian salterns. FEMS Microbiol. Ecol. 2016, 92, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Finstad, K.M.; Probst, A.J.; Thomas, B.C.; Andersen, G.L.; Demergasso, C.; Echeverría, A.; Amundson, R.G.; Banfield, J.F. Microbial community structure and the persistence of cyanobacterial populations in salt crusts of the hyperarid Atacama desert from genome-resolved metagenomics. Front. Microbiol. 2017, 8, 1435. [Google Scholar] [CrossRef] [PubMed]
- Gomariz, M.; Martínez-García, M.; Santos, F.; Rodriguez, F.; Capella-Gutiérrez, S.; Gabaldón, T.; Rosselló-Móra, R.; Meseguer, I.; Antón, J. From community approaches to single-cell genomics: The discovery of ubiquitous hyperhalophilic Bacteroidetes generalists. ISME J. 2015, 9, 16–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-García, M.; Santos, F.; Moreno-Paz, M.; Parro, V.; Antón, J. Unveiling viral-host interactions within the “microbial dark matter”. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vogt, J.C.; Abed, R.M.M.; Albach, D.C.; Palinska, K.A. Bacterial and archaeal diversity in hypersaline cyanobacterial mats along a transect in the intertidal flats of the Sultanate of Oman. Microb. Ecol. 2018, 75, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Zhaxybayeva, O.; Stepanauskas, R.; Mohan, N.R.; Papke, R.T. Cell sorting analysis of geographically separated hypersaline environments. Extremophiles 2013, 17, 265–275. [Google Scholar] [CrossRef] [PubMed]
- De la Haba, R.R.; Sánchez-Porro, C.; Márquez, M.C.; Ventosa, A. Taxonomy of halophiles. In Extremophiles Handboook; Springer: Berlin/Heidelberg, Germany, 2010; pp. 255–309. ISBN 978-4-431-53897-4. [Google Scholar]
- Shi, Y.; Adams, J.M.; Ni, Y.; Yang, T.; Jing, X.; Chen, L. The biogeography of soil archaeal communities on the eastern Tibetan Plateau. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Legault, B.A.; Lopez-Lopez, A.; Alba-Casado, J.C.; Doolittle, W.F.; Bolhuis, H.; Rodriguez-Valera, F.; Papke, R.T. Environmental genomics of Haloquadratum walsbyi in a saltern crystallizer indicates a large pool of accessory genes in an otherwise coherent species. BMC Genom. 2006, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Enache, M.; Popescu, G.; Itoh, T.; Kamekura, M. Halophilic microorganisms from man-made and natural hypersaline environments: Physiology, ecology, and biotechnological potential. In Adaption of Microbial Life to Environmental Extremes: Novel Research Results and Application; Stan-Lotter, H., Fendrihan, S., Eds.; Springer: Vienna, Austria, 2012; pp. 173–197. ISBN 978-3-211-99691-1. [Google Scholar]
- Ma, B.; Gong, J. A meta-analysis of the publicly available bacterial and archaeal sequence diversity in saline soils. World J. Microbiol. Biotechnol. 2013, 29, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.-Y.; Cohoon, M.; Disz, T.; Edwards, R.; Fonstein, M.; Frank, E.D.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, V.; Øvreås, L. Microbial diversity, life strategies, and adaptations to life in extreme soils. In Microbiology of Extreme Soils; Dion, P., Nautiyal, S., Eds.; Springer: Berlin, Germany, 2008; pp. 15–43. [Google Scholar]
- Dechesne, A.; Wang, G.; Gulez, G.; Or, D.; Smets, B.F. Hydration-controlled bacterial motility and dispersal on surfaces. Proc. Natl. Acad. Sci. USA 2010, 107, 14369–14372. [Google Scholar] [CrossRef] [PubMed]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 2010, 8, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, G.A.; Morgan, J.A.W.; Whipps, J.M.; Saunders, J.R. The role of motility in the in vitro attachment of Pseudomonas putida PaW8 to wheat roots. FEMS Microbiol. Ecol. 2001, 35, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Limoli, D.H.; Jones, C.J.; Wozniak, D.J.; Cruz, S. Bacterial extracellular polysaccharides in biofilm formation and function. Microbiol. Spectr. 2015, 3, 1–30. [Google Scholar] [CrossRef]
- Abrudan, M.I.; Smakman, F.; Grimbergen, A.J.; Westhoff, S.; Miller, E.L.; van Wezel, G.P.; Rozen, D.E. Socially mediated induction and suppression of antibiosis during bacterial coexistence. Proc. Natl. Acad. Sci. USA 2015, 112, 11054–11059. [Google Scholar] [CrossRef] [PubMed]
- Tyc, O.; Song, C.; Dickschat, J.S.; Vos, M.; Garbeva, P. The ecological role of volatile and soluble secondary metabolites produced by soil bacteria. Trends Microbiol. 2018, 25, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Aanderud, Z.T.; Vert, J.C.; Lennon, J.T.; Magnusson, T.W.; Breakwell, D.P.; Harker, A.R. Bacterial dormancy is more prevalent in freshwater than hypersaline lakes. Front. Microbiol. 2016, 7, 853. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. Microbial life at high salt concentrations: Phylogenetic and metabolic diversity. Saline Syst. 2008, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Ciulla, R.A.; Diaz, M.R.; Taylor, B.F.; Roberts, M.F. Organic osmolytes in aerobic bacteria from Mono Lake, an alkaline, moderately hypersaline environment. Appl. Environ. Microbiol. 1997, 63, 220–226. [Google Scholar] [PubMed]
- Nesme, J.; Achouak, W.; Agathos, S.N.; Bailey, M.; Baldrian, P.; Brunel, D.; Frostegård, Å.; Heulin, T.; Jansson, J.K.; Jurkevitch, E.; et al. Back to the future of soil metagenomics. Front. Microbiol. 2016, 7, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, E.A.; Seitzer, P.M.; Tritt, A.; Larsen, D.; Krusor, M.; Yao, A.I.; Wu, D.; Madern, D.; Eisen, J.A.; Darling, A.E.; et al. Phylogenetically driven sequencing of extremely halophilic archaea reveals strategies for static and dynamic osmo-response. PLoS Genet. 2014, 10, e1004784. [Google Scholar] [CrossRef] [PubMed]
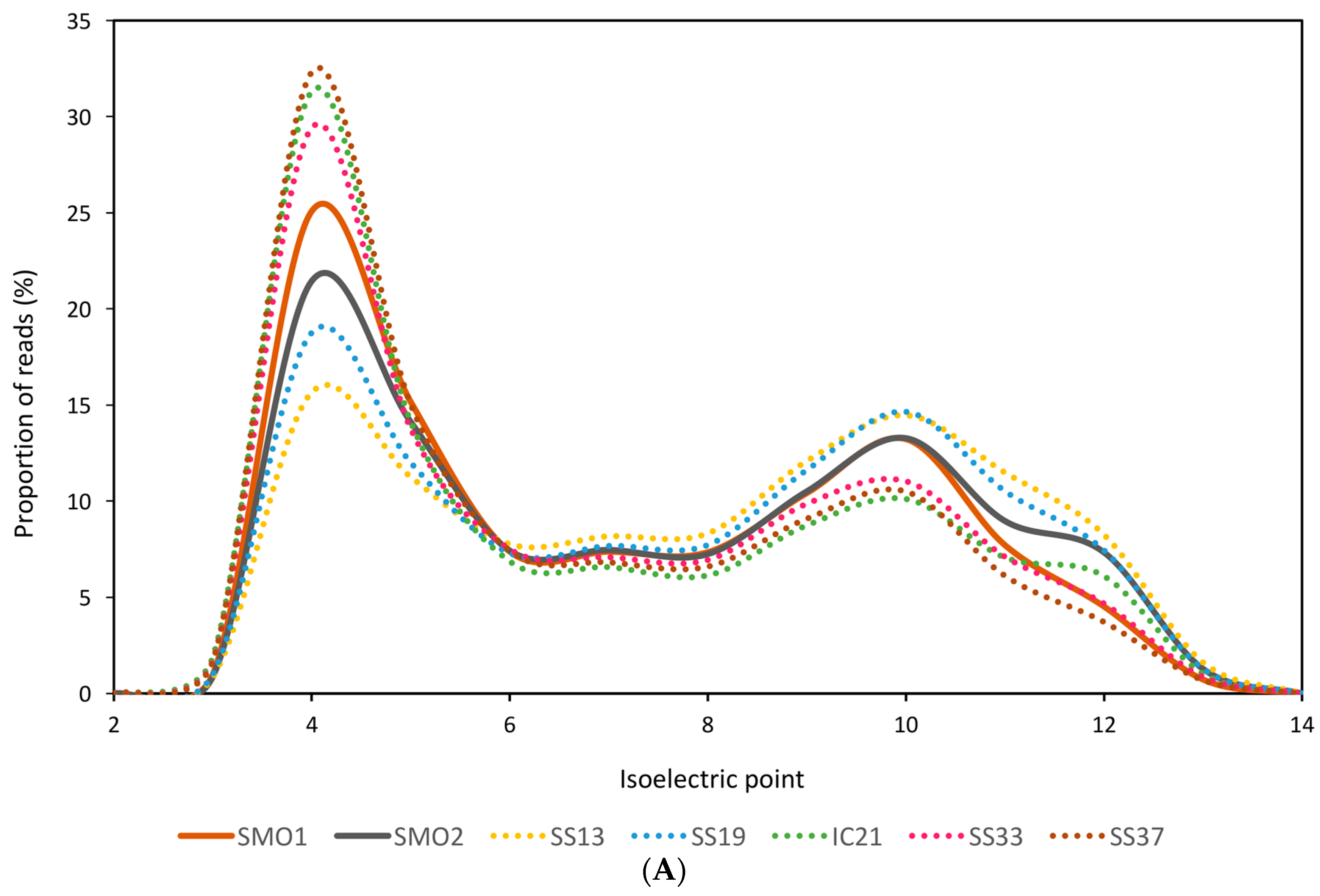
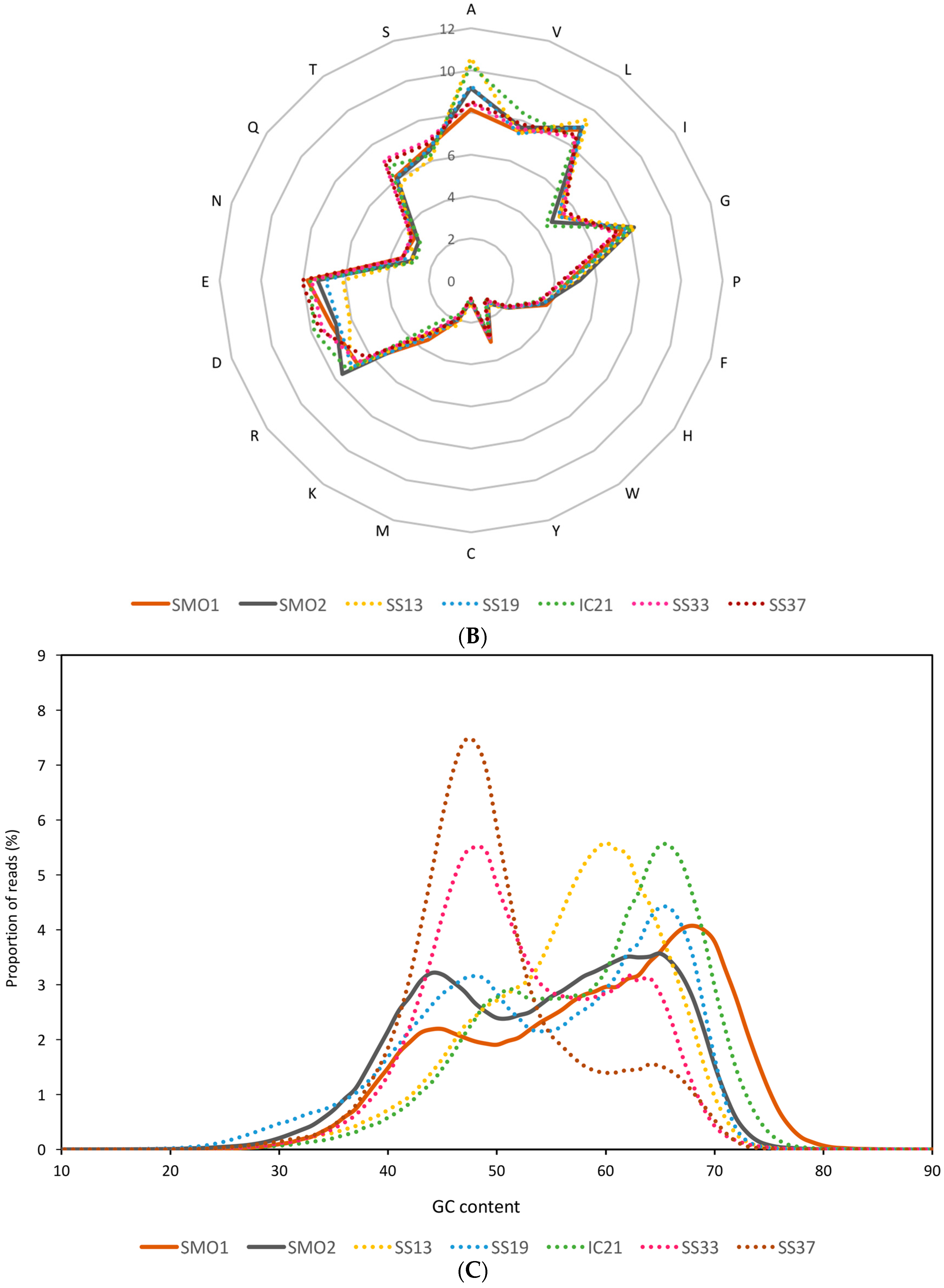
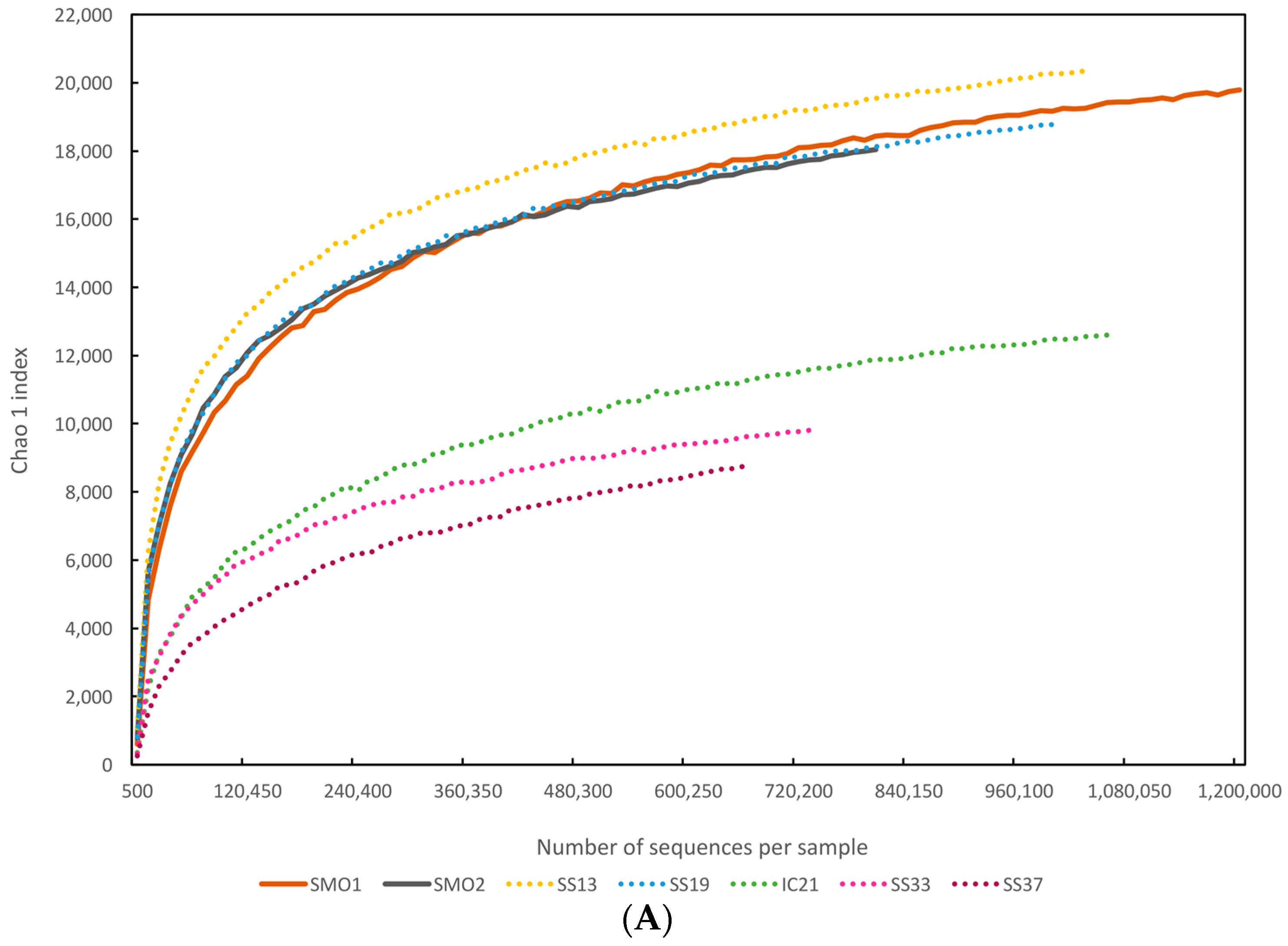
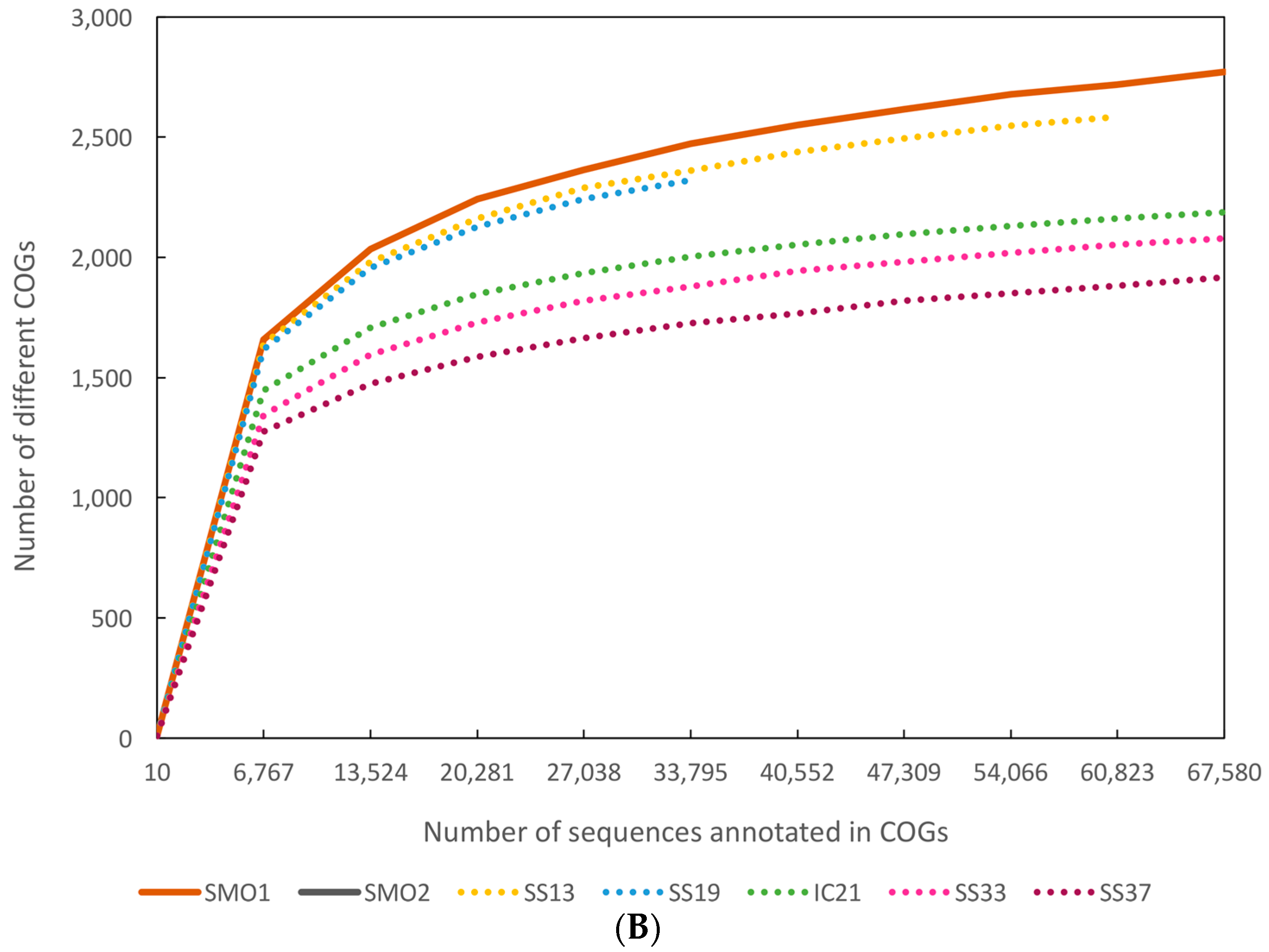

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database Name | Habitat | Salinity | pH | No. of Sequences | Avg. Read Length (bp) | Percent of Assembled Reads (%) | No. of Contigs >1 kb/N50 (bp) | Ref. |
|---|---|---|---|---|---|---|---|---|
| SMO1 | Saline soil | 24.0 dS/m | 7.8 | 1,289,630 | 628 | 26.7 | 25,001/1857 | [59] |
| SMO2 | Saline soil | 54.4 dS/m | 8.9 | 839,941 | 629 | |||
| SS13 | Saltern | 13% NaCl | 8.0 | 1,481,803 | 305 | 63.7 | 8193/2280 | [23] |
| SS19 | Saltern | 19% NaCl | 8.0 | 1,241,633 | 361 | 65.3 | 12,742/2429 | [22] |
| SS33 | Saltern | 33% NaCl | 7.0 | 963,381 | 367 | 59.0 | 6040/2069 | [23] |
| SS37 | Saltern | 37% NaCl | 7.1 | 736,936 | 417 | 67.0 | 6589/2336 | [22] |
| IC21 | Saltern | 21% NaCl | 7.5 | 1,191,373 | 397 | 69.2 | 9270/2793 | [56] |
| SMO1 Database | SMO2 Database | ||
|---|---|---|---|
| Haloarcula | 12.7% | Salinimicrobium | 9.2% |
| Halorubrum | 10.3% | Salinigranum | 9.0% |
| Fodinibius | 9.0% | Halolamina | 8.7% |
| Halolamina | 7.4% | Haloarcula | 8.4% |
| Salinigranum | 7.1% | Unclassified Halobacteria | 6.3% |
| Unclassified Halobacteria | 6.7% | Halobellus | 4.6% |
| Salinibacter | 4.1% | Unclassified Gammaproteobacteria | 4.1% |
| Gracilimonas | 3.4% | Gracilimonas | 3.0% |
| Halohasta | 3.4% | Halorubrum | 2.7% |
| Halapricum | 2.7% | Halomicroarcula | 2.2% |
| Halobellus | 2.5% | Marinobacter | 2.2% |
| Natronomonas | 2.5% | Natronomonas | 2.2% |
| Halonotius | 2.1% | Pseudidiomarina | 2.2% |
| Haloplanus | 2.1% | Altererythrobacter | 1.9% |
| Halorientalis | 1.9% | Halomarina | 1.9% |
| Halorubellus | 1.8% | Haloplanus | 1.9% |
| Halomicroarcula | 1.6% | Halorientalis | 1.9% |
| Salinimicrobium | 1.6% | Fodinibius | 1.6% |
| Unclassified Ectothiorhodospiraceae | 1.2% | Halomonas | 1.6% |
| Unclassified Gammaproteobacteria | 1.2% | Nocardiopsis | 1.6% |
| Halalkalicoccus | 1.1% | Unclassified Flavobacteriaceae | 1.4% |
| Halomonas | 1.1% | Idiomarina | 1.1% |
| Others | 12.7% | Others | 20.4% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vera-Gargallo, B.; Ventosa, A. Metagenomic Insights into the Phylogenetic and Metabolic Diversity of the Prokaryotic Community Dwelling in Hypersaline Soils from the Odiel Saltmarshes (SW Spain). Genes 2018, 9, 152. https://doi.org/10.3390/genes9030152
Vera-Gargallo B, Ventosa A. Metagenomic Insights into the Phylogenetic and Metabolic Diversity of the Prokaryotic Community Dwelling in Hypersaline Soils from the Odiel Saltmarshes (SW Spain). Genes. 2018; 9(3):152. https://doi.org/10.3390/genes9030152
Chicago/Turabian StyleVera-Gargallo, Blanca, and Antonio Ventosa. 2018. "Metagenomic Insights into the Phylogenetic and Metabolic Diversity of the Prokaryotic Community Dwelling in Hypersaline Soils from the Odiel Saltmarshes (SW Spain)" Genes 9, no. 3: 152. https://doi.org/10.3390/genes9030152